
Structure vs. Function of TRIB1—Myeloid Neoplasms and Beyond


Abstract
Simple Summary
Abstract
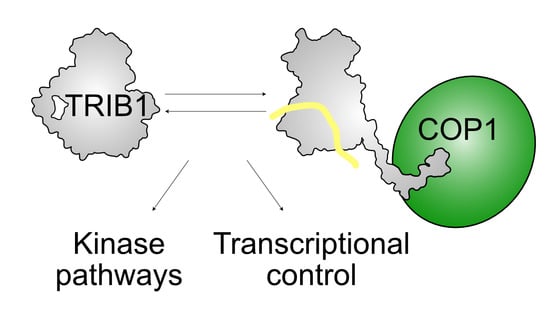
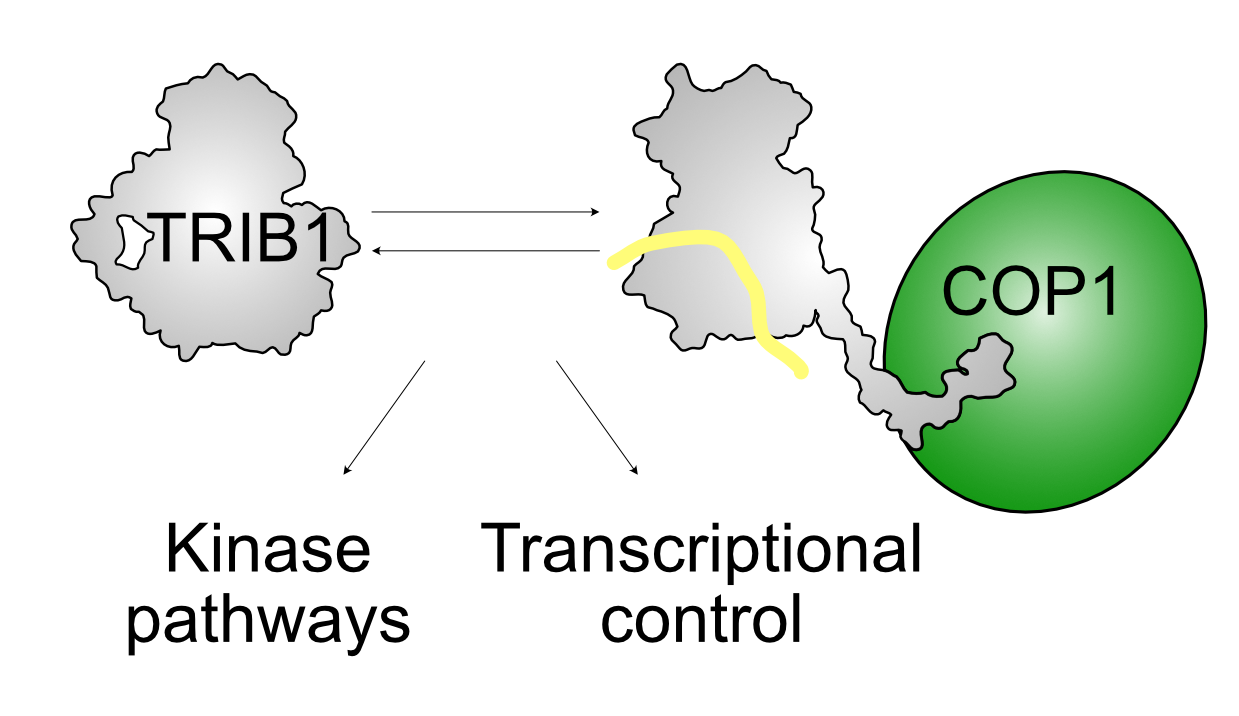{kind=link}
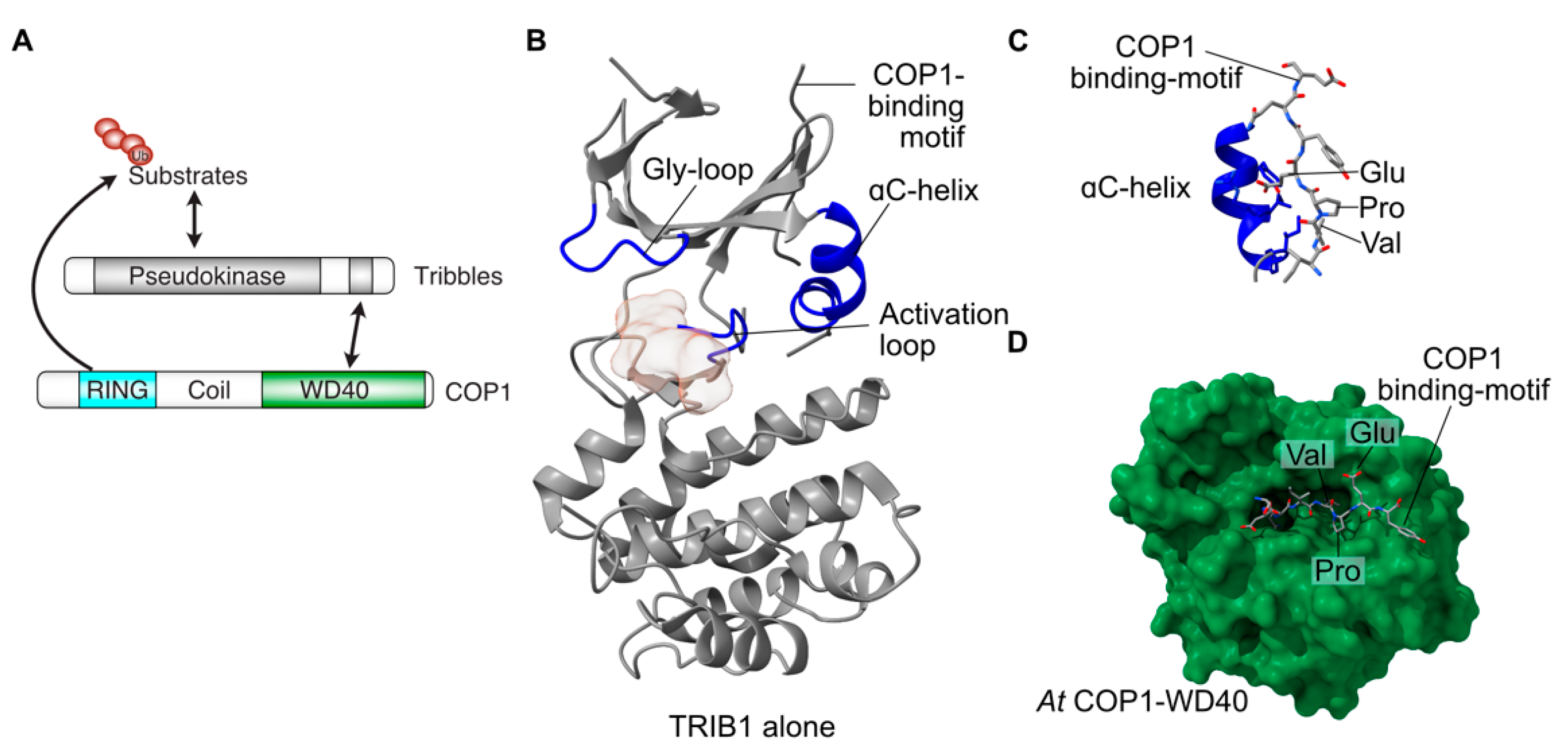{kind=link}
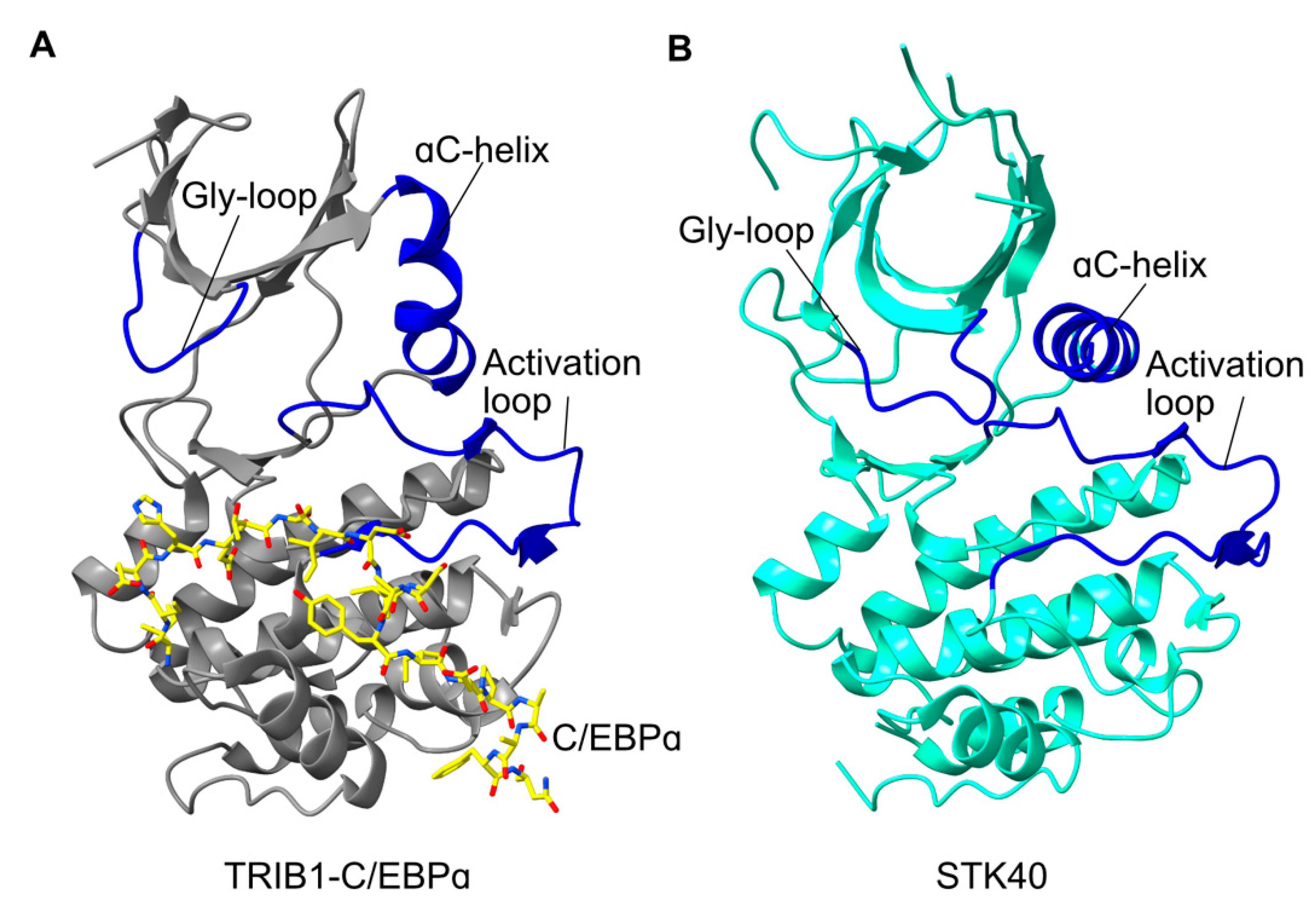{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Molecular Mechanism of Tribbles Function
2.1. Overall Tribbles Structure
2.2. Tribbles Pseudokinase Domain Structure
2.3. Co-ordination of Substrate- and COP1-Binding
2.4. Structural Conservation of Tribbles Proteins
3. Cancer-Relevant Pathways Regulated by TRIB1
3.1. Proposed Interaction Partners of Tribbles and TRIB1
3.2. TRIB1 and MAP Kinase Pathway Regulation
3.3. TRIB1 and AKT in NF-κB Regulation
3.4. TRIB1 in JAK/STAT Signalling
3.5. TRIB1 in Retinoic Acid Signalling
4. TRIB1 Function in Cancer Development and Therapy
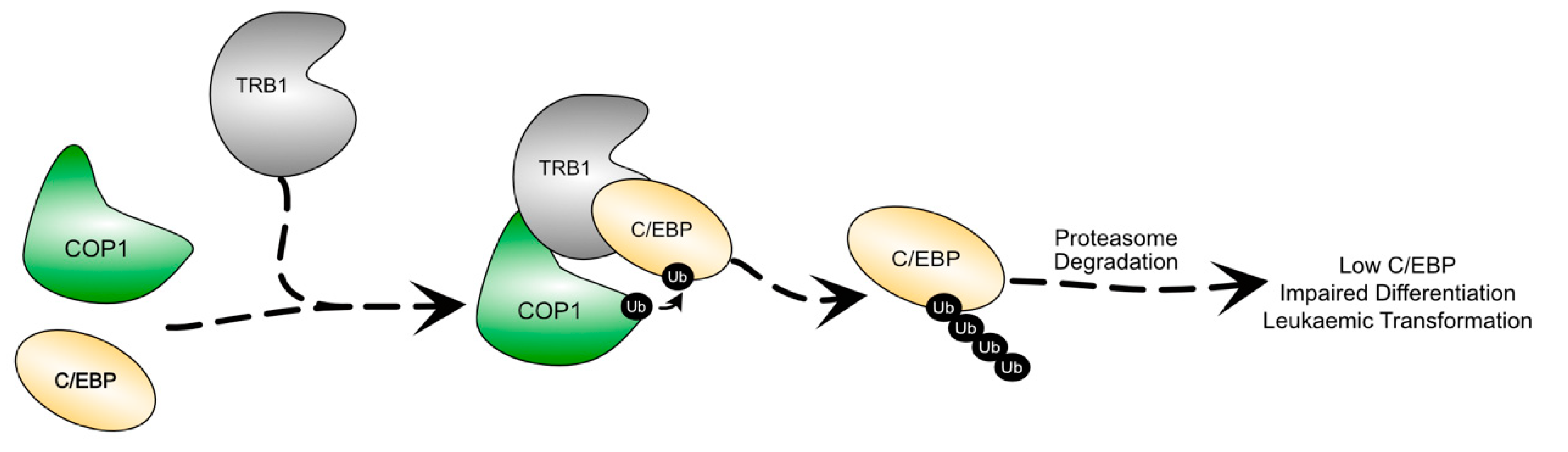
4.1. TRIB1 in Myeloid Neoplasms
4.2. TRIB1 in Solid Tumours
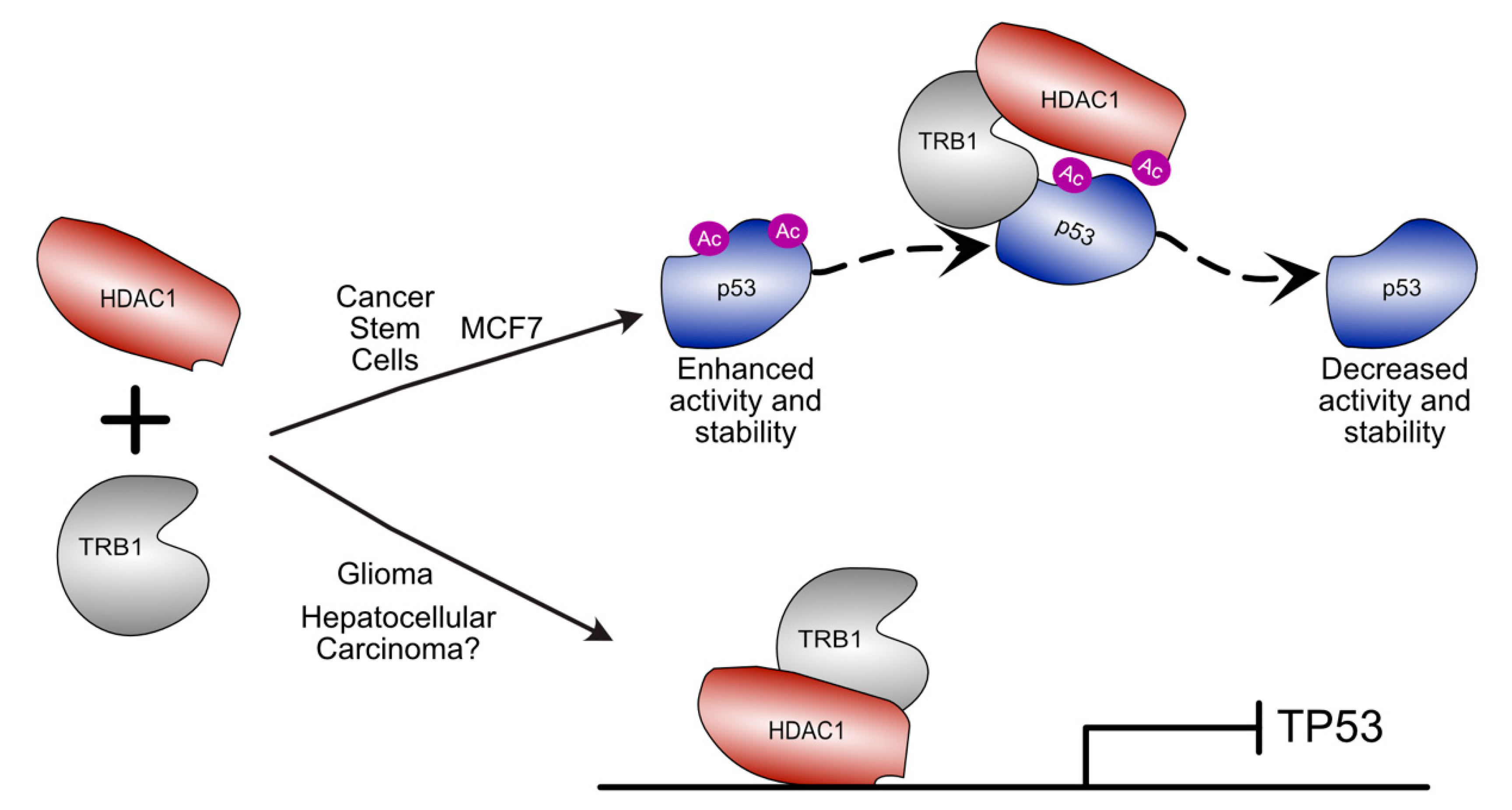
4.2.1. Regulation of p53
4.2.2. Association with the Oncogenic c-MYC
4.2.3. Regulation of Cell Cycle Progression in Breast Cancer
4.2.4. Regulation of the Tumour Microenvironment
4.3. TRIB1 and Treatment Resistance
5. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Mace, P.D.; Murphy, J.M. There’s more to Death than Life: Non-Catalytic Functions in Kinase and Pseudokinase Signaling. J. Biol. Chem. 2021, 100705. [Google Scholar] [CrossRef] [PubMed]
- Kwon, A.; Scott, S.; Taujale, R.; Yeung, W.; Kochut, K.J.; Eyers, P.A.; Kannan, N. Tracing the Origin and Evolution of Pseudokinases across the Tree of Life. Sci. Signal. 2019, 12, eaav3810. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Mace, P.D.; Eyers, P.A. Live and Let Die: Insights into Pseudoenzyme Mechanisms from Structure. Curr. Opin. Struct. Biol. 2017, 47, 95–104. [Google Scholar] [CrossRef]
- Kung, J.E.; Jura, N. Prospects for Pharmacological Targeting of Pseudokinases. Nat. Rev. Drug Discov. 2019, 4, 177. [Google Scholar] [CrossRef]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef]
- Salome, M.; Hopcroft, L.; Keeshan, K. Inverse and Correlative Relationships between TRIBBLES Genes Indicate Non-Redundant Functions during Normal and Malignant Hemopoiesis. Exp. Hematol. 2018, 66, 63–78.e13. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T. The Role of Trib1 in Myeloid Leukaemogenesis and Differentiation. Biochem. Soc. Trans. 2015, 43, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Naiki, T.; Saijou, E.; Miyaoka, Y.; Sekine, K.; Miyajima, A. TRB2, a Mouse Tribbles Ortholog, Suppresses Adipocyte Differentiation by Inhibiting AKT and C/EBPβ*. J. Biol. Chem. 2007, 282, 24075–24082. [Google Scholar] [CrossRef]
- Hegedus, Z.; Czibula, A.; Kiss-Toth, E. Tribbles: A Family of Kinase-like Proteins with Potent Signalling Regulatory Function. Cell Signal. 2007, 19, 238–250. [Google Scholar] [CrossRef]
- Kiss-Toth, E.; Wyllie, D.H.; Holland, K.; Marsden, L.; Jozsa, V.; Oxley, K.M.; Polgar, T.; Qwarnstrom, E.E.; Dower, S.K. Functional Mapping and Identification of Novel Regulators for the Toll/Interleukin-1 Signalling Network by Transcription Expression Cloning. Cell Signal. 2006, 18, 202–214. [Google Scholar] [CrossRef]
- Murphy, J.M.; Nakatani, Y.; Jamieson, S.A.; Dai, W.; Lucet, I.S.; Mace, P.D. Molecular Mechanism of CCAAT-Enhancer Binding Protein Recruitment by the TRIB1 Pseudokinase. Structure 2015, 23, 2111–2121. [Google Scholar] [CrossRef]
- Keeshan, K.; Bailis, W.; Dedhia, P.H.; Vega, M.E.; Shestova, O.; Xu, L.; Toscano, K.; Uljon, S.N.; Blacklow, S.C.; Pear, W.S. Transformation by Tribbles Homolog 2 (Trib2) Requires Both the Trib2 Kinase Domain and COP1 Binding. Blood 2010, 116, 4948–4957. [Google Scholar] [CrossRef]
- Jamieson, S.A.; Ruan, Z.; Burgess, A.E.; Curry, J.R.; McMillan, H.D.; Brewster, J.L.; Dunbier, A.K.; Axtman, A.D.; Kannan, N.; Mace, P.D. Substrate Binding Allosterically Relieves Autoinhibition of the Pseudokinase TRIB1. Sci. Signal. 2018, 11, eaau0597. [Google Scholar] [CrossRef]
- Durzynska, I.; Xu, X.; Adelmant, G.; Ficarro, S.B.; Marto, J.A.; Sliz, P.; Uljon, S.; Blacklow, S.C. STK40 Is a Pseudokinase That Binds the E3 Ubiquitin Ligase COP1. Structure 2017, 25, 287–294. [Google Scholar] [CrossRef]
- Salomé, M.; Magee, A.; Yalla, K.; Chaudhury, S.; Sarrou, E.; Carmody, R.J.; Keeshan, K. A Trib2-P38 Axis Controls Myeloid Leukaemia Cell Cycle and Stress Response Signalling. Cell Death Dis. 2018, 9, 443. [Google Scholar] [CrossRef]
- Yokoyama, T.; Toki, T.; Aoki, Y.; Kanezaki, R.; Park, M.J.; Kanno, Y.; Takahara, T.; Yamazaki, Y.; Ito, E.; Hayashi, Y.; et al. Identification of TRIB1 R107L Gain-of-Function Mutation in Human Acute Megakaryocytic Leukemia. Blood 2012, 119, 2608–2611. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.Y.; Guan, H.; Czibula, A.; King, A.R.; Eder, K.; Heath, E.; Suvarna, S.K.; Dower, S.K.; Wilson, A.G.; Francis, S.E.; et al. Human Tribbles-1 Controls Proliferation and Chemotaxis of Smooth Muscle Cells via MAPK Signaling Pathways*. J. Biol. Chem. 2007, 282, 18379–18387. [Google Scholar] [CrossRef] [PubMed]
- Kiss-Toth, E.; Bagstaff, S.M.; Sung, H.Y.; Jozsa, V.; Dempsey, C.; Caunt, J.C.; Oxley, K.M.; Wyllie, D.H.; Polgar, T.; Harte, M.; et al. Human Tribbles, a Protein Family Controlling Mitogen-Activated Protein Kinase Cascades. J. Biol. Chem. 2004, 279, 42703–42708. [Google Scholar] [CrossRef]
- Otsuki, L.; Brand, A.H. Cell Cycle Heterogeneity Directs the Timing of Neural Stem Cell Activation from Quiescence. Science 2018, 360, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, R.; Xing, H.; Mirzoeva, O.K.; Sarde, P.; Curtis, C.; Feiler, H.S.; McDonagh, P.; Gray, J.W.; Khalil, I.; Korn, W.M. Bayesian Network Inference Modeling Identifies TRIB1 as a Novel Regulator of Cell-Cycle Progression and Survival in Cancer Cells. Cancer Res. 2017, 77, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Sebo, Z.; Pence, L.; Dobens, L.L. Drosophila Tribbles Antagonizes Insulin Signaling-Mediated Growth and Metabolism via Interactions with Akt Kinase. PLoS ONE 2014, 9, e109530. [Google Scholar] [CrossRef]
- Zareen, N.; Biswas, S.C.; Greene, L.A. A Feed-Forward Loop Involving Trib3, Akt and FoxO Mediates Death of NGF-Deprived Neurons. Cell Death Differ. 2013, 20, 1719–1730. [Google Scholar] [CrossRef][Green Version]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A Tribbles Homolog That Inhibits Akt/PKB Activation by Insulin in Liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef]
- Qi, L.; Heredia, J.E.; Altarejos, J.Y.; Screaton, R.; Goebel, N.; Niessen, S.; MacLeod, I.X.; Liew, C.W.; Kulkarni, R.N.; Bain, J.; et al. TRB3 Links the E3 Ubiquitin Ligase COP1 to Lipid Metabolism. Science 2006, 312, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Kato, J.-Y.; Nakamae, I.; Yoneda-Kato, N. COP1 Targets C/EBPα for Degradation and Induces Acute Myeloid Leukemia via Trib1. Blood 2013, 122, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Dedhia, P.H.; Keeshan, K.; Uljon, S.; Xu, L.; Vega, M.E.; Shestova, O.; Zaks-Zilberman, M.; Romany, C.; Blacklow, S.C.; Pear, W.S. Differential Ability of Tribbles Family Members to Promote Degradation of C/EBPalpha and Induce Acute Myelogenous Leukemia. Blood 2010, 116, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Keeshan, K.; He, Y.; Wouters, B.J.; Shestova, O.; Xu, L.; Sai, H.; Rodriguez, C.G.; Maillard, I.; Tobias, J.W.; Valk, P.; et al. Tribbles Homolog 2 Inactivates C/EBPalpha and Causes Acute Myelogenous Leukemia. Cancer Cell 2006, 10, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Bailey, F.P.; Byrne, D.P.; Oruganty, K.; Eyers, C.E.; Novotny, C.J.; Shokat, K.M.; Kannan, N.; Eyers, P.A. The Tribbles 2 (TRB2) Pseudokinase Binds to ATP and Autophosphorylates in a Metal-Independent Manner. Biochem. J. 2015, 467, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Uljon, S.; Xu, X.; Durzynska, I.; Stein, S.; Adelmant, G.; Marto, J.A.; Pear, W.S.; Blacklow, S.C. Structural Basis for Substrate Selectivity of the E3 Ligase COP1. Structure 2016, 24, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, D.M.; Byrne, D.P.; Yeung, W.; Shrestha, S.; Bailey, F.P.; Ferries, S.; Eyers, C.E.; Keeshan, K.; Wells, C.; Drewry, D.H.; et al. Covalent Inhibitors of EGFR Family Protein Kinases Induce Degradation of Human Tribbles 2 (TRIB2) Pseudokinase in Cancer Cells. Sci. Signal. 2018, 11, eaat7951. [Google Scholar] [CrossRef]
- Sakai, S.; Miyajima, C.; Uchida, C.; Itoh, Y.; Hayashi, H.; Inoue, Y. Tribbles-Related Protein Family Members as Regulators or Substrates of the Ubiquitin-Proteasome System in Cancer Development. Curr. Cancer Drug Targets 2016, 16, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, N.; Pang, B.; Tong, D.; Sun, D.; Sun, H.; Zhang, C.; Sun, W.; Meng, X.; Bai, J.; et al. TRIB1 Promotes Colorectal Cancer Cell Migration and Invasion through Activation MMP-2 via FAK/Src and ERK Pathways. Oncotarget 2017, 8, 47931–47942. [Google Scholar] [CrossRef] [PubMed]
- Rome, K.S.; Stein, S.J.; Kurachi, M.; Petrovic, J.; Schwartz, G.W.; Mack, E.A.; Uljon, S.; Wu, W.W.; DeHart, A.G.; McClory, S.E.; et al. Trib1 Regulates T Cell Differentiation during Chronic Infection by Restraining the Effector Program. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Mack, E.A.; Stein, S.J.; Rome, K.S.; Xu, L.; Wertheim, G.B.; Melo, R.C.N.; Pear, W.S. Trib1 Regulates Eosinophil Lineage Commitment and Identity by Restraining the Neutrophil Program. Blood 2019, 133, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.J.; Mack, E.A.; Rome, K.S.; Pear, W.S. Tribbles in Normal and Malignant Haematopoiesis. Biochem. Soc. Trans. 2015, 43, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kidoya, H.; Naito, H.; Yamamoto, M.; Takemura, N.; Nakagawa, K.; Yoshioka, Y.; Morii, E.; Takakura, N.; Takeuchi, O.; et al. Critical Role of Trib1 in Differentiation of Tissue-Resident M2-like Macrophages. Nature 2013, 495, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Wang, G.; Wang, G.; Zhuang, J.; He, S.; Song, Y.; Ni, J.; Xia, W.; Wang, J. The Oncogenic Role of Tribbles 1 in Hepatocellular Carcinoma Is Mediated by a Feedback Loop Involving MicroRNA-23a and P53. Front. Physiol. 2017, 8, 789. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, C.; Inoue, Y.; Hayashi, H. Pseudokinase Tribbles 1 (TRB1) Negatively Regulates Tumor-Suppressor Activity of P53 through P53 Deacetylation. Biol. Pharm. Bull. 2015, 38, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, Y.; Nakayama, K.; Ogawa, A.; Makishima, S.; Boonvisut, S.; Hirao, A.; Iwasaki, Y.; Yada, T.; Yanagisawa, Y.; Miyashita, H.; et al. TRIB1 Downregulates Hepatic Lipogenesis and Glycogenesis via Multiple Molecular Interactions. J. Mol. Endocrinol. 2014, 52, 145–158. [Google Scholar] [CrossRef]
- Imajo, M.; Nishida, E. Human Tribbles Homolog 1 Functions as a Negative Regulator of Retinoic Acid Receptor. Genes Cells 2010, 15, 1089–1097. [Google Scholar] [CrossRef]
- Makishima, S.; Boonvisut, S.; Ishizuka, Y.; Watanabe, K.; Nakayama, K.; Iwamoto, S. Sin3A-Associated Protein, 18 KDa, a Novel Binding Partner of TRIB1, Regulates MTTP Expression. J. Lipid Res. 2015, 56, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Kanno, Y.; Yamazaki, Y.; Takahara, T.; Miyata, S.; Nakamura, T. Trib1 Links the MEK1/ERK Pathway in Myeloid Leukemogenesis. Blood 2010, 116, 2768–2775. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Ren, Y.; Zhang, J.; Chen, J.; Zhou, W.; Guo, W.; Wang, X.; Chen, H.; Li, M.; et al. Cisplatin-Enriching Cancer Stem Cells Confer Multidrug Resistance in Non-Small Cell Lung Cancer via Enhancing TRIB1/HDAC Activity. Cell Death Dis. 2017, 8, e2746. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Wu, W.; Zhang, Q.; Sun, Y.; Cui, Y.; Wu, F.; Wei, X.; Qi, G.; Liang, X.; Tang, F.; et al. Inhibition of Tribbles Protein-1 Attenuates Radioresistance in Human Glioma Cells. Sci. Rep. 2015, 5, 15961. [Google Scholar] [CrossRef]
- Yoshino, S.; Yokoyama, T.; Sunami, Y.; Takahara, T.; Nakamura, A.; Yamazaki, Y.; Tsutsumi, S.; Aburatani, H.; Nakamura, T. Trib1 Promotes Acute Myeloid Leukemia Progression by Modulating the Transcriptional Programs of Hoxa9. Blood 2020, 137, 75–88. [Google Scholar] [CrossRef]
- Liang, K.L.; O’Connor, C.; Veiga, J.P.; McCarthy, T.V.; Keeshan, K. TRIB2 Regulates Normal and Stress-Induced Thymocyte Proliferation. Cell Discov. 2016, 2, 15050. [Google Scholar] [CrossRef]
- Hill, R.; Madureira, P.A.; Ferreira, B.; Baptista, I.; Machado, S.; Colaço, L.; dos Santos, M.; Liu, N.; Dopazo, A.; Ugurel, S.; et al. TRIB2 Confers Resistance to Anti-Cancer Therapy by Activating the Serine/Threonine Protein Kinase AKT. Nat. Commun. 2017, 8, 14687. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional Regulation of Macrophage Polarization: Enabling Diversity with Identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Arndt, L.; Dokas, J.; Gericke, M.; Kutzner, C.E.; Müller, S.; Jeromin, F.; Thiery, J.; Burkhardt, R. Tribbles Homolog 1 Deficiency Modulates Function and Polarization of Murine Bone Marrow-Derived Macrophages. J. Biol. Chem. 2018, 293, 11527–11536. [Google Scholar] [CrossRef]
- Connolly, R.M.; Nguyen, N.K.; Sukumar, S. Molecular Pathways: Current Role and Future Directions of the Retinoic Acid Pathway in Cancer Prevention and Treatment. Clin. Cancer Res. 2013, 19, 1651–1659. [Google Scholar] [CrossRef]
- Keeshan, K.; Vieugué, P.; Chaudhury, S.; Rishi, L.; Gaillard, C.; Liang, L.; Garcia, E.; Nakamura, T.; Omidvar, N.; Kogan, S.C. Co-Operative Leukemogenesis in Acute Myeloid Leukemia and Acute Promyelocytic Leukemia Reveals C/EBPα as a Common Target of TRIB1 and PML/RARA. Haematologica 2016, 101, 1228–1236. [Google Scholar] [CrossRef]
- Keeshan, K.; Santilli, G.; Corradini, F.; Perrotti, D.; Calabretta, B. Transcription Activation Function of C/EBPalpha Is Required for Induction of Granulocytic Differentiation. Blood 2003, 102, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Nerlov, C. The C/EBP Family of Transcription Factors: A Paradigm for Interaction between Gene Expression and Proliferation Control. Trends Cell Biol. 2007, 17, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Gery, S.; Gombart, A.F.; Yi, W.S.; Koeffler, C.; Hofmann, W.-K.; Koeffler, H.P. Transcription Profiling of C/EBP Targets Identifies Per2 as a Gene Implicated in Myeloid Leukemia. Blood 2005, 106, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Calkhoven, C.F.; Müller, C.; Leutz, A. Translational Control of C/EBPalpha and C/EBPbeta Isoform Expression. Genes Dev. 2000, 14, 1920–1932. [Google Scholar] [PubMed]
- Rørth, P.; Szabo, K.; Texido, G. The Level of C/EBP Protein Is Critical for Cell Migration during Drosophila Oogenesis and Is Tightly Controlled by Regulated Degradation. Mol. Cell 2000, 6, 23–30. [Google Scholar] [CrossRef]
- Jin, G.; Yamazaki, Y.; Takuwa, M.; Takahara, T.; Kaneko, K.; Kuwata, T.; Miyata, S.; Nakamura, T. Trib1 and Evi1 Cooperate with Hoxa and Meis1 in Myeloid Leukemogenesis. Blood 2007, 109, 3998–4005. [Google Scholar] [CrossRef]
- Yokoyama, T.; Nakamura, T. Tribbles in Disease: Signaling Pathways Important for Cellular Function and Neoplastic Transformation. Cancer Sci. 2011, 102, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Lin, L.; Liu, S.; Qin, M.; He, S.; Zhu, L.; Huang, J. Ginkgo Biloba Extract Inhibits Metastasis and ERK/Nuclear Factor Kappa B (NF-ΚB) Signaling Pathway in Gastric Cancer. Med. Sci. Monit. 2019, 25, 6836–6845. [Google Scholar] [CrossRef]
- Liu, Z.-Z.; Han, Z.-D.; Liang, Y.-K.; Chen, J.-X.; Wan, S.; Zhuo, Y.-J.; Cai, Z.-D.; Deng, Y.-L.; Lin, Z.-Y.; Mo, R.-J.; et al. TRIB1 Induces Macrophages to M2 Phenotype by Inhibiting IKB-Zeta in Prostate Cancer. Cell. Signal. 2019, 59, 152–162. [Google Scholar] [CrossRef]
- Mashima, T.; Soma-Nagae, T.; Migita, T.; Kinoshita, R.; Iwamoto, A.; Yuasa, T.; Yonese, J.; Ishikawa, Y.; Seimiya, H. TRIB1 Supports Prostate Tumorigenesis and Tumor-Propagating Cell Survival by Regulation of Endoplasmic Reticulum Chaperone Expression. Cancer Res. 2014, 74, 4888–4897. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Nakamae, I.; Kato, J.; Yokoyama, T.; Ito, H.; Yoneda-Kato, N. Myeloid Leukemia Factor 1 Stabilizes Tumor Suppressor C/EBPα to Prevent Trib1-Driven Acute Myeloid Leukemia. Blood Adv. 2017, 1, 1682–1693. [Google Scholar] [CrossRef]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An Online Survival Analysis Tool to Rapidly Assess the Effect of 22,277 Genes on Breast Cancer Prognosis Using Microarray Data of 1,809 Patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Cermelli, S.; Jang, I.S.; Bernard, B.; Grandori, C. Synthetic Lethal Screens as a Means to Understand and Treat MYC-Driven Cancers. Cold Spring Harb. Perspect. Med. 2014, 4, a014209. [Google Scholar] [CrossRef]
- Toyoshima, M.; Toyoshima, M.; Howie, H.L.; Howie, H.L.; Imakura, M.; Imakura, M.; Walsh, R.M.; Walsh, R.M.; Annis, J.E.; Annis, J.E.; et al. Functional Genomics Identifies Therapeutic Targets for MYC-Driven Cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 9545–9550. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R. a Transcriptional Amplification in Tumor Cells with Elevated C-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. C-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and Repression by Oncogenic MYC Shape Tumour-Specific Gene Expression Profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Giaccia, A.J. Harnessing Synthetic Lethal Interactions in Anticancer Drug Discovery. Nat. Rev. Drug Discov. 2011, 10, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Farc, O.; Cristea, V. An Overview of the Tumor Microenvironment, from Cells to Complex Networks (Review). Exp. Ther. Med. 2021, 21, 96. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal. Transduct Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Jurj, A.; Pop, L.-A.; Zanoaga, O.; Ciocan-Cârtiţă, C.A.; Cojocneanu, R.; Moldovan, C.; Raduly, L.; Pop-Bica, C.; Trif, M.; Irimie, A.; et al. New Insights in Gene Expression Alteration as Effect of Paclitaxel Drug Resistance in Triple Negative Breast Cancer Cells. Cell. Physiol. Biochem. 2020, 54, 648–664. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N.; et al. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McMillan, H.D.; Keeshan, K.; Dunbier, A.K.; Mace, P.D. Structure vs. Function of TRIB1—Myeloid Neoplasms and Beyond. Cancers 2021, 13, 3060. https://doi.org/10.3390/cancers13123060
McMillan HD, Keeshan K, Dunbier AK, Mace PD. Structure vs. Function of TRIB1—Myeloid Neoplasms and Beyond. Cancers. 2021; 13(12):3060. https://doi.org/10.3390/cancers13123060
Chicago/Turabian StyleMcMillan, Hamish D, Karen Keeshan, Anita K Dunbier, and Peter D Mace. 2021. "Structure vs. Function of TRIB1—Myeloid Neoplasms and Beyond" Cancers 13, no. 12: 3060. https://doi.org/10.3390/cancers13123060
APA StyleMcMillan, H. D., Keeshan, K., Dunbier, A. K., & Mace, P. D. (2021). Structure vs. Function of TRIB1—Myeloid Neoplasms and Beyond. Cancers, 13(12), 3060. https://doi.org/10.3390/cancers13123060