
The Oncogene MYCN Modulates Glycolytic and Invasive Genes to Enhance Cell Viability and Migration in Human Retinoblastoma

 , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Cell Culture
2.3. Immunoblotting
2.4. Immunohistochemistry
2.5. Lentiviral Mediated shRNA Knockdown
2.6. Determination of Cell Viability
2.7. Apoptosis Assay
2.8. Cell Cycle Analysis
2.9. Glucose Uptake
2.10. Lactate Assay
2.11. Measurement of Reactive Oxygen Species (ROS)
2.12. Drug Combination Assay
2.13. Wound Healing Assay
2.14. Microarray Analysis
2.14.1. Data Interpretation and Identification of Differentially Expressed Genes (DEGs)
2.14.2. Gene Ontology (GO), Pathway and Function Enrichment Analysis of DEGs
2.15. RNA Isolation and Validation of Identified Genes
2.16. Gelatin Zymography
2.17. Statistical Analysis
3. Results
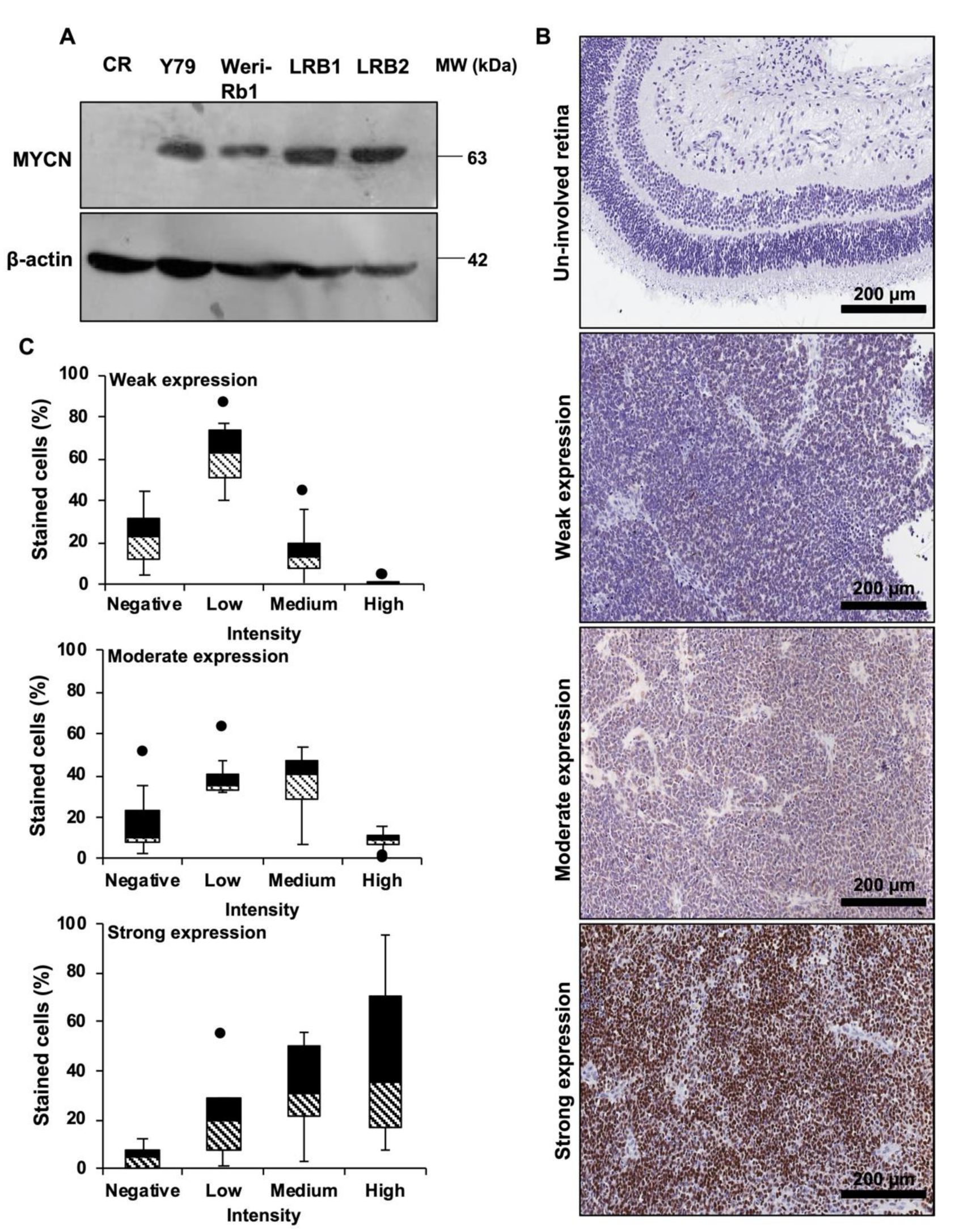
3.1. Overexpression of MYCN in Cell Lines and Enucleated Patient Specimens of Human Retinoblastoma
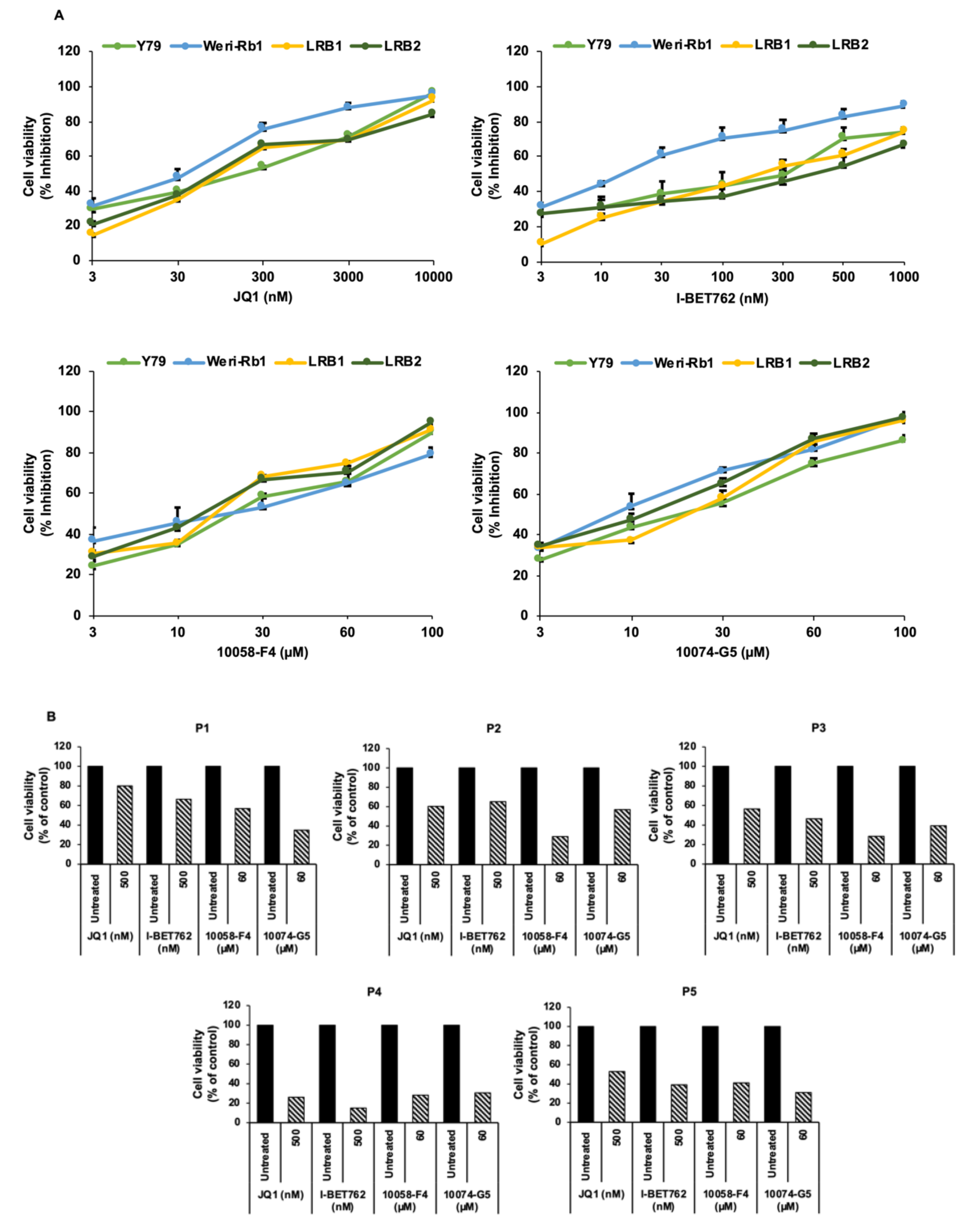
3.2. Pharmacological Inhibition of MYCN Led to Decreased RB Cell Viability
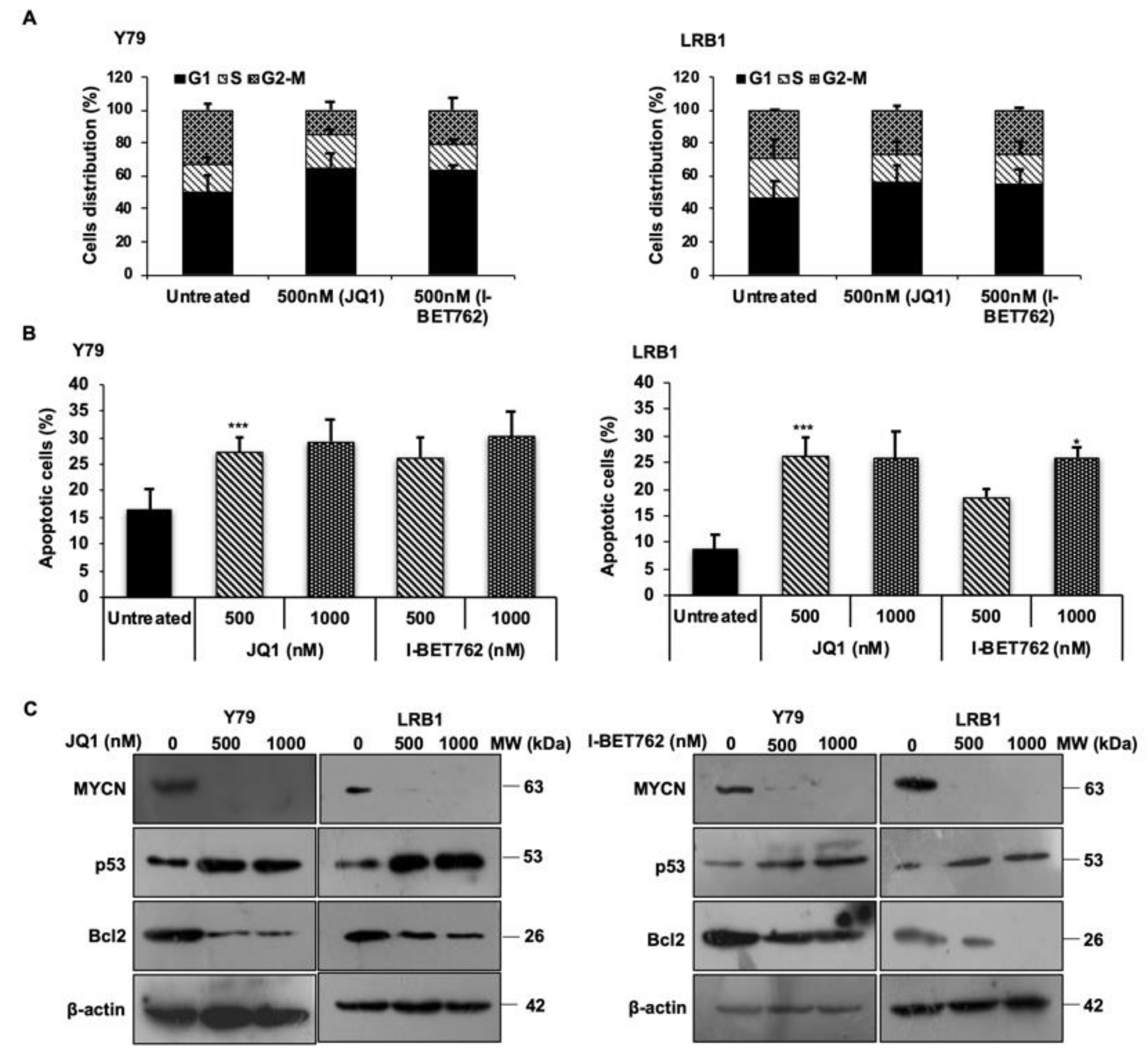
3.3. MYCN Inhibition Induces Cell Cycle Arrest at G0/G1 Phase and Triggers Apoptosis in RB Cells
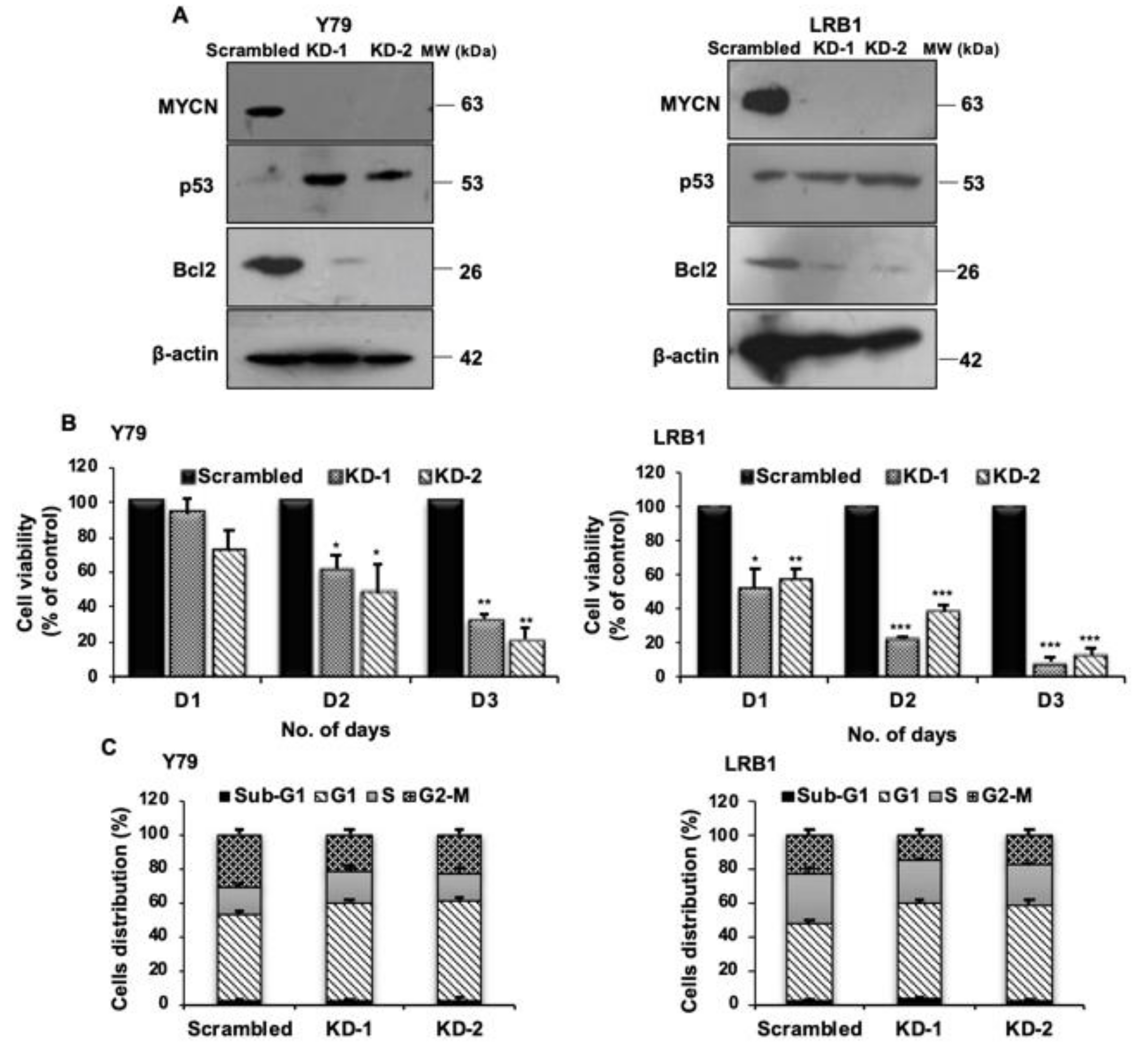
3.4. shRNA Mediated Knockdown of MYCN Inhibits Cell Proliferation in RB
3.5. Microarray Analysis upon MYCN Knockdown
3.5.1. Identification of Differentially Expressed Genes (DEGs), Gene Ontology (GO), Enriched Functions and Pathway Analysis
3.5.2. Screening of the Identified DEGs Related to Glucose Metabolism and Migration
Validation of Identified Metabolic Genes
Effect of MYCN Inhibition on Metabolic Parameters
MYCN Regulates Genes Involved in Cellular Migration and Metastasis
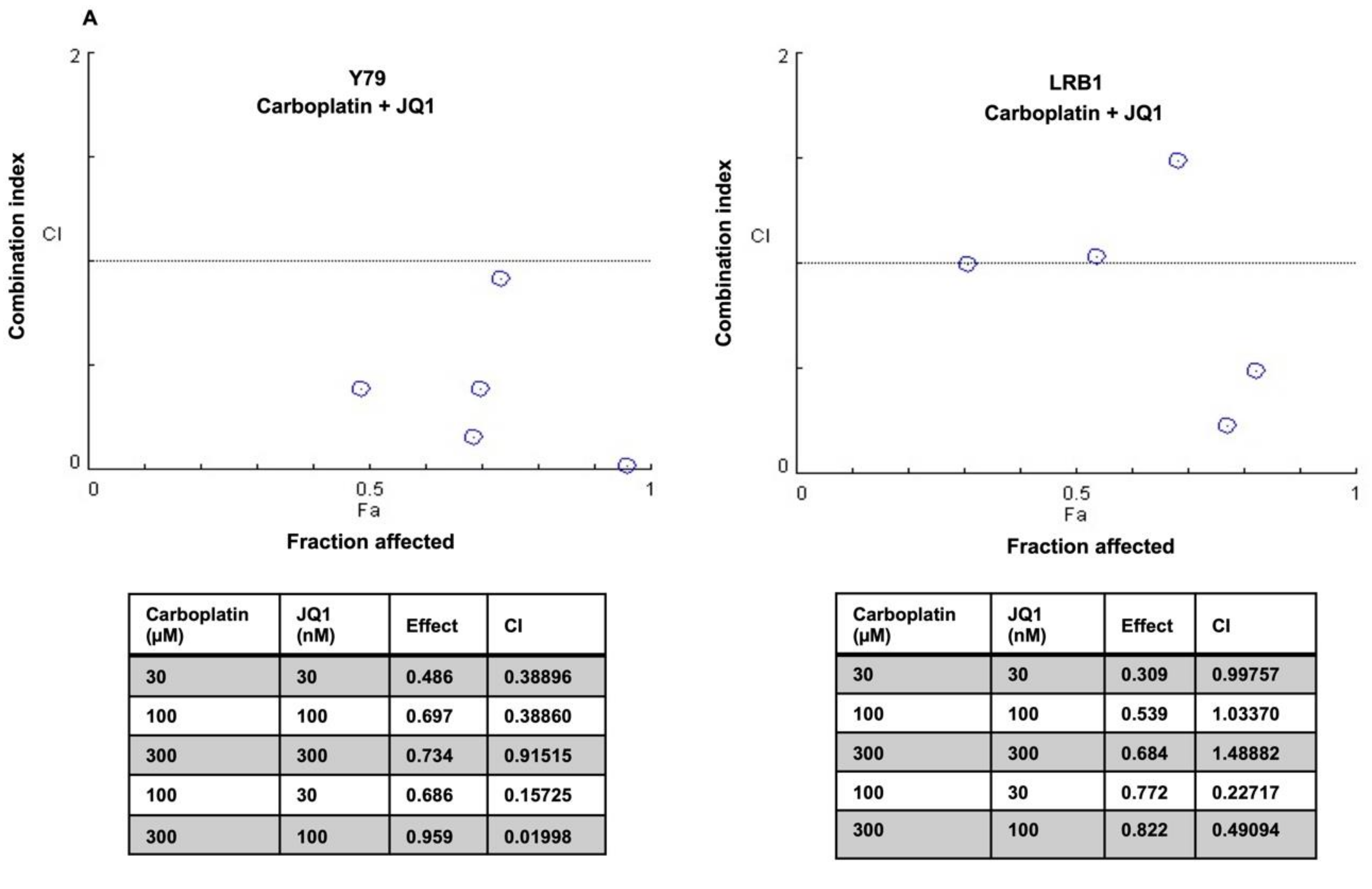
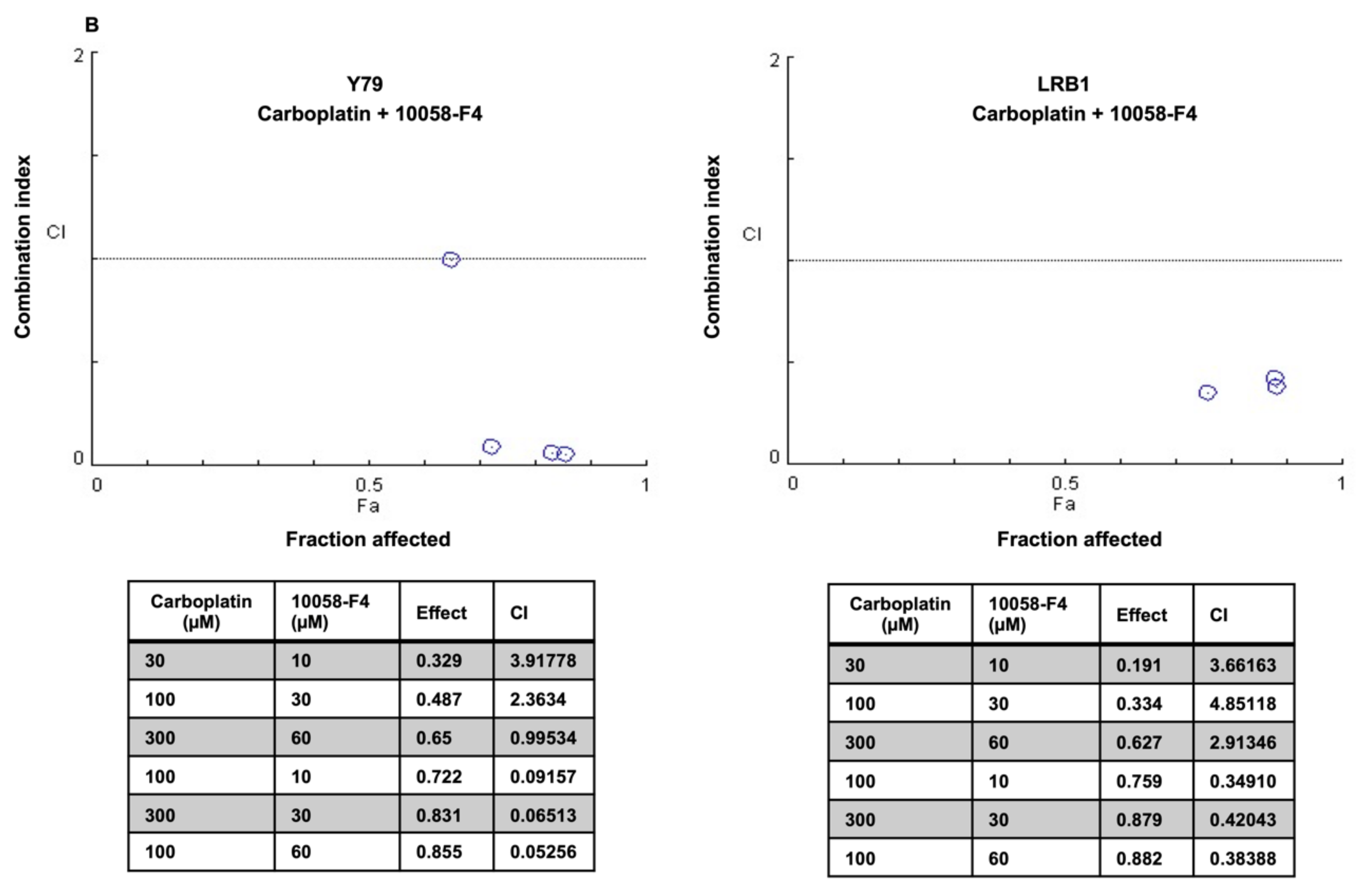
3.6. Drug Combination Effects of MYCN Inhibitors and Carboplatin on Retinoblastoma Cell Viability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shields, C.L.; Shields, J.A. Diagnosis and Management of Retinoblastoma. Cancer Control 2004, 11, 317–327. [Google Scholar] [CrossRef]
- Dryja, T.P.; Friend, S.; Weinberg, R.A. Genetic Sequences That Predispose to Retinoblastoma and Osteosarcoma. Symp. Fundam. Cancer Res. 1986, 39, 115–119. [Google Scholar]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A Human DNA Segment with Properties of the Gene That Predisposes to Retinoblastoma and Osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef]
- Rushlow, D.E.; Mol, B.M.; Kennett, J.Y.; Yee, S.; Pajovic, S.; Thériault, B.L.; Prigoda-Lee, N.L.; Spencer, C.; Dimaras, H.; Corson, T.W.; et al. Characterisation of Retinoblastomas without RB1 Mutations: Genomic, Gene Expression, and Clinical Studies. Lancet Oncol. 2013, 14, 327–334. [Google Scholar] [CrossRef]
- Zugbi, S.; Ganiewich, D.; Bhattacharyya, A.; Aschero, R.; Ottaviani, D.; Sampor, C.; Cafferata, E.G.; Mena, M.; Sgroi, M.; Winter, U.; et al. Clinical, Genomic, and Pharmacological Study of MYCN-Amplified RB1 Wild-Type Metastatic Retinoblastoma. Cancers 2020, 12, 2714. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, D.W. The Retinoblastoma Tumor-Suppressor Gene, the Exception That Proves the Rule. Oncogene 2006, 25, 5233–5243. [Google Scholar] [CrossRef] [PubMed]
- Dimaras, H.; Khetan, V.; Halliday, W.; Orlic, M.; Prigoda, N.L.; Piovesan, B.; Marrano, P.; Corson, T.W.; Eagle, R.C.; Squire, J.A.; et al. Loss of RB1 Induces Non-Proliferative Retinoma: Increasing Genomic Instability Correlates with Progression to Retinoblastoma. Hum. Mol. Genet. 2008, 17, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Thériault, B.L.; Dimaras, H.; Gallie, B.L.; Corson, T.W. The Genomic Landscape of Retinoblastoma: A Review. Clin. Exp. Ophthalmol. 2014, 42, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Stenfelt, S.; Blixt, M.K.E.; All-Ericsson, C.; Hallböök, F.; Boije, H. Heterogeneity in Retinoblastoma: A Tale of Molecules and Models. Clin. Transl. Med. 2017, 6, 42. [Google Scholar] [CrossRef]
- Afshar, A.R.; Pekmezci, M.; Bloomer, M.M.; Cadenas, N.J.; Stevers, M.; Banerjee, A.; Roy, R.; Olshen, A.B.; van Ziffle, J.; Onodera, C.; et al. Next-Generation Sequencing of Retinoblastoma Identifies Pathogenic Alterations beyond RB1 Inactivation That Correlate with Aggressive Histopathologic Features. Ophthalmology 2020, 127, 804–813. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, Metabolism, Cell Growth, and Tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.K.; Huang, J.; Ma, A.; Kretzner, L.; Alt, F.W.; Eisenman, R.N.; Weintraub, H. Binding of Myc Proteins to Canonical and Noncanonical DNA Sequences. Mol. Cell. Biol. 1993, 13, 5216–5224. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, A.; Schwab, M. The MYCN/Max Protein Complex in Neuroblastoma. Short Review. Eur. J. Cancer 1995, 31, 516–519. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H. The N-Myc Oncogene: Maximizing Its Targets, Regulation, and Therapeutic Potential. Mol. Cancer Res. 2014, 12, 815–822. [Google Scholar] [CrossRef]
- Giroux, S.; Charron, J. Defective Development of the Embryonic Liver in N-Myc-Deficient Mice. Dev. Biol. 1998, 195, 16–28. [Google Scholar] [CrossRef][Green Version]
- Kalkat, M.; de Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Lee, W.H.; Murphree, A.L.; Benedict, W.F. Expression and Amplification of the N-Myc Gene in Primary Retinoblastoma. Nature 1984, 309, 458–460. [Google Scholar] [CrossRef]
- McEvoy, J.; Nagahawatte, P.; Finkelstein, D.; Richards-Yutz, J.; Valentine, M.; Ma, J.; Mullighan, C.; Song, G.; Chen, X.; Wilson, M.; et al. RB1 Gene Inactivation by Chromothripsis in Human Retinoblastoma. Oncotarget 2014, 5, 438–450. [Google Scholar] [CrossRef]
- Bowles, E.; Corson, T.W.; Bayani, J.; Squire, J.A.; Wong, N.; Lai, P.B.S.; Gallie, B.L. Profiling Genomic Copy Number Changes in Retinoblastoma beyond Loss of RB1. Genes Chromosomes Cancer 2007, 46, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Michael Bishop, J. Amplification of N-Myc in Untreated Human Neuroblastomas Correlates with Advanced Disease Stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Biassoni, V.; Gandola, L.; Garrè, M.L.; Gatta, G.; Giangaspero, F.; Poggi, G.; Rutkowski, S. Childhood Medulloblastoma. Crit. Rev. Oncol. /Hematol. 2016, 105, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.A.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3 Mutations Drive Pediatric Glioblastoma through Upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.G.; Yeh, R.F.; Ray, A.; Wang, N.J.; Smirnov, I.; Yu, M.; Hariono, S.; Silber, J.; Feiler, H.S.; Gray, J.W.; et al. Comparative Analyses of Gene Copy Number and MRNA Expression in Glioblastoma Multiforme Tumors and Xenografts. Neuro-oncology 2009, 11, 477–487. [Google Scholar] [CrossRef]
- Williamson, D.; Lu, Y.J.; Gordon, T.; Sciot, R.; Kelsey, A.; Fisher, C.; Poremba, C.; Anderson, J.; Pritchard-Jones, K.; Shipley, J. Relationship between MYCN Copy Number and Expression in Rhabdomyosarcomas and Correlation with Adverse Prognosis in the Alveolar Subtype. J. Clin. Oncol. 2005, 23, 880–888. [Google Scholar] [CrossRef]
- Fielitz, K.; Althoff, K.; de Preter, K.; Nonnekens, J.; Ohli, J.; Elges, S.; Hartmann, W.; Klöppel, G.; Knösel, T.; Schulte, M.; et al. Characterization of Pancreatic Glucagon-Producing Tumors and Pituitary Gland Tumors in Transgenic Mice Overexpressing MYCN in HGFAP-Positive Cells. Oncotarget 2016, 7, 74415–74426. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting Oncogenic Myc as a Strategy for Cancer Treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef]
- Sradhanjali, S.; Tripathy, D.; Rath, S.; Mittal, R.; Reddy, M.M. Overexpression of Pyruvate Dehydrogenase Kinase 1 in Retinoblastoma: A Potential Therapeutic Opportunity for Targeting Vitreous Seeds and Hypoxic Regions. PLoS ONE 2017, 12, e0177744. [Google Scholar] [CrossRef]
- Borah, N.A.; Sradhanjali, S.; Barik, M.R.; Jha, A.; Tripathy, D.; Kaliki, S.; Rath, S.; Raghav, S.K.; Patnaik, S.; Mittal, R.; et al. Aurora Kinase B Expression, Its Regulation and Therapeutic Targeting in Human Retinoblastoma. Investig. Ophthalmol. Vis. Sci. 2021, 62, 16. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Schwermer, M.; Hiber, M.; Dreesmann, S.; Rieb, A.; Theißen, J.; Herold, T.; Schramm, A.; Temming, P.; Steenpass, L. Comprehensive Characterization of RB1 Mutant and MYCN Amplified Retinoblastoma Cell Lines. Exp. Cell Res. 2019, 375, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Kaewkhaw, R.; Rojanaporn, D. Retinoblastoma: Etiology, Modeling, and Treatment. Cancers 2020, 12, 2304. [Google Scholar] [CrossRef]
- Xu, X.L.; Fang, Y.; Lee, T.C.; Forrest, D.; Gregory-Evans, C.; Almeida, D.; Liu, A.; Jhanwar, S.C.; Abramson, D.H.; Cobrinik, D. Retinoblastoma Has Properties of a Cone Precursor Tumor and Depends Upon Cone-Specific MDM2 Signaling. Cell 2009, 137, 1018–1031. [Google Scholar] [CrossRef]
- Lillington, D.M.; Goff, L.K.; Kingston, J.E.; Onadim, Z.; Price, E.; Domizio, P.; Young, B.D. High Level Amplification of N-MYC Is Not Associated with Adverse Histology or Outcome in Primary Retinoblastoma Tumours. Br. J. Cancer 2002, 87, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of Multiple Copies of the N- Myc Oncogene with Rapid Progression of Neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Wu, N.; Jia, D.; Bates, B.; Basom, R.; Eberhart, C.G.; MacPherson, D. A Mouse Model of MYCN-Driven Retinoblastoma Reveals MYCN-Independent Tumor Reemergence. J. Clin. Investig. 2017, 127, 888–898. [Google Scholar] [CrossRef]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition. Cancer Discov. 2013, 3, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Larsson, K.; Frenzel, A.; Oliynyk, G.; Zirath, H.; Prochownik, E.V.; Westwood, N.J.; Henriksson, M.A. Targeting of the MYCN Protein with Small Molecule C-MYC Inhibitors. PLoS ONE 2014, 9, e97285. [Google Scholar] [CrossRef] [PubMed]
- Zirath, H.; Frenzel, A.; Oliynyk, G.; Segerström, L.; Westermark, U.K.; Larsson, K.; Persson, M.M.; Hultenby, K.; Lehtiö, J.; Einvik, C.; et al. MYC Inhibition Induces Metabolic Changes Leading to Accumulation of Lipid Droplets in Tumor Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10258–10263. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.S.; Williams, C.R.; Royce, D.B.; Pioli, P.A.; Sporn, M.B.; Liby, K.T. Bromodomain Inhibitors, JQ1 and I-BET 762, as Potential Therapies for Pancreatic Cancer. Cancer Lett. 2017, 394, 76–87. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of Glucose Transporter 1 and Glycolytic Gene Expression by C-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of Myc E-Box Phylogenetic Footprints in Glycolytic Genes by Chromatin Immunoprecipitation Assays. Mol. Cell. Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef] [PubMed]
- Sradhanjali, S.; Reddy, M.M. Inhibition of Pyruvate Dehydrogenase Kinase as a Therapeutic Strategy against Cancer. Curr. Top. Med. Chem. 2018, 18, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and Mitochondrial Respiration: A Neglected Connection. Cancer Res. 2015, 75, 3685–3686. [Google Scholar] [CrossRef]
- Xiao, J.; Yang, W.; Xu, B.; Zhu, H.; Zou, J.; Su, C.; Rong, J.; Wang, T.; Chen, Z. Expression of Fibronectin in Esophageal Squamous Cell Carcinoma and Its Role in Migration 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. BMC Cancer 2018, 18, 976. [Google Scholar] [CrossRef]
- Cai, X.; Liu, C.; Zhang, T.N.; Zhu, Y.W.; Dong, X.; Xue, P. Down-Regulation of FN1 Inhibits Colorectal Carcinogenesis by Suppressing Proliferation, Migration, and Invasion. J. Cell. Biochem. 2018, 119, 4717–4728. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Nakayama, H.; Nagata, M.; Yoshida, R.; Kawahara, K.; Hirosue, A.; Tanaka, T.; Yuno, A.; Matsuoka, Y.; Kojima, T.; et al. Overexpression of Fibronectin Confers Cell Adhesion-Mediated Drug Resistance (CAM-DR) against 5-FU in Oral Squamous Cell Carcinoma Cells. Int. J. Oncol. 2014, 44, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Sponziello, M.; Rosignolo, F.; Celano, M.; Maggisano, V.; Pecce, V.; de Rose, R.F.; Lombardo, G.E.; Durante, C.; Filetti, S.; Damante, G.; et al. Fibronectin-1 Expression Is Increased in Aggressive Thyroid Cancer and Favors the Migration and Invasion of Cancer Cells. Mol. Cell. Endocrinol. 2016, 431, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Y.; Lu, L.; Yang, L.; Yin, S.; Wang, Y.; Qi, Z.; Meng, J.; Zang, R.; Yang, G. Fibrillin-1, Induced by Aurora-A but Inhibited by BRCA2, Promotes Ovarian Cancer Metastasis. Oncotarget 2015, 6, 6670–6683. [Google Scholar] [CrossRef] [PubMed]
- Cierna, Z.; Mego, M.; Jurisica, I.; Machalekova, K.; Chovanec, M.; Miskovska, V.; Svetlovska, D.; Kalavska, K.; Rejlekova, K.; Kajo, K.; et al. Fibrillin-1 (FBN-1) a New Marker of Germ Cell Neoplasia in Situ. BMC Cancer 2016, 16, 597. [Google Scholar] [CrossRef]
- Li, L.N.; Zhou, X.; Gu, Y.; Yan, J. Prognostic Value of MMP-9 in Ovarian Cancer: A Meta-Analysis. Asian Pac. J. Cancer Prev. 2013, 14, 4107–4113. [Google Scholar] [CrossRef]
- Shao, W.; Wang, W.; Xiong, X.G.; Cao, C.; Yan, T.D.; Chen, G.; Chen, H.; Yin, W.; Liu, J.; Gu, Y.; et al. Prognostic Impact of MMP-2 and MMP-9 Expression in Pathologic Stage IA Non-Small Cell Lung Cancer. J. Surg. Oncol. 2011, 104, 841–846. [Google Scholar] [CrossRef]
- Liang, S.; Chang, L. Serum Matrix Metalloproteinase-9 Level as a Biomarker for Colorectal Cancer: A Diagnostic Meta-Analysis. Biomark. Med. 2018, 12, 393–402. [Google Scholar] [CrossRef]
- Joseph, C.; Alsaleem, M.; Orah, N.; Narasimha, P.L.; Miligy, I.M.; Kurozumi, S.; Ellis, I.O.; Mongan, N.P.; Green, A.R.; Rakha, E.A. Elevated MMP9 Expression in Breast Cancer Is a Predictor of Shorter Patient Survival. Breast Cancer Res. Treat. 2020, 182, 267–282. [Google Scholar] [CrossRef]
- Webb, A.H.; Gao, B.T.; Goldsmith, Z.K.; Irvine, A.S.; Saleh, N.; Lee, R.P.; Lendermon, J.B.; Bheemreddy, R.; Zhang, Q.; Brennan, R.C.; et al. Inhibition of MMP-2 and MMP-9 Decreases Cellular Migration, and Angiogenesis in in Vitro Models of Retinoblastoma. BMC Cancer 2017, 17, 434. [Google Scholar] [CrossRef]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 2021, 26, 1981. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Pérez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN Protein in Health and Disease. Genes 2017, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Aubry, A.; Pearson, J.D.; Huang, K.; Livne-bar, I.; Ahmad, M.; Jagadeesan, M.; Khetan, V.; Ketela, T.; Brown, K.R.; Yu, T.; et al. Functional Genomics Identifies New Synergistic Therapies for Retinoblastoma. Oncogene 2020, 39, 5338–5357. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Total | Positive | Negative | Statistical Test | p-Value | Remarks | |
|---|---|---|---|---|---|---|---|
| Age at diagnosis (Months) | Median | 36 | 24 | Student’s t test | 0.42 | NS | |
| Mean | 33.61 ± 24.30 | 28.90 ± 16.41 | |||||
| Range | 1–96 | 12–84 | |||||
| Gender | Male | 37 | 24 | 13 | Chi-square test | 0.56 | NS |
| Female | 21 | 12 | 9 | ||||
| Laterality | Unilateral | 52 | 31 | 21 | Chi-square test | 0.25 | NS |
| Bilateral | 6 | 5 | 1 | ||||
| Chemotherapy | Yes | 16 | 8 | 8 | Chi-square test | 0.24 | NS |
| No | 42 | 28 | 14 | ||||
| Choroid invasion | Present | 28 | 18 | 10 | Chi-square test | 0.73 | NS |
| Absent | 30 | 18 | 12 | ||||
| Optic nerve invasion | Present | 34 | 18 | 16 | Chi-square test | 0.08 | NS |
| Absent | 24 | 18 | 6 | ||||
| Anterior chamber invasion | Present | 9 | 4 | 5 | Chi-square test | 0.23 | NS |
| Absent | 49 | 32 | 17 | ||||
| Scleral invasion | Present | 9 | 5 | 4 | Chi-square test | 0.66 | NS |
| Absent | 49 | 31 | 18 | ||||
| Differentiation | Well | 7 | 4 | 3 | Chi-square test | 0.95 | NS |
| Moderate | 11 | 7 | 4 | ||||
| Poor | 34 | 21 | 13 | ||||
| Undifferentiated | 6 | 4 | 2 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sradhanjali, S.; Rout, P.; Tripathy, D.; Kaliki, S.; Rath, S.; Modak, R.; Mittal, R.; Chowdary, T.K.; Reddy, M.M. The Oncogene MYCN Modulates Glycolytic and Invasive Genes to Enhance Cell Viability and Migration in Human Retinoblastoma. Cancers 2021, 13, 5248. https://doi.org/10.3390/cancers13205248
Sradhanjali S, Rout P, Tripathy D, Kaliki S, Rath S, Modak R, Mittal R, Chowdary TK, Reddy MM. The Oncogene MYCN Modulates Glycolytic and Invasive Genes to Enhance Cell Viability and Migration in Human Retinoblastoma. Cancers. 2021; 13(20):5248. https://doi.org/10.3390/cancers13205248
Chicago/Turabian StyleSradhanjali, Swatishree, Padmalochan Rout, Devjyoti Tripathy, Swathi Kaliki, Suryasnata Rath, Rahul Modak, Ruchi Mittal, Tirumala Kumar Chowdary, and Mamatha M. Reddy. 2021. "The Oncogene MYCN Modulates Glycolytic and Invasive Genes to Enhance Cell Viability and Migration in Human Retinoblastoma" Cancers 13, no. 20: 5248. https://doi.org/10.3390/cancers13205248
APA StyleSradhanjali, S., Rout, P., Tripathy, D., Kaliki, S., Rath, S., Modak, R., Mittal, R., Chowdary, T. K., & Reddy, M. M. (2021). The Oncogene MYCN Modulates Glycolytic and Invasive Genes to Enhance Cell Viability and Migration in Human Retinoblastoma. Cancers, 13(20), 5248. https://doi.org/10.3390/cancers13205248