
Silencing CDCA8 Suppresses Hepatocellular Carcinoma Growth and Stemness via Restoration of ATF3 Tumor Suppressor and Inactivation of AKT/β–Catenin Signaling

 , ,
, ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Big Data Analysis of CDCA8 Expression and Its Prognostic Value in HCC
2.2. Silencing of CDCA8 Inhibits HCC Cell Growth, Colony Formation and Migration
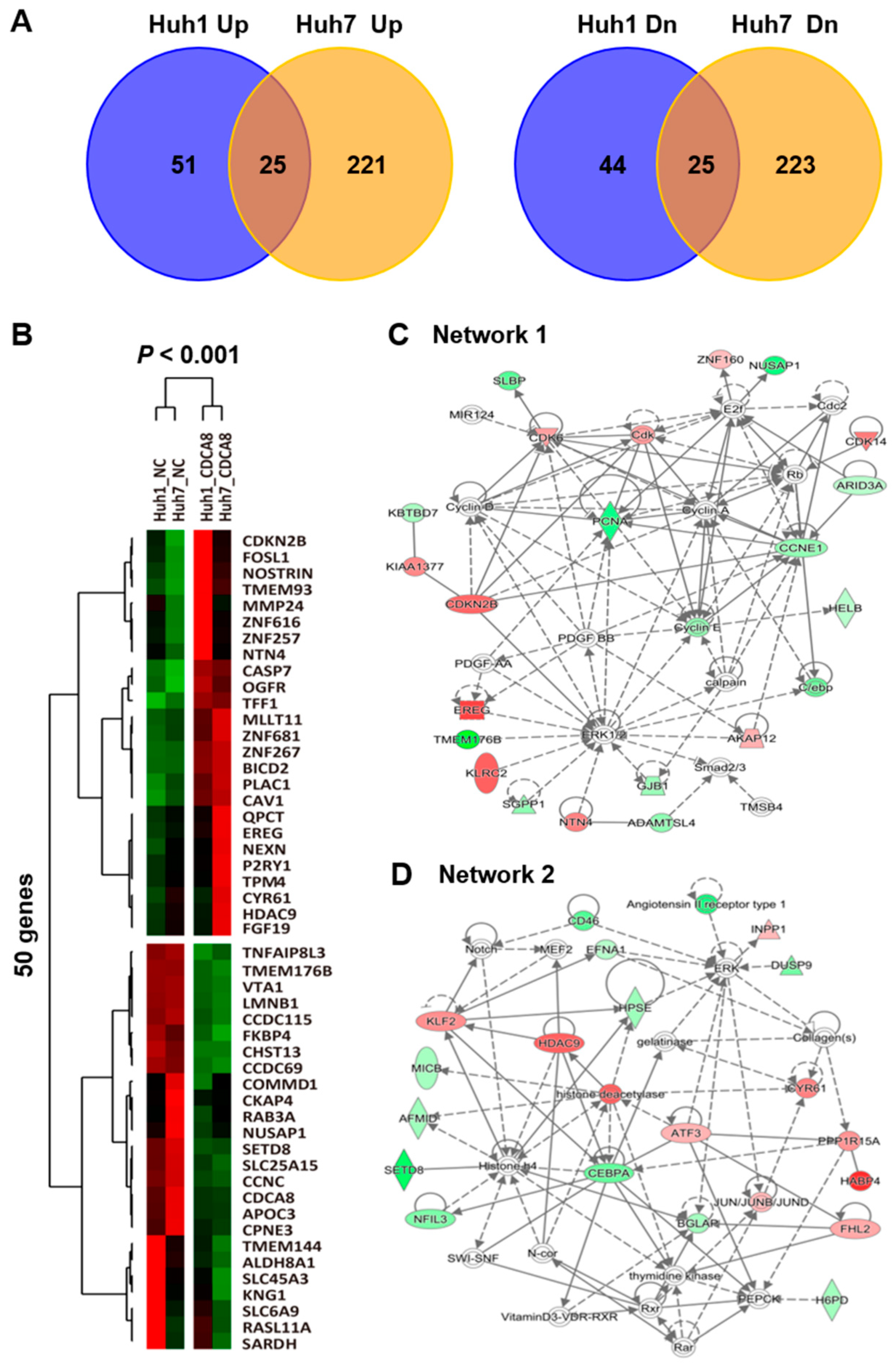
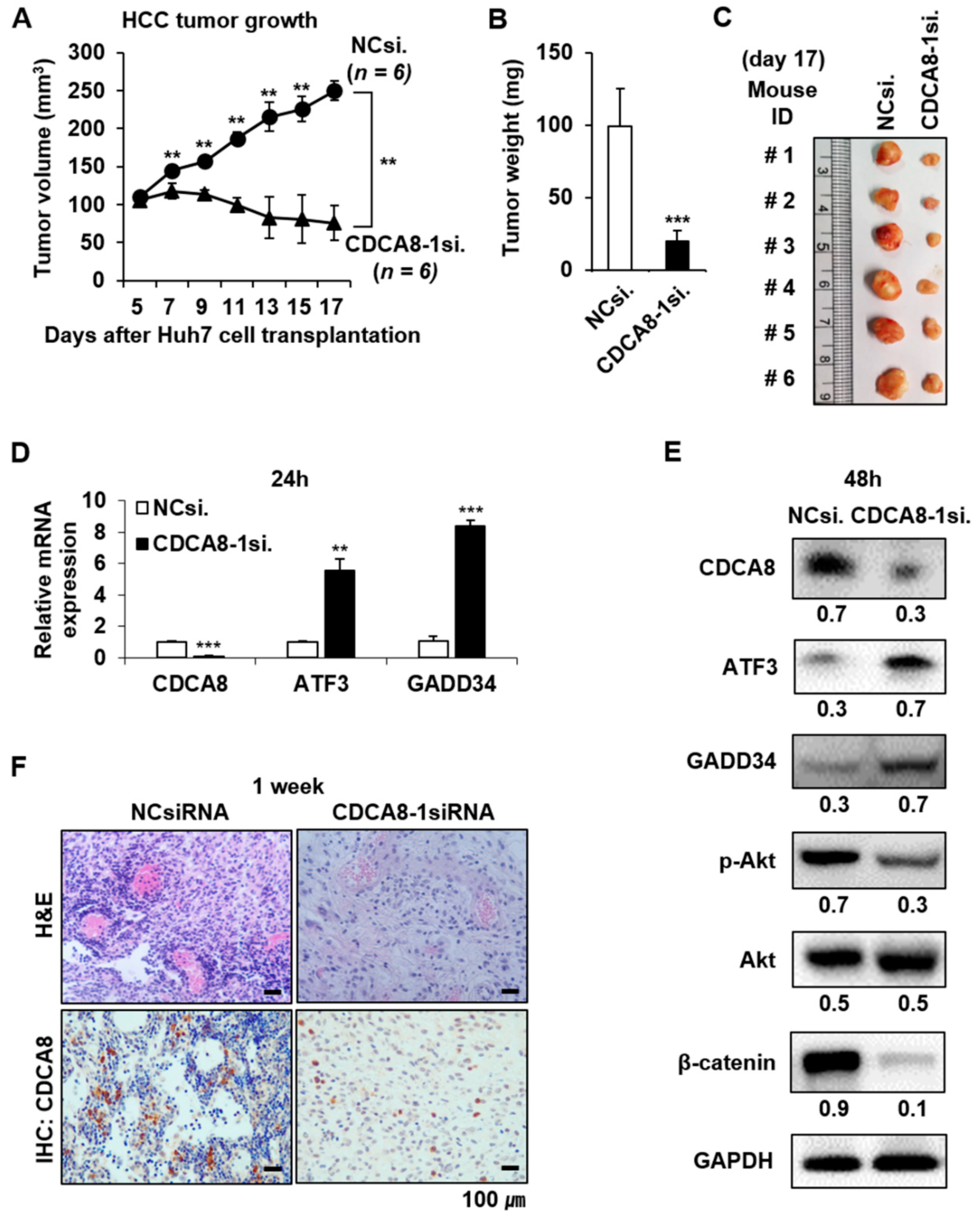
2.3. CDCA8 Knockdown Upregulates ATF3 and GADD34 Tumor Suppressors to Exert Apoptotic Progression
2.4. CDCA8 Knockdown Inactivates AKT/β–Catenin Signaling and Reduces Stemness
2.5. Targeting CDCA8 Suppresses HCC Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines and siRNA Transfection
4.2. Cell Proliferation and Clonogenic Assay
4.3. Quantitative RT-PCR
4.4. Cell Cycle Analysis
4.5. Wound Healing Assay
4.6. Sphere Forming Assay
4.7. RNA Sequencing and Functional Network Analysis
4.8. Magnetic Cell Sorting and Flow Cytometry
4.9. Plasmid Transfection and Overexpression of CDCA8
4.10. In Vivo Tumor Growth
4.11. Western Blot Analysis
4.12. Immunohistochemistry
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Roberts, L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H. Hepatocellular carcinoma development in cirrhosis. Best Pract. Res. Clin. Gastroenterol. 2007, 21, 161–173. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef]
- Befeler, A.S.; Di Bisceglie, A.M. Hepatocellular carcinoma: Diagnosis and treatment. Gastroenterology 2002, 122, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef]
- Roberts, L.R.; Gores, G.J. Hepatocellular carcinoma: Molecular pathways and new therapeutic targets. Semin. Liver Dis. 2005, 25, 212–225. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Gassmann, R.; Carvalho, A.; Henzing, A.J.; Ruchaud, S.; Hudson, D.F.; Honda, R.; Nigg, E.A.; Gerloff, D.L.; Earnshaw, W.C. Borealin: A novel chromosomal passenger required for stability of the bipolar mitotic spindle. J. Cell Biol. 2004, 166, 179–191. [Google Scholar] [CrossRef]
- Ruchaud, S.; Carmena, M.; Earnshaw, W.C. Chromosomal passengers: Conducting cell division. Nat. Rev. Mol. Cell Biol. 2007, 8, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Z.; Bao, X.; Fang, Y.; Ni, P.; Chen, Q.; Zhang, W.; Deng, A. Borealin/Dasra B is overexpressed in colorectal cancers and contributes to proliferation of cancer cells. Med. Oncol. 2014, 31, 248. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Miao, C.X.; Xu, X.M.; Liu, L.J.; Gu, Y.F.; Zhou, D.; Chen, L.S.; Lin, G.; Lu, G.X. Transcriptional activation of human CDCA8 gene regulated by transcription factor NF-Y in embryonic stem cells and cancer cells. J. Biol. Chem. 2015, 290, 22423–22434. [Google Scholar] [CrossRef] [PubMed]
- Won, C.; Kim, B.H.; Yi, E.H.; Choi, K.J.; Kim, E.K.; Jeong, J.M.; Lee, J.H.; Jang, J.J.; Yoon, J.H.; Jeong, W.I.; et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology 2015, 62, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Kittler, R.; Pelletier, L.; Heninger, A.K.; Slabicki, M.; Theis, M.; Miroslaw, L.; Poser, I.; Lawo, S.; Grabner, H.; Kozak, K.; et al. Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat. Cell Biol. 2007, 9, 1401–1412. [Google Scholar] [CrossRef]
- Xie, J.; Xie, Y.M.; Chen, B.; Pan, F.; Guo, J.C.; Zhao, Q.; Shen, J.H.; Wu, Z.Y.; Wu, J.Y.; Xu, L.Y.; et al. ATF3 functions as a novel tumor suppressor with prognostic significance in esophageal squamous cell carcinoma. Oncotarget 2014, 5, 8569–8582. [Google Scholar] [CrossRef]
- Morton, E.; Macrae, I.M.; McCabe, C.; Brown, S.M.; White, F. Identification of the growth arrest and DNA damage protein GADD34 in the normal human heart and demonstration of alterations in expression following myocardial ischaemia. Int. J. Cardiol. 2006, 107, 126–129. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Farook, J.M.; Shields, J.; Tawfik, A.; Markand, S.; Sen, T.; Smith, S.B.; Brann, D.; Dhandapani, K.M.; Sen, N. GADD34 induces cell death through inactivation of Akt following traumatic brain injury. Cell Death Dis. 2013, 4, e754. [Google Scholar] [CrossRef]
- Chin, Y.R.; Toker, A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal. 2009, 21, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.J.; Lin, W.H.; Chang, Y.W.; Wei, K.C.; Liang, C.J.; Chen, S.C.; Lee, J.L. Polarized cell migration induces cancer type-specific CD133/integrin/Src/Akt/GSK3β/β-catenin signaling required for maintenance of cancer stem cell properties. Oncotarget 2016, 7, 52613. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.W.; Wo, J.Y.W.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.L.; Chen, T.H.; Wang, C.F.; Chiang, Y.H.; Huang, Y.L.; Wong, F.H.; Chou, C.K.; Chen, C.M. Borealin/Dasra B is a cell cycle-regulated chromosomal passenger protein and its nuclear accumulation is linked to poor prognosis for human gastric cancer. Exp. Cell Res. 2006, 312, 962–973. [Google Scholar] [CrossRef]
- Hayama, S.; Daigo, Y.; Yamabuki, T.; Hirata, D.; Kato, T.; Miyamoto, M.; Ito, T.; Tsuchiya, E.; Kondo, S.; Nakamura, Y. Phosphorylation and activation of cell division cycle associated 8 by aurora kinase B plays a significant role in human lung carcinogenesis. Cancer Res. 2007, 67, 4113–4122. [Google Scholar] [CrossRef]
- Hackl, C.; Lang, S.A.; Moser, C.; Mori, A.; Fichtner-Feigl, S.; Hellerbrand, C.; Dietmeier, W.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Activating transcription factor-3 (ATF3) functions as a tumor suppressor in colon cancer and is up-regulated upon heat-shock protein 90 (Hsp90) inhibition. BMC Cancer 2010, 10, 668. [Google Scholar] [CrossRef]
- Chen, C.C.; Young, J.L.; Monzon, R.I.; Chen, N.; Todorovic, V.; Lau, L.F. Cytotoxicity of TNFalpha is regulated by integrin-mediated matrix signaling. EMBO J. 2007, 26, 1257–1267. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Kanneganti, T.D. Caspase-7: A protease involved in apoptosis and inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 21–24. [Google Scholar] [CrossRef]
- Fernandez-Zapico, M.E.; Lomberk, G.A.; Tsuji, S.; DeMars, C.J.; Bardsley, M.R.; Lin, Y.H.; Almada, L.L.; Han, J.J.; Mukhopadhyay, D.; Ordog, T.; et al. A functional family-wide screening of SP/KLF proteins identifies a subset of suppressors of KRAS-mediated cell growth. Biochem. J. 2011, 435, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Kayed, H.; Bekasi, S.; Keleg, S.; Michalski, C.W.; Giese, T.; Friess, H.; Kleef, J. BGLAP is expressed in pancreatic cancer cells and increases their growth and invasion. Mol. Cancer 2007, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, C.; Cormier-Daire, V. The ADAMTS(L) family and human genetic disorders. Hum. Mol. Genet. 2011, 20, R163–R167. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.L.; Zheng, L.X.; Baldwin, A.; Ohta, T.; Hurt, M.M.; Marzluff, W.F. Stem-loop binding protein, the protein that binds the 3’ end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms. Mol. Cell Biol. 2000, 20, 4188–4198. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Van Tine, B.A.; Wei, Y.; Garrett, M.D.; Nelson, D.; Adams, P.D.; Wang, J.; Qin, J.; Chow, L.T.; Harper, J.W. Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev. 2000, 14, 2298–2313. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Factor, V.M.; Thorgeirsson, S.S. Epigenetic regulation of cancer stem cells in liver cancer: Current concepts and clinical implications. J. Hepatol. 2010, 53, 568–577. [Google Scholar] [CrossRef]
- Ma, S. Biology and clinical implications of CD133+ liver cancer stem cells. Exp. Cell Res. 2013, 319, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef]
- Baik, I.H.; Jo, G.H.; Seo, D.; Ko, M.J.; Cho, C.H.; Lee, M.G.; Lee, Y.H. Knockdown of RPL9 expression inhibits colorectal carcinoma growth via the inactivation of Id-1/NF-κB signaling axis. Int. J. Oncol. 2016, 49, 1953–1962. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, T.; Ko, M.J.; Seo, Y.-R.; Jung, S.-J.; Seo, D.; Park, S.-Y.; Park, K.U.; Kim, K.S.; Kim, M.; Seo, J.H.; et al. Silencing CDCA8 Suppresses Hepatocellular Carcinoma Growth and Stemness via Restoration of ATF3 Tumor Suppressor and Inactivation of AKT/β–Catenin Signaling. Cancers 2021, 13, 1055. https://doi.org/10.3390/cancers13051055
Jeon T, Ko MJ, Seo Y-R, Jung S-J, Seo D, Park S-Y, Park KU, Kim KS, Kim M, Seo JH, et al. Silencing CDCA8 Suppresses Hepatocellular Carcinoma Growth and Stemness via Restoration of ATF3 Tumor Suppressor and Inactivation of AKT/β–Catenin Signaling. Cancers. 2021; 13(5):1055. https://doi.org/10.3390/cancers13051055
Chicago/Turabian StyleJeon, Taewon, Min Ji Ko, Yu-Ri Seo, Soo-Jung Jung, Daekwan Seo, So-Young Park, Keon Uk Park, Kwang Seok Kim, Mikyung Kim, Ji Hae Seo, and et al. 2021. "Silencing CDCA8 Suppresses Hepatocellular Carcinoma Growth and Stemness via Restoration of ATF3 Tumor Suppressor and Inactivation of AKT/β–Catenin Signaling" Cancers 13, no. 5: 1055. https://doi.org/10.3390/cancers13051055
APA StyleJeon, T., Ko, M. J., Seo, Y.-R., Jung, S.-J., Seo, D., Park, S.-Y., Park, K. U., Kim, K. S., Kim, M., Seo, J. H., Park, I.-C., Kim, M.-J., Bae, J.-H., Song, D.-K., Cho, C. H., Lee, J.-H., & Lee, Y.-H. (2021). Silencing CDCA8 Suppresses Hepatocellular Carcinoma Growth and Stemness via Restoration of ATF3 Tumor Suppressor and Inactivation of AKT/β–Catenin Signaling. Cancers, 13(5), 1055. https://doi.org/10.3390/cancers13051055