
Data-Driven Mathematical Model of Osteosarcoma



Abstract
Simple Summary
Abstract
1. Introduction
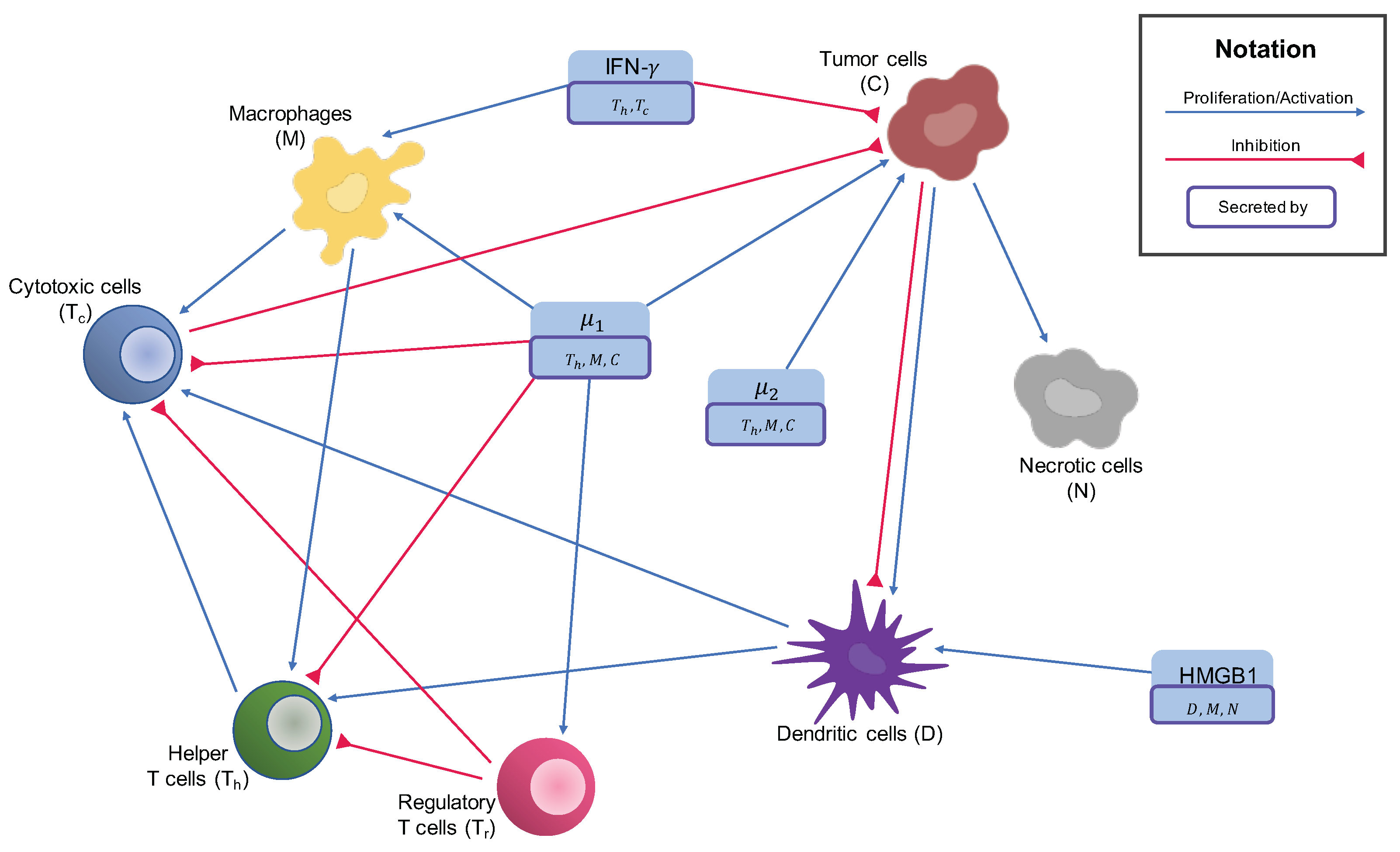
2. Materials and Methods
2.1. Cytokines
2.2. Cells in the Tumor Microenvironment
2.2.1. Macrophages
2.2.2. T Cells and NK Cells
2.2.3. Dendritic Cells
2.2.4. Cancer Cells
2.2.5. Necrotic Cells
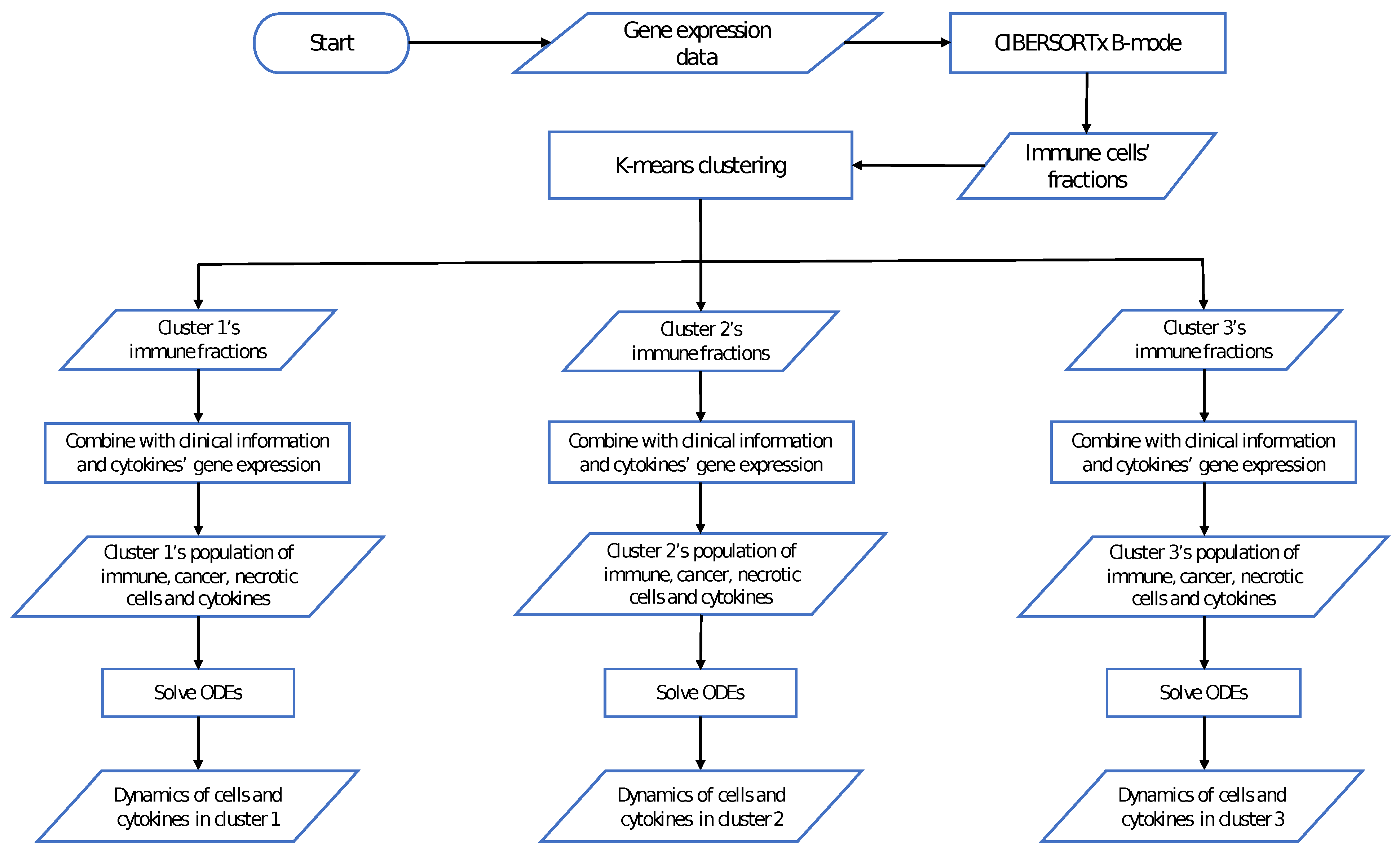
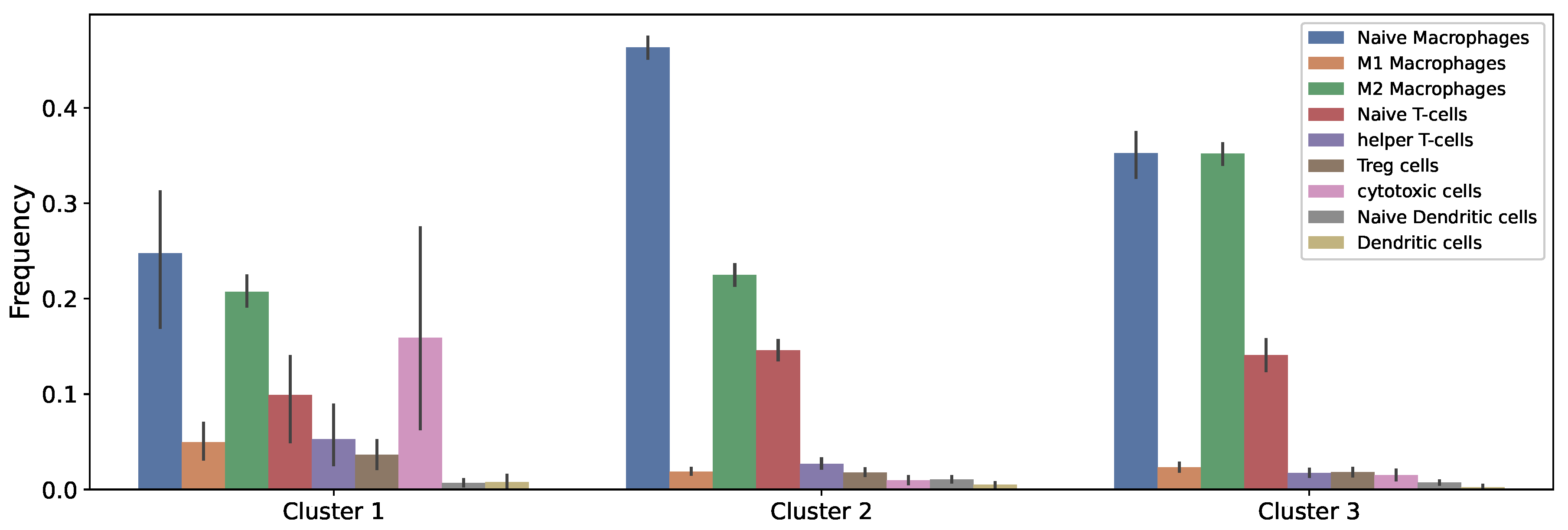
2.3. Data of the Model
2.4. Parameter Estimation
2.5. Non-Dimensionalization
2.6. Sensitivity Analysis
3. Results
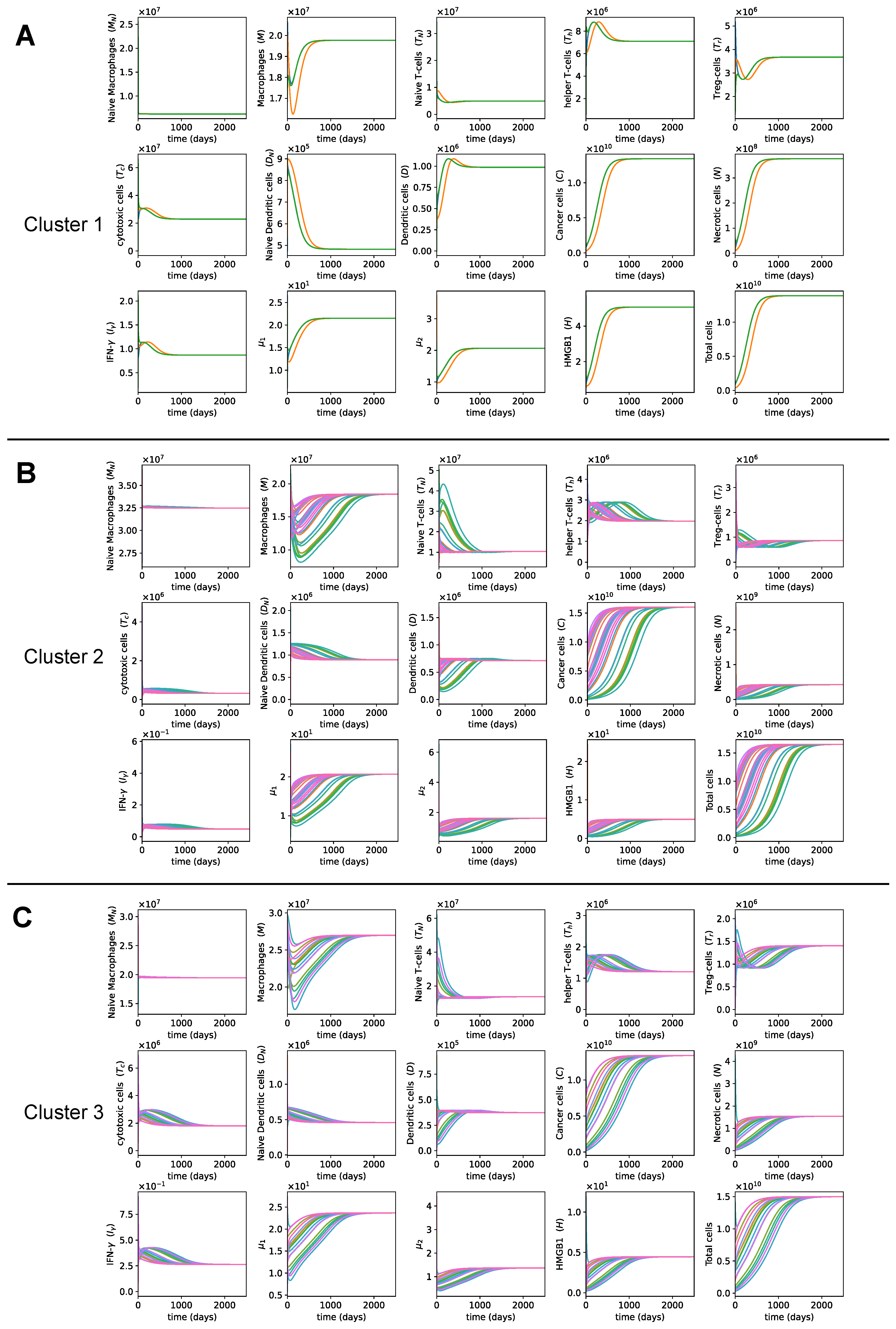
3.1. Dynamics of the Tumor Microenvironment
3.2. Sensitivity Analysis
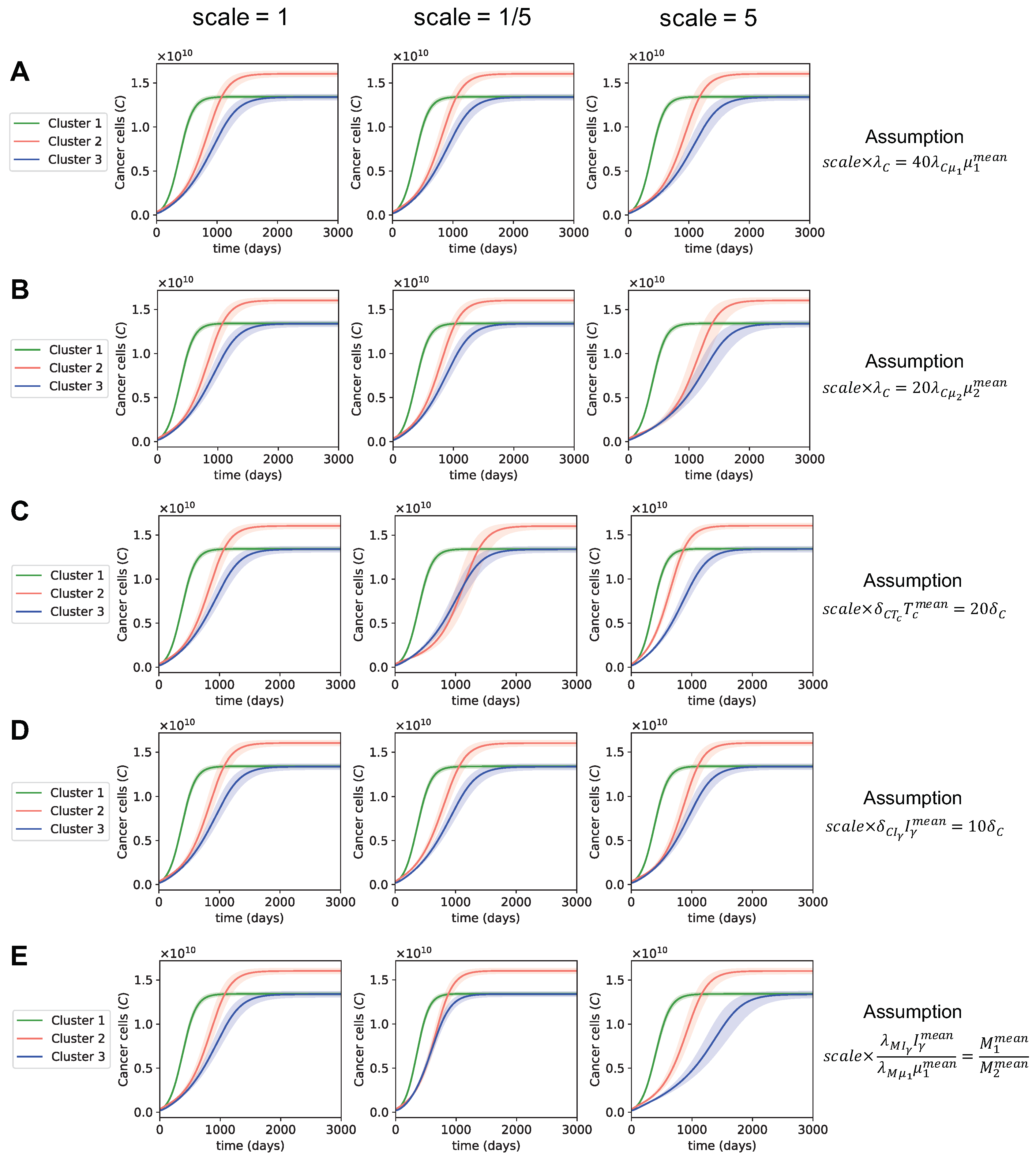
3.3. Dynamics with Varying Assumptions
3.4. Dynamics with Different Initial Conditions
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TARGET | Therapeutically Applicable Research to Generate Effective Treatments |
| GEO | Gene Expression Omnibus |
| ODE | Ordinary differential equation |
| HMGB1 | High mobility group box 1 |
| UCSC | University of California Santa Cruz |
| TIIC | Tumor infiltrating immune cell |
| NK | Natural killer |
Appendix A. System Analysis
Appendix A.1. System of ODEs
Appendix A.2. Positivity
Appendix A.3. Boundedness
Appendix A.3.1. Macrophages
Appendix A.3.2. T-Cells
Appendix A.3.3. Dendritic Cells
Appendix A.3.4. Cancer Cells
Appendix A.3.5. Interferon-γ
Appendix A.3.6. Remaining Variables
Appendix B. Derivation of the Parameter Set
Appendix B.1. Additional Assumptions
Appendix B.2. Parameter Values and Sources

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Cluster 1 | Cluster 2 | Cluster 3 | Source |
|---|---|---|---|---|
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| [146] | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| [147,148,149] | ||||
| [150,151] | ||||
| [152] | ||||
| Estimated | ||||
| Estimated | ||||
| [153] | ||||
| [154] | ||||
| Estimated | ||||
| Estimated | ||||
| [153] | ||||
| [155] | ||||
| Estimated | ||||
| [156] | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| [157] | ||||
| [158,159,160,161] | ||||
| [162,163] | ||||
| [164] | ||||
| Estimated | ||||
| Estimated | ||||
| Estimated | ||||
| Scaling factor | ||||
| Scaling factor | ||||
| Scaling factor | ||||
| Scaling factor | ||||
| Scaling factor |
Appendix C. Non-Dimensionalization
Appendix D. Dynamics of the Tumor Microenvironment with Cross-Cluster Initial Conditions

References
- Available online: https://www.cancer.org/cancer/osteosarcoma/about/key-statistics.html (accessed on 16 April 2021).
- Ottaviani, G.; Jaffe, N. The Epidemiology of Osteosarcoma. Cancer Treat Res. 2009, 3–13. [Google Scholar] [CrossRef]
- Yang, Y.; Han, L.; He, Z.; Li, X.; Yang, S.; Yang, J.; Zhang, Y.; Li, D.; Yang, Y.; Yang, Z. Advances in limb salvage treatment of osteosarcoma. J. Bone Oncol. 2018, 10, 36–40. [Google Scholar] [CrossRef]
- PDQ Pediatric Treatment Editorial Board. Osteosarcoma Treatment (PDQ®): Patient Version. In PDQ Cancer Information Summaries [Internet]; National Cancer Institute: Bethesda MD, USA, 2002. [Google Scholar]
- Marchandet, L.; Lallier, M.; Charrier, C.; Baud’huin, M.; Ory, B.; Lamoureux, F. Mechanisms of Resistance to Conventional Therapies for Osteosarcoma. Cancers 2021, 13, 683. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Gao, Z.; Xu, H.; Zhang, Z.; Fu, P. A meta-analysis of randomized control trials of surgical methods with osteosarcoma outcomes. J. Orthop. Surg. Res. 2017, 12, 5. [Google Scholar] [CrossRef]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M.; et al. Osteosarcoma: The Addition of Muramyl Tripeptide to Chemotherapy Improves Overall Survival—A Report From the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.; Pala, L.; Bagnardi, V.; De Pas, T.; Martinetti, M.; Viale, G.; Gelber, R.D.; Goldhirsch, A. Cancer immunotherapy efficacy and patients’ sex: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 737–746. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef]
- Schwarz, R.; Bruland, O.; Cassoni, A.; Schomberg, P.; Bielack, S. The Role of Radiotherapy in Oseosarcoma. Cancer Treat Res. 2009, 147–164. [Google Scholar] [CrossRef]
- Sahuı, R.K.; Sharma, A.K.; Patel, S.; Kalaı, P.; Goyal, A.; Patro, S.K. Sternal Mass with Respiratory Compromise in a 10-year-old Child. J. Bone Soft Tissue Tumors 2019, 2, 2–3. [Google Scholar] [CrossRef]
- Hiraoka, M.; Jo, S.; Dodo, Y.; Ono, K.; Takahashi, M.; Nishida, H.; Abe, M. Clinical results of radiofrequency hyperthermia combined with radiation in the treatment of radioresistant cancers. Cancer 1984, 54, 2898–2904. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Ma, B.A.; Qiu, X.C.; Li, Y.L.; Ye, J.; Zhou, Y. Preliminary report on treatment of bone tumors with microwave-induced hyperthermia. Bioelectromagnetics 1996, 17, 218–222. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Ma, B.A.; Zhou, Y.; Zhang, M.H.; Hao, X.B. Bone tumors of the extremities or pelvis treated by microwave-induced hyperthermia. Clin. Orthop. Relat. Res. 2003, 406, 165–175. [Google Scholar] [CrossRef]
- Farzin, A.; Fathi, M.; Emadi, R. Multifunctional magnetic nanostructured hardystonite scaffold for hyperthermia, drug delivery and tissue engineering applications. Mater. Sci. Eng. C 2017, 70, 21–31. [Google Scholar] [CrossRef]
- Fanti, A.; Lodi, M.B.; Vacca, G.; Mazzarella, G. Numerical Investigation of Bone Tumor Hyperthermia Treatment Using Magnetic Scaffolds. IEEE J. Electromagn. RF Microwaves Med. Biol. 2018, 2, 294–301. [Google Scholar] [CrossRef]
- Lodi, M.B.; Fanti, A.; Muntoni, G.; Mazzarella, G. A Multiphysic Model for the Hyperthermia Treatment of Residual Osteosarcoma Cells in Upper Limbs Using Magnetic Scaffolds. IEEE J. Multiscale Multiphys. Comput. 2019, 4, 337–347. [Google Scholar] [CrossRef]
- Prudowsky, Z.D.; Yustein, J.T. Recent Insights into Therapy Resistance in Osteosarcoma. Cancers 2020, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. Innate Immune Recognition of Cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Shirai, T.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Igarashi, K.; Tsuchiya, H. Current and Emerging Targets in Immunotherapy for Osteosarcoma. J. Oncol. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Z.; Li, B.; Wang, S.; Chen, T.; Ye, Z. Innate immune cells: A potential and promising cell population for treating osteosarcoma. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef]
- Wang, K.; Vella, A.T. Regulatory T Cells and Cancer: A Two-Sided Story. Immunol. Investig. 2016, 45, 797–812. [Google Scholar] [CrossRef]
- Wang, Z.; Li, B.; Ren, Y.; Ye, Z. T-cell-based immunotherapy for osteosarcoma: Challenges and opportunities. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Corthay, A. How do regulatory t cells work? Scand. J. Immunol. 2009, 70, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef]
- Heymann, M.F.; Lézot, F.; Heymann, D. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell. Immunol. 2019, 343, 103711. [Google Scholar] [CrossRef] [PubMed]
- Cersosimo, F.; Lonardi, S.; Bernardini, G.; Telfer, B.; Mandelli, G.E.; Santucci, A.; Vermi, W.; Giurisato, E. Tumor-associated macrophages in osteosarcoma: From mechanisms to therapy. Int. J. Mol. Sci. 2020, 21, 5207. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, G.; Chen, R.; Hua, Y.; Cai, Z. Mesenchymal stem cells in the osteosarcoma microenvironment: Their biological properties, influence on tumor growth, and therapeutic implications. Stem Cell Res. Ther. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Corre, I.; Verrecchia, F.; Crenn, V.; Redini, F.; Trichet, V. The Osteosarcoma Microenvironment: A Complex But Targetable Ecosystem. Cells 2020, 9, 976. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Kawaguchi, S.; Torigoe, T.; Asanuma, H.; Nakazawa, E.; Shimozawa, K.; Nabeta, Y.; Kimura, S.; Kaya, M.; Nagoya, S.; et al. Prognostic significance of HLA class I expression in osteosarcoma defined by anti-pan HLA class I monoclonal antibody, EMR8-5. Cancer Sci. 2006, 97, 1374–1380. [Google Scholar] [CrossRef]
- Tarek, N.; Lee, D.A. Natural killer cells for osteosarcoma. Curr. Adv. Osteosarcoma 2014, 341–353. [Google Scholar] [CrossRef]
- Li, Z. Potential of human γδ T cells for immunotherapy of osteosarcoma. Mol. Biol. Rep. 2013, 40, 427–437. [Google Scholar] [CrossRef]
- Shahriyari, L.; Komarova, N.L. Symmetric vs. asymmetric stem cell divisions: An adaptation against cancer? PLoS ONE 2013, 8, e76195. [Google Scholar] [CrossRef] [PubMed]
- Shahriyari, L.; Komarova, N.L. The role of the bi-compartmental stem cell niche in delaying cancer. Phys. Biol. 2015, 12, 055001. [Google Scholar] [CrossRef]
- Shahriyari, L.; Mahdipour-Shirayeh, A. Modeling dynamics of mutants in heterogeneous stem cell niche. Phys. Biol. 2017, 14. [Google Scholar] [CrossRef]
- Bollas, A.; Shahriyari, L. The role of backward cell migration in two-hit mutants’ production in the stem cell niche. PLoS ONE 2017, 12, 2017. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.; Enderling, H. Mathematical Models of Cancer: When to Predict Novel Therapies, and When Not to. Bull. Math. Biol. 2019, 81, 3722–3731. [Google Scholar] [CrossRef]
- Chamseddine, I.M.; Rejniak, K.A. Hybrid modeling frameworks of tumor development and treatment. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- MOREIRA, J.; DEUTSCH, A. Cellular automaton models of tumor development: A critical review. Adv. Complex Syst. 2002, 5, 247–267. [Google Scholar] [CrossRef]
- Lowengrub, J.S.; Frieboes, H.B.; Jin, F.; Chuang, Y.L.; Li, X.; Macklin, P.; Wise, S.M.; Cristini, V. Nonlinear modelling of cancer: Bridging the gap between cells and tumours. Nonlinearity 2010, 23, R1–R91. [Google Scholar] [CrossRef]
- Shahriyari, L. Cell dynamics in tumour environment after treatments. J. R. Soc. Interface 2017, 14, 20160977. [Google Scholar] [CrossRef]
- Ji, B.; Chen, J.; Zhen, C.; Yang, Q.; Yu, N. Mathematical modelling of the role of Endo180 network in the development of metastatic bone disease in prostate cancer. Comput. Biol. Med. 2020, 117, 103619. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cervantes, I.; Morales, M.A.; Agustín-Serrano, R.; Cardenas-García, M.; Pérez-Luna, P.V.; Arroyo-Reyes, B.L.; Maldonado-García, A. Polylactic acid/sodium alginate/hydroxyapatite composite scaffolds with trabecular tissue morphology designed by a bone remodeling model using 3D printing. J. Mater. Sci. 2019, 9478–9496. [Google Scholar] [CrossRef]
- Burova, I.; Peticone, C.; De Silva Thompson, D.; Knowles, J.C.; Wall, I.; Shipley, R.J. A parameterised mathematical model to elucidate osteoblast cell growth in a phosphate-glass microcarrier culture. J. Tissue Eng. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Haghiralsadat, F.; Amoabediny, G.; Naderinezhad, S.; Nazmi, K.; De Boer, J.P.; Zandieh-Doulabi, B.; Forouzanfar, T.; Helder, M.N. EphA2 Targeted Doxorubicin-Nanoliposomes for Osteosarcoma Treatment. Pharm. Res. 2017, 34, 2891–2900. [Google Scholar] [CrossRef]
- Lui, G.; Treluyer, J.M.; Fresneau, B.; Piperno-Neumann, S.; Gaspar, N.; Corradini, N.; Gentet, J.C.; Marec Berard, P.; Laurence, V.; Schneider, P.; et al. A Pharmacokinetic and Pharmacogenetic Analysis of Osteosarcoma Patients Treated With High-Dose Methotrexate: Data From the OS2006/Sarcoma-09 Trial. J. Clin. Pharmacol. 2018, 58, 1541–1549. [Google Scholar] [CrossRef]
- Kather, J.N.; Poleszczuk, J.; Suarez-Carmona, M.; Krisam, J.; Charoentong, P.; Valous, N.A.; Weis, C.A.; Tavernar, L.; Leiss, F.; Herpel, E.; et al. In Silico Modeling of Immunotherapy and Stroma-Targeting Therapies in Human Colorectal Cancer. Cancer Res. 2017, 77, 6442–6452. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, S.A.; Rambely, A.S. A New ODE-Based Model for Tumor Cells and Immune System Competition. Mathematics 2020, 8, 1285. [Google Scholar] [CrossRef]
- de Pillis, L.; Savage, H.; Radunskaya, A. Mathematical model of colorectal cancer with monoclonal antibody treatments. arXiv 2013, arXiv:1312.3023. [Google Scholar]
- Kirshtein, A.; Akbarinejad, S.; Hao, W.; Le, T.; Su, S.; Aronow, R.A.; Shahriyari, L. Data Driven Mathematical Model of Colon Cancer Progression. J. Clin. Med. 2020, 9, 3947. [Google Scholar] [CrossRef]
- Le, T.; Su, S.; Shahriyari, L. Immune Classification of Osteosarcoma. Math. Biosci. Eng. 2021, 18, 1–16. [Google Scholar] [CrossRef]
- Byrne, H.M.; Cox, S.M.; Kelly, C. Macrophage-tumour interactions: In vivo dynamics. Discret. Contin. Dyn. Syst. B 2004, 4, 81. [Google Scholar]
- de Pillis, L.; Caldwell, T.; Sarapata, E.; Williams, H. Mathematical modeling of regulatory T cell effects on renal cell carcinoma treatment. Discret. Contin. Dyn. Syst. B 2013, 18, 915. [Google Scholar]
- Robertson-Tessi, M.; El-Kareh, A.; Goriely, A. A mathematical model of tumor–immune interactions. J. Theor. Biol. 2012, 294, 56–73. [Google Scholar] [CrossRef]
- Robertson-Tessi, M.; El-Kareh, A.; Goriely, A. A model for effects of adaptive immunity on tumor response to chemotherapy and chemoimmunotherapy. J. Theor. Biol. 2015, 380, 569–584. [Google Scholar] [CrossRef]
- de Pillis, L.; Renee Fister, K.; Gu, W.; Collins, C.; Daub, M.; Gross, D.; Moore, J.; Preskill, B. Mathematical model creation for cancer chemo-immunotherapy. Comput. Math. Methods Med. 2009, 10, 165–184. [Google Scholar] [CrossRef]
- Heymann, M.F.; Heymann, D. Immune Environment and Osteosarcoma. Colloids Surf. A Physicochem. Eng. 2012, 38. [Google Scholar] [CrossRef]
- Kelleher, F.C.; O’Sullivan, H. Monocytes, Macrophages, and Osteoclasts in Osteosarcoma. J. Adolesc. Young Adult Oncol. 2017, 6, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Whelan, J.; Patterson, D.; Perisoglou, M.; Bielack, S.; Marina, N.; Smeland, S.; Bernstein, M. The role of interferons in the treatment of osteosarcoma. Pediatr. Blood Cancer 2010, 54, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Dumitriu, I.E.; Baruah, P.; Manfredi, A.A.; Bianchi, M.E.; Rovere-Querini, P. HMGB1: Guiding immunity from within. Trends Immunol. 2005, 26, 381–387. [Google Scholar] [CrossRef]
- Rovere-Querini, P.; Capobianco, A.; Scaffidi, P.; Valentinis, B.; Catalanotti, F.; Giazzon, M.; Dumitriu, I.E.; Müller, S.; Iannacone, M.; Traversari, C.; et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004, 5, 825–830. [Google Scholar] [CrossRef]
- Yang, J.; Ma, Z.; Wang, Y.; Wang, Z.; Tian, Y.; Du, Y.; Bian, W.; Duan, Y.; Liu, J. Necrosis of osteosarcoma cells induces the production and release of high-mobility group box 1 protein. Exp. Ther. Med. 2018, 15, 461–466. [Google Scholar] [CrossRef]
- Parker, K.H.; Sinha, P.; Horn, L.A.; Clements, V.K.; Yang, H.; Li, J.; Tracey, K.J.; Ostrand-Rosenberg, S. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014, 74, 5723–5733. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zhang, Q.; Zeh, H.J.; Lotze, M.T.; Tang, D. HMGB1 in cancer: Good, bad, or both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef]
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous danger signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef]
- Ranzato, E.; Martinotti, S.; Patrone, M. Emerging roles for HMGB1 protein in immunity, inflammation, and cancer. Immunotargets Ther. 2015, 101. [Google Scholar] [CrossRef]
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36–44. [Google Scholar] [CrossRef]
- Pahl, J.H.; Kwappenberg, K.M.; Varypataki, E.M.; Santos, S.J.; Kuijjer, M.L.; Mohamed, S.; Wijnen, J.T.; van Tol, M.J.D.; Cleton-Jansen, A.-M.; Schilham, M.W.; et al. Macrophages inhibit human osteosarcoma cell growth after activation with the bacterial cell wall derivative liposomal muramyl tripeptide in combination with interferon-(gamma). J. Exp. Clin. Cancer Res. 2014, 33, 1–13. [Google Scholar] [CrossRef]
- Jacobson, N.G.; Szabo, S.J.; Weber-Nordt, R.M.; Zhong, Z.; Schreiber, R.D.; Darnell, J.E., Jr.; Murphy, K.M. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat) 3 and Stat4. J. Exp. Med. 1995, 181, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.A.; Li, M.O. TGF-β: Guardian of T cell function. J. Immunol. 2013, 191, 3973–3979. [Google Scholar] [CrossRef] [PubMed]
- Lafont, V.; Sanchez, F.; Laprevotte, E.; Michaud, H.A.; Gros, L.; Eliaou, J.F.; Bonnefoy, N. Plasticity of γδ T cells: Impact on the anti-tumor response. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.J.; Ornelles, D.A.; Mitchell, L.M.; Brzoza-Lewis, K.L.; Hiltbold, E.M. IL-12 produced by dendritic cells augments CD8+ T cell activation through the production of the chemokines CCL1 and CCL17. J. Immunol. 2008, 181, 8576–8584. [Google Scholar] [CrossRef]
- Li, L.; Jay, S.M.; Wang, Y.; Wu, S.W.; Xiao, Z. IL-12 stimulates CTLs to secrete exosomes capable of activating bystander CD8+ T cells. Sci. Rep. 2017, 7, 13365. [Google Scholar] [CrossRef]
- Dyson, K.A.; Stover, B.D.; Grippin, A.; Mendez-Gomez, H.R.; Lagmay, J.; Mitchell, D.A.; Sayour, E.J. Emerging trends in immunotherapy for pediatric sarcomas. J. Hematol. Oncol. 2019, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lamora, A.; Talbot, J.; Mullard, M.; Brounais-Le Royer, B.; Redini, F.; Verrecchia, F. TGF-β Signaling in Bone Remodeling and Osteosarcoma Progression. J. Clin. Med. 2016, 5, 96. [Google Scholar] [CrossRef] [PubMed]
- Tsukumo, S.I.; Yasutomo, K. Regulation of CD8+ T cells and antitumor immunity by Notch signaling. Front. Immunol. 2018, 9, 101. [Google Scholar] [CrossRef]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Hochberg, M. Self-regulation of growth in three dimensions. J. Exp. Med. 1973, 138, 745–753. [Google Scholar] [CrossRef]
- Enderling, H.; Sunassee, E.; Caudell, J.J. Predicting patient-specific radiotherapy responses in head and neck cancer to personalize radiation dose fractionation. bioRxiv 2019, 630806. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Le, T.; Aronow, R.A.; Kirshtein, A.; Shahriyari, L. A review of digital cytometry methods: Estimating the relative abundance of cell types in a bulk of cells. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Su, S.; Akbarinejad, S.; Shahriyari, L. Immune classification of clear cell renal cell carcinoma. Sci. Rep. 2021, 11, 4338. [Google Scholar] [CrossRef] [PubMed]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.; Bürger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.; Lankester, A.C.; et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: A rationale for treatment with macrophage activating agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Arthur, D.; Vassilvitskii, S. K-means++: The advantages of careful seeding. In Proceedings of the 18th Annual ACM-SIAM Symposium on Discrete Algorithms, New Orleans, LA, USA, 7–9 January 2007. [Google Scholar]
- Yuan, C.; Yang, H. Research on K-value selection method of K-means clustering algorithm. J Multidiscip. Sci. J. 2019, 2, 226–235. [Google Scholar] [CrossRef]
- Kasalak, Ö.; Overbosch, J.; Glaudemans, A.W.; Boellaard, R.; Jutte, P.C.; Kwee, T.C. Primary tumor volume measurements in Ewing sarcoma: MRI inter-and intraobserver variability and comparison with FDG-PET. Acta Oncol. 2018, 57, 534–540. [Google Scholar] [CrossRef]
- Grimer, R.J. Size matters for sarcomas! Ann. R. Coll. Surg. Engl. 2006, 88, 519–524. [Google Scholar] [CrossRef]
- Qiu, Z.Y.; Cui, Y.; Wang, X.M. Natural bone tissue and its biomimetic. In Mineralized Collagen Bone Graft Substitutes; Woodhead Publishing: Cambridge, UK, 2019; pp. 1–22. [Google Scholar]
- Hao, W.; Crouser, E.D.; Friedman, A. Mathematical model of sarcoidosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16065–16070. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Friedman, A. Mathematical model on Alzheimer’s disease. BMC Syst. Biol. 2016, 10, 108. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Zi, Z. Sensitivity analysis approaches applied to systems biology models. IET Syst. Biol. 2011, 5, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Heiss, F.; Winschel, V. Likelihood approximation by numerical integration on sparse grids. J. Econom. 2008, 144, 62–80. [Google Scholar] [CrossRef]
- Gerstner, T.; Griebel, M. Numerical integration using sparse grids. Numer. Algorithms 1998, 18, 209–232. [Google Scholar] [CrossRef]
- Fu, C.; Jiang, A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef] [PubMed]
- Kim, R. Cancer immunoediting: From immune surveillance to immune escape. Cancer Immunother. 2007, 9–27. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Werner, H.M.J.; Mills, G.B.; Ram, P.T. Cancer Systems Biology: A peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014, 11, 167–176. [Google Scholar] [CrossRef]
- Hoffman, F.; Gavaghan, D.; Osborne, J.; Barrett, I.; You, T.; Ghadially, H.; Sainson, R.; Wilkinson, R.; Byrne, H. A mathematical model of antibody-dependent cellular cytotoxicity (ADCC). J. Theor. Biol. 2018, 436, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Mahasa, K.J.; Ouifki, R.; Eladdadi, A.; de Pillis, L. Mathematical model of tumor–immune surveillance. J. Theor. Biol. 2016, 404, 312–330. [Google Scholar] [CrossRef]
- den Breems, N.Y.; Eftimie, R. The re-polarisation of M2 and M1 macrophages and its role on cancer outcomes. J. Theor. Biol. 2016, 390, 23–39. [Google Scholar] [CrossRef]
- Frascoli, F.; Kim, P.S.; Hughes, B.D.; Landman, K.A. A dynamical model of tumour immunotherapy. Math. Biosci. 2014, 253, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Chappell, M.; Chelliah, V.; Cherkaoui, M.; Derks, G.; Dumortier, T.; Evans, N.; Ferrarini, M.; Fornari, C.; Ghazal, P.; Guerriero, M.; et al. Mathematical modelling for combinations of immuno-oncology and anti-cancer therapies. In Proceedings of the Report QSP UK Meet, Macclesfield, UK, 14–17 September 2015; pp. 1–15. [Google Scholar]
- Kaur, G.; Ahmad, N. On study of immune response to tumor cells in prey-predator system. Int. Sch. Res. Not. 2014, 2014, 346597. [Google Scholar] [CrossRef] [PubMed]
- de Pillis, L.; Gallegos, A.; Radunskaya, A. A model of dendritic cell therapy for melanoma. Front. Oncol. 2013, 3, 56. [Google Scholar]
- López, Á.G.; Seoane, J.M.; Sanjuán, M.A. A validated mathematical model of tumor growth including tumor–host interaction, cell-mediated immune response and chemotherapy. Bull. Math. Biol. 2014, 76, 2884–2906. [Google Scholar] [CrossRef]
- Wilkie, K.P.; Hahnfeldt, P. Modeling the dichotomy of the immune response to cancer: Cytotoxic effects and tumor-promoting inflammation. Bull. Math. Biol. 2017, 79, 1426–1448. [Google Scholar] [CrossRef]
- Bekisz, S.; Geris, L. Cancer modeling: From mechanistic to data-driven approaches, and from fundamental insights to clinical applications. J. Comput. Sci. 2020, 46, 101198. [Google Scholar] [CrossRef]
- Heymann, M.F.; Heymann, D. Immune environment and osteosarcoma. In Osteosarcoma-Biology, Behavior and Mechanisms; InTech: London, UK, 2017; pp. 105–120. [Google Scholar]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Advances in immune checkpoint inhibitors for bone sarcoma therapy. J. Bone Oncol. 2019, 15, 100221. [Google Scholar] [CrossRef] [PubMed]
- Fritzsching, B.; Fellenberg, J.; Moskovszky, L.; Sápi, Z.; Krenacs, T.; Machado, I.; Poeschl, J.; Lehner, B.; Szendroi, M.; Bosch, A.L.; et al. CD8+/FOXP3+-ratio in osteosarcoma microenvironment separates survivors from non-survivors: A multicenter validated retrospective study. Oncoimmunology 2015, 4, e990800. [Google Scholar] [CrossRef]
- Asano, Y.; Kashiwagi, S.; Goto, W.; Kurata, K.; Noda, S.; Takashima, T.; Onoda, N.; Tanaka, S.; Ohsawa, M.; Hirakawa, K. Tumour-infiltrating CD8 to FOXP3 lymphocyte ratio in predicting treatment responses to neoadjuvant chemotherapy of aggressive breast cancer. Br. J. Surg. 2016, 103, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Nirei, T.; Sunami, E.; Nagawa, H.; Kitayama, J. Density of CD4 (+) and CD8 (+) T lymphocytes in biopsy samples can be a predictor of pathological response to chemoradiotherapy (CRT) for rectal cancer. Radiat. Oncol. 2011, 6, 1–6. [Google Scholar] [CrossRef]
- Riemann, D.; Cwikowski, M.; Turzer, S.; Giese, T.; Grallert, M.; Schütte, W.; Seliger, B. Blood immune cell biomarkers in lung cancer. Clin. Exp. Immunol. 2019, 195, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Riemann, D.; Hase, S.; Fischer, K.; Seliger, B. Granulocyte-to-dendritic cell-ratio as marker for the immune monitoring in patients with renal cell carcinoma. Clin. Transl. Med. 2014, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Sherratt, J.A. Modelling the macrophage invasion of tumours: Effects on growth and composition. Math. Med. Biol. J. IMA 1998, 15, 165–185. [Google Scholar] [CrossRef]
- de Pillis, L.; Radunskaya, A. A mathematical tumor model with immune resistance and drug therapy: An optimal control approach. Comput. Math. Methods Med. 2001, 3, 79–100. [Google Scholar] [CrossRef]
- de Pillis, L.; Radunskaya, A. The dynamics of an optimally controlled tumor model: A case study. Math. Comput. Model. 2003, 37, 1221–1244. [Google Scholar] [CrossRef]
- Voss, A.; Voss, J. A fast numerical algorithm for the estimation of diffusion model parameters. J. Math. Psychol. 2008, 52, 1–9. [Google Scholar] [CrossRef]
- Parra-Rojas, C.; Hernandez-Vargas, E.A. PDEparams: Parameter fitting toolbox for partial differential equations in python. Bioinformatics 2020, 36, 2618–2619. [Google Scholar] [CrossRef]
- Vyshemirsky, V.; Girolami, M. BioBayes: A software package for Bayesian inference in systems biology. Bioinformatics 2008, 24, 1933–1934. [Google Scholar] [CrossRef] [PubMed]
- Xun, X.; Cao, J.; Mallick, B.; Maity, A.; Carroll, R.J. Parameter estimation of partial differential equation models. J. Am. Stat. Assoc. 2013, 108, 1009–1020. [Google Scholar] [CrossRef]
- Tran, L.; Allen, C.T.; Xiao, R.; Moore, E.; Davis, R.; Park, S.J.; Spielbauer, K.; Van Waes, C.; Schmitt, N.C. Cisplatin alters antitumor immunity and synergizes with PD-1/PD-L1 inhibition in head and neck squamous cell carcinoma. Cancer Immunol. Res. 2017, 5, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Crum, E.D. Effect of cisplatin upon expression of in vivo immune tumor resistance. Cancer Immunol. Immunother. 1993, 36, 18–24. [Google Scholar] [CrossRef]
- Yin, Y.; Hu, Q.; Xu, C.; Qiao, Q.; Qin, X.; Song, Q.; Peng, Y.; Zhao, Y.; Zhang, Z. Co-delivery of doxorubicin and interferon-γ by thermosensitive nanoparticles for cancer immunochemotherapy. Mol. Pharm. 2018, 15, 4161–4172. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Tanaka, K.; Itonaga, I.; Iwasaki, T.; Miyazaki, M.; Ikeda, S.; Tsumura, H. Dendritic cells combined with doxorubicin induces immunogenic cell death and exhibits antitumor effects for osteosarcoma. Oncol. Lett. 2016, 11, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Waguespack, M.; Barker, S.A.; Li, S. Doxorubicin Directs the Accumulation of Interleukin-12–Induced IFNγ into Tumors for Enhancing STAT1–Dependent Antitumor Effect. Clin. Cancer Res. 2007, 13, 4252–4260. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grossman, S.A.; Ye, X.; Lesser, G.; Sloan, A.; Carraway, H.; Desideri, S.; Piantadosi, S. Immunosuppression in Patients with High-Grade Gliomas Treated with Radiation and Temozolomide. Clin. Cancer Res. 2011, 17, 5473–5480. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, H.d.A.; Villar, R.C. Radiotherapy and immune response: The systemic effects of a local treatment. Clinics 2018, 73. [Google Scholar] [CrossRef] [PubMed]
- Yagawa, Y.; Tanigawa, K.; Kobayashi, Y.; Yamamoto, M. Cancer immunity and therapy using hyperthermia with immunotherapy, radiotherapy, chemotherapy, and surgery. J. Cancer Metastasis Treat. 2017, 3, 218–230. [Google Scholar] [CrossRef]
- Spratt, J.S., Jr. The rates of growth of skeletal sarcomas. Cancer 1965, 18, 14–24. [Google Scholar] [CrossRef]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.A.; Bigley, V.; Flavell, R.A.; Gilroy, D.W.; Asquith, B.; et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Allers, C.; Sugimoto, C.; Ahmed, N.; Fujioka, H.; Kim, W.K.; Didier, E.S.; Kuroda, M.J. Rapid turnover and high production rate of myeloid cells in adult rhesus macaques with compensations during aging. J. Immunol. 2018, 200, 4059–4067. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Hao, W.; Friedman, A. Serum upar as biomarker in breast cancer recurrence: A mathematical model. PLoS ONE 2016, 11, e0153508. [Google Scholar] [CrossRef] [PubMed]
- Farber, D.L.; Yudanin, N.A.; Restifo, N.P. Human memory T cells: Generation, compartmentalization and homeostasis. Nat. Rev. Immunol. 2014, 14, 24–35. [Google Scholar] [CrossRef]
- De Boer, R.J.; Homann, D.; Perelson, A.S. Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J. Immunol. 2003, 171, 3928–3935. [Google Scholar] [CrossRef]
- Vukmanovic-Stejic, M.; Zhang, Y.; Cook, J.E.; Fletcher, J.M.; McQuaid, A.; Masters, J.E.; Rustin, M.H.; Taams, L.S.; Beverley, P.C.; Macallan, D.C.; et al. Human CD4+ CD25 hi Foxp3+ regulatory T cells are derived by rapid turnover of memory populations in vivo. J. Clin. Investig. 2006, 116, 2423–2433. [Google Scholar] [CrossRef]
- Cella, M.; Engering, A.; Pinet, V.; Pieters, J.; Lanzavecchia, A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature 1997, 388, 782–787. [Google Scholar] [CrossRef]
- Diao, J.; Winter, E.; Cantin, C.; Chen, W.; Xu, L.; Kelvin, D.; Phillips, J.; Cattral, M.S. In situ replication of immediate dendritic cell (DC) precursors contributes to conventional DC homeostasis in lymphoid tissue. J. Immunol. 2006, 176, 7196–7206. [Google Scholar] [CrossRef]
- Foon, K.A.; Sherwin, S.A.; Abrams, P.G.; Stevenson, H.C.; Holmes, P.; Maluish, A.E.; Oldham, R.K.; Herberman, R.B. A phase I trial of recombinant gamma interferon in patients with cancer. Cancer Immunol. Immunother. 1985, 20, 193–197. [Google Scholar] [CrossRef]
- Fuentes-Calvo, I.; Martínez-Salgado, C. TGFB1 (transforming growth factor, beta 1). In Atlas of Genetics and Cytogenetics in Oncology and Haematology; 2013; Available online: http://atlasgeneticsoncology.org/Genes/GC_TGFB1.html (accessed on 3 December 2020).
- Saxena, A.; Khosraviani, S.; Noel, S.; Mohan, D.; Donner, T.; Hamad, A.R.A. Interleukin-10 paradox: A potent immunoregulatory cytokine that has been difficult to harness for immunotherapy. Cytokine 2015, 74, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Conlon, P.; Tyler, S.; Grabstein, K.; Morrissey, P. Interleukin-4 (B-cell stimulatory factor-1) augments the in vivo generation of cytotoxic cells in immunosuppressed animals. Biotechnol. Ther. 1989, 1, 31–41. [Google Scholar] [PubMed]
- Khodoun, M.; Lewis, C.; Yang, J.Q.; Orekov, T.; Potter, C.; Wynn, T.; Mentink-Kane, M.; Hershey, G.K.K.; Wills-Karp, M.; Finkelman, F.D. Differences in expression, affinity, and function of soluble (s) IL-4Rα and sIL-13Rα2 suggest opposite effects on allergic responses. J. Immunol. 2007, 179, 6429–6438. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Storfer-Isser, A.; Kirchner, H.L.; Johnson, N.; Jenny, N.; Tracy, R.P.; Redline, S. Soluble interleukin 6 receptor: A novel marker of moderate to severe sleep-related breathing disorder. Arch. Intern. Med. 2006, 166, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Balestrino, M. Cytokine Imbalances in Multiple Sclerosis: A Computer Simulation. Master’s Thesis, Cornell University, Ithaca, NY, USA, 2009. [Google Scholar]
- Zandarashvili, L.; Sahu, D.; Lee, K.; Lee, Y.S.; Singh, P.; Rajarathnam, K.; Iwahara, J. Real-time kinetics of high-mobility group box 1 (HMGB1) oxidation in extracellular fluids studied by in situ protein NMR spectroscopy. J. Biol. Chem. 2013, 288, 11621–11627. [Google Scholar] [CrossRef] [PubMed]







| Variable | Name | Description |
|---|---|---|
| Naive T-cells | ||
| Helper T-cells | ||
| Cytotoxic cells | includes CD8+ T-cells and NK cells | |
| Regulatory T-cells | ||
| Naive dendritic cells | ||
| D | Activated dendritic cells | antigen presenting cells |
| Naive macrophages | includes naive macrophages and monocytes | |
| M | Macrophages | includes M1 macrophages and M2 macrophages |
| C | Cancer cells | |
| N | Nectrotic cells | |
| H | HMGB1 | |
| Cytokines group | includes effects of TGF-, IL-4, IL-10 and IL-13 | |
| Cytokines group | includes effects of IL-6 and IL-17 | |
| IFN- |
| Cluster | |||||
|---|---|---|---|---|---|
| 1 | |||||
| 2 | |||||
| 3 | |||||
| 1 | |||||
| 2 | |||||
| 3 | |||||
| 1 | 0.868 | 21.510 | 2.067 | 5.076 | |
| 2 | 0.049 | 20.714 | 1.611 | 4.948 | |
| 3 | 0.263 | 23.663 | 1.371 | 4.453 |
| Cluster | |||||||
|---|---|---|---|---|---|---|---|
| 1 | 2.367 | 1.005 | 0.019 | 0.794 | 0.764 | 0.828 | 1.122 |
| 2 | 0.954 | 0.753 | 1.299 | 1.451 | 2.313 | 0.062 | 0.071 |
| 3 | 0.866 | 1.104 | 0.572 | 0.340 | 0.484 | 0 | 1.643 |
| 1 | 0 | 0.020 | 0.160 | 2.394 | 1.104 | 1.806 | 1.059 |
| 2 | 0.693 | 0.005 | 0.018 | 0.859 | 1.307 | 3.259 | 0.988 |
| 3 | 0 | 0.014 | 0.0008 | 0.276 | 1.030 | 1.296 | 1.284 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, T.; Su, S.; Kirshtein, A.; Shahriyari, L. Data-Driven Mathematical Model of Osteosarcoma. Cancers 2021, 13, 2367. https://doi.org/10.3390/cancers13102367
Le T, Su S, Kirshtein A, Shahriyari L. Data-Driven Mathematical Model of Osteosarcoma. Cancers. 2021; 13(10):2367. https://doi.org/10.3390/cancers13102367
Chicago/Turabian StyleLe, Trang, Sumeyye Su, Arkadz Kirshtein, and Leili Shahriyari. 2021. "Data-Driven Mathematical Model of Osteosarcoma" Cancers 13, no. 10: 2367. https://doi.org/10.3390/cancers13102367
APA StyleLe, T., Su, S., Kirshtein, A., & Shahriyari, L. (2021). Data-Driven Mathematical Model of Osteosarcoma. Cancers, 13(10), 2367. https://doi.org/10.3390/cancers13102367