Natural Killer Cell Activation Receptor NKp30 Oligomerization Depends on Its N-Glycosylation

 , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Results
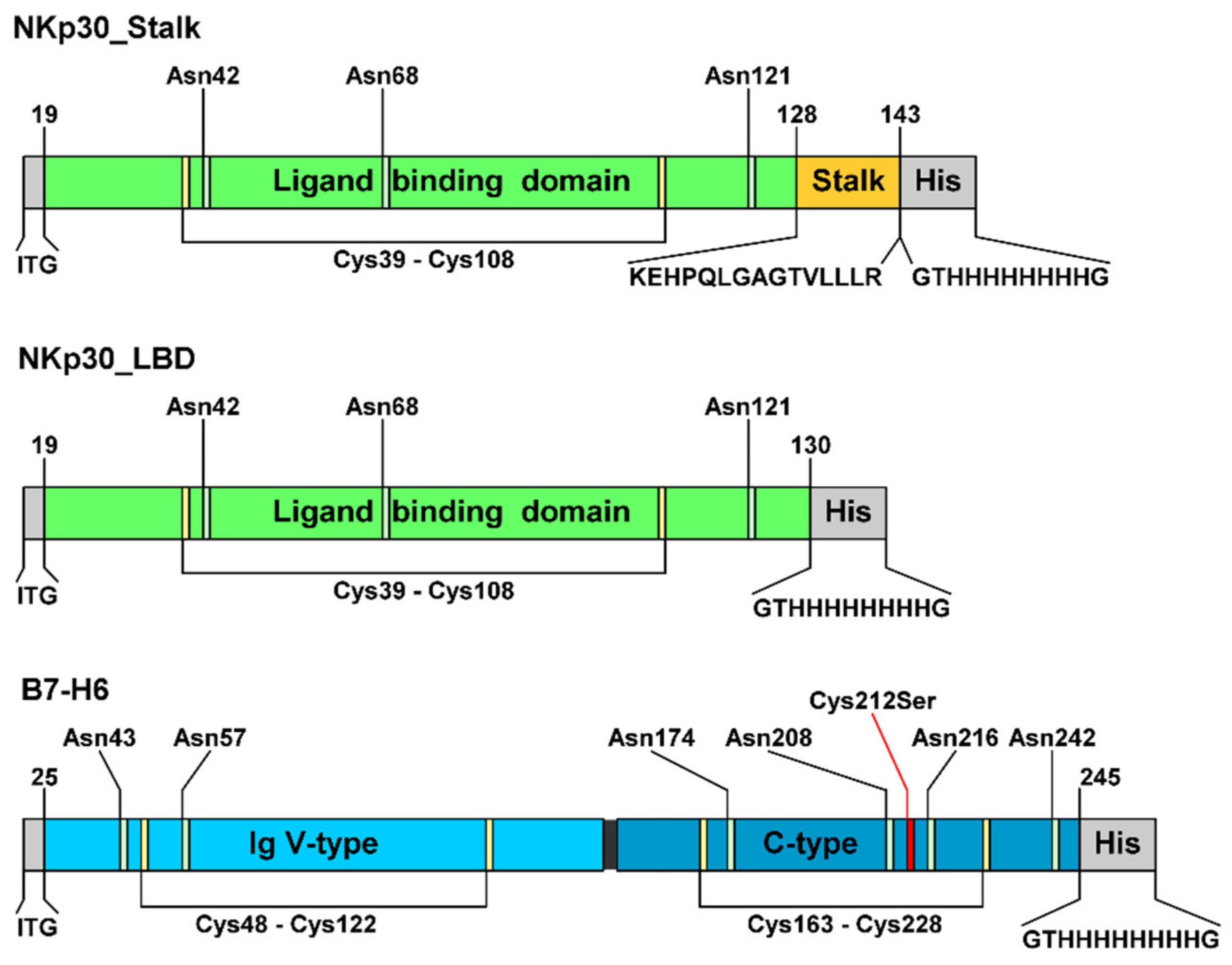
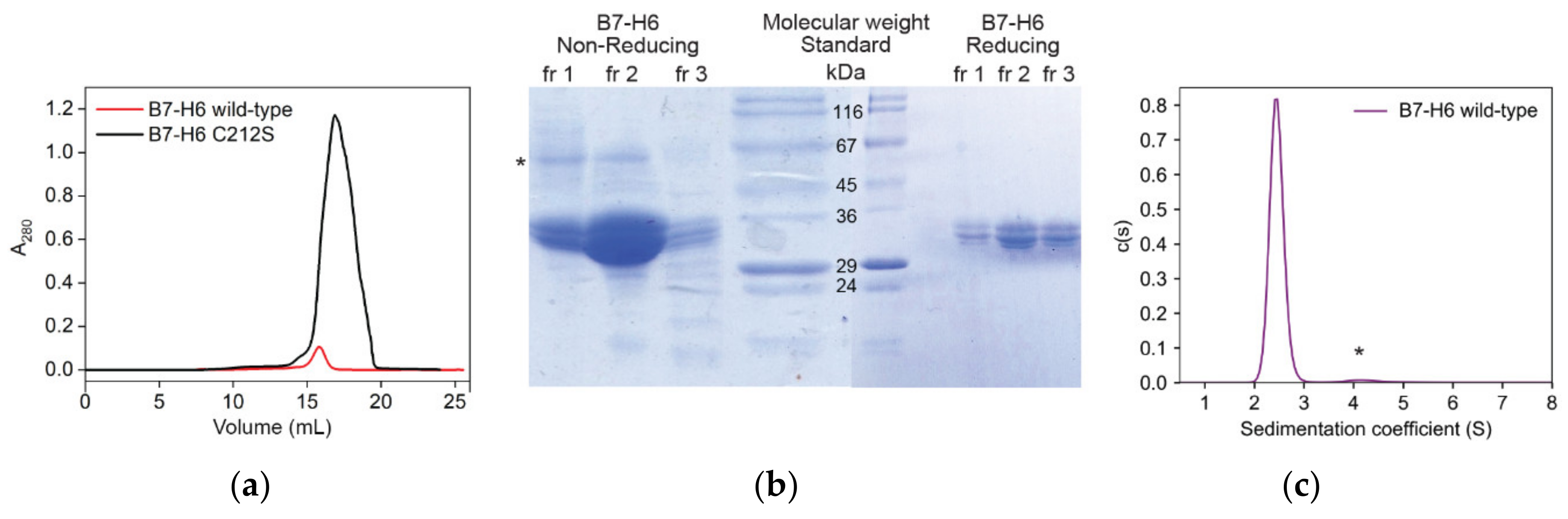
2.1. Recombinant Expression and Purification of Stable, Soluble, Glycosylated NKp30, and B7-H6 Proteins
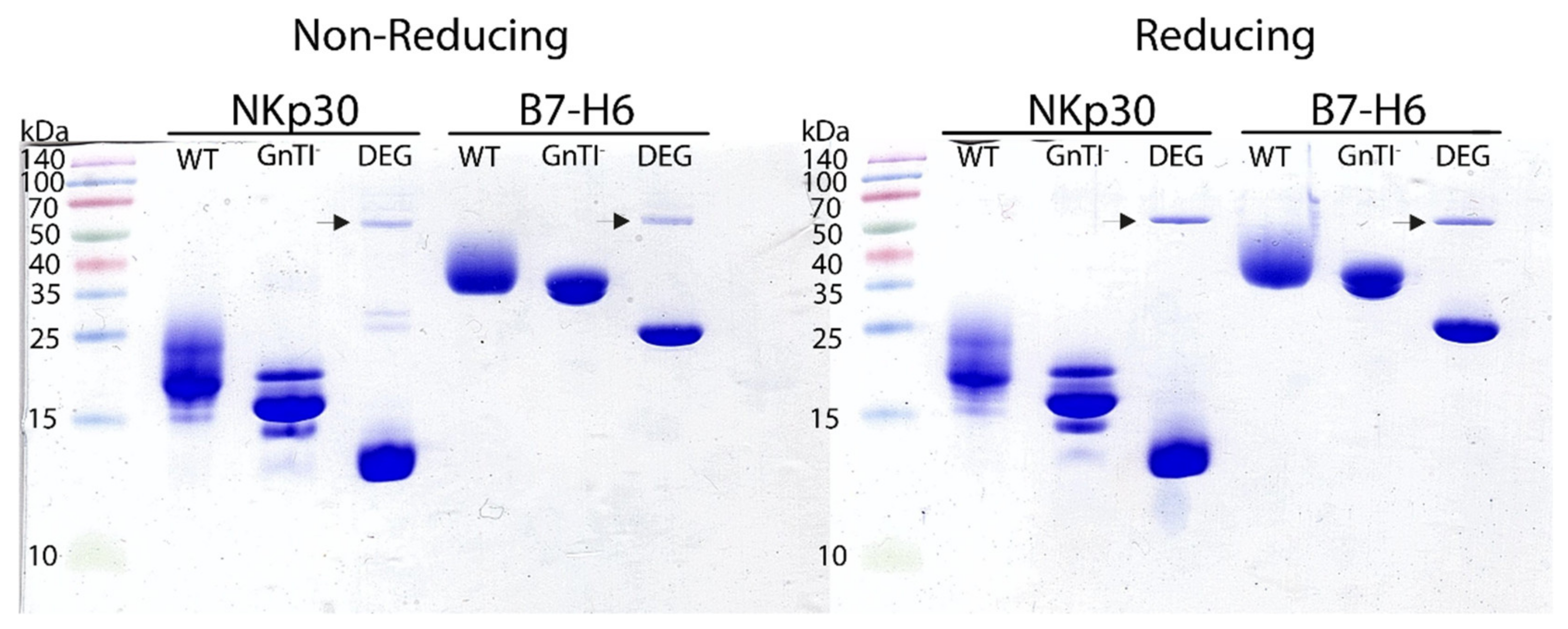
2.2. Protein Deglycosylation
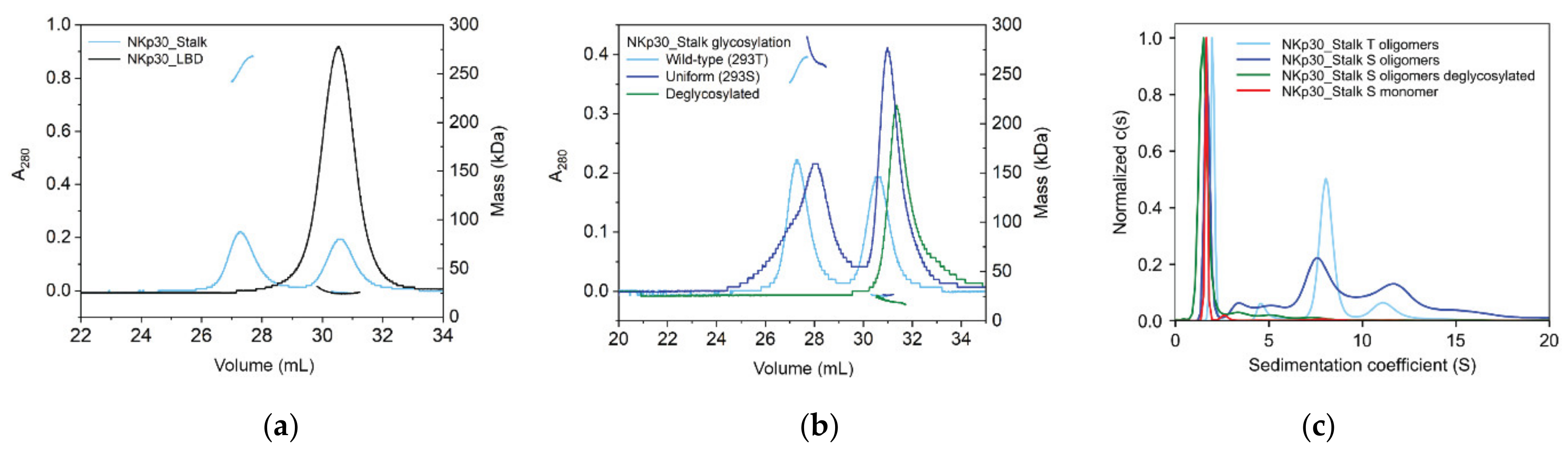
2.3. NKp30 Glycosylation Promotes Its Oligomerization
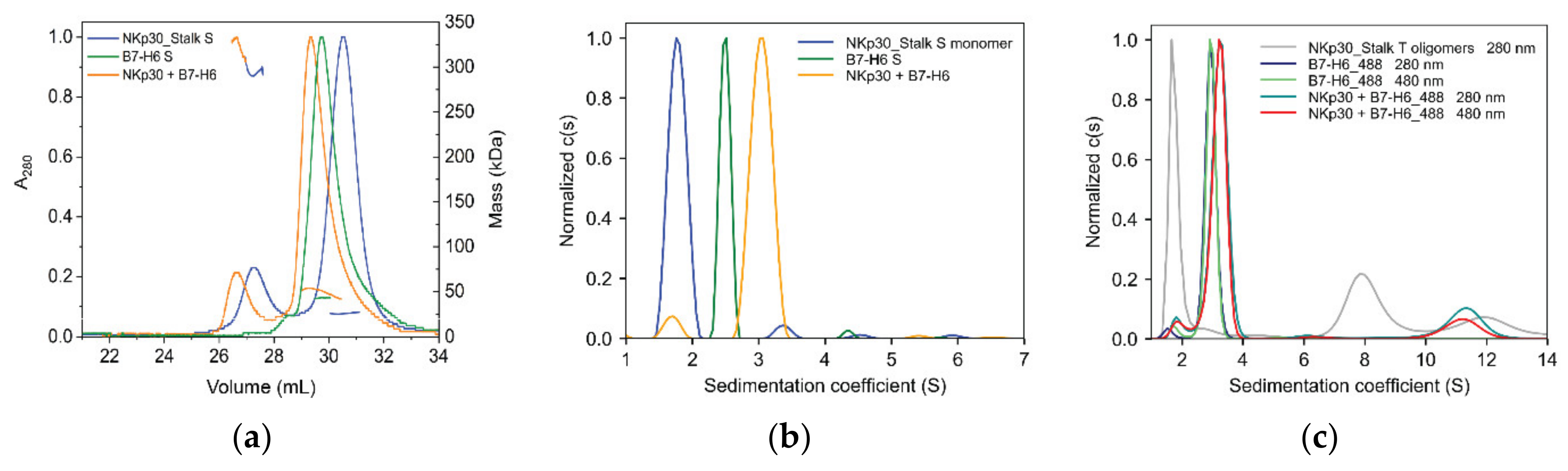
2.4. B7-H6 Forms Equimolar Complex with Monomeric NKp30, But Not with Its Oligomeric Form
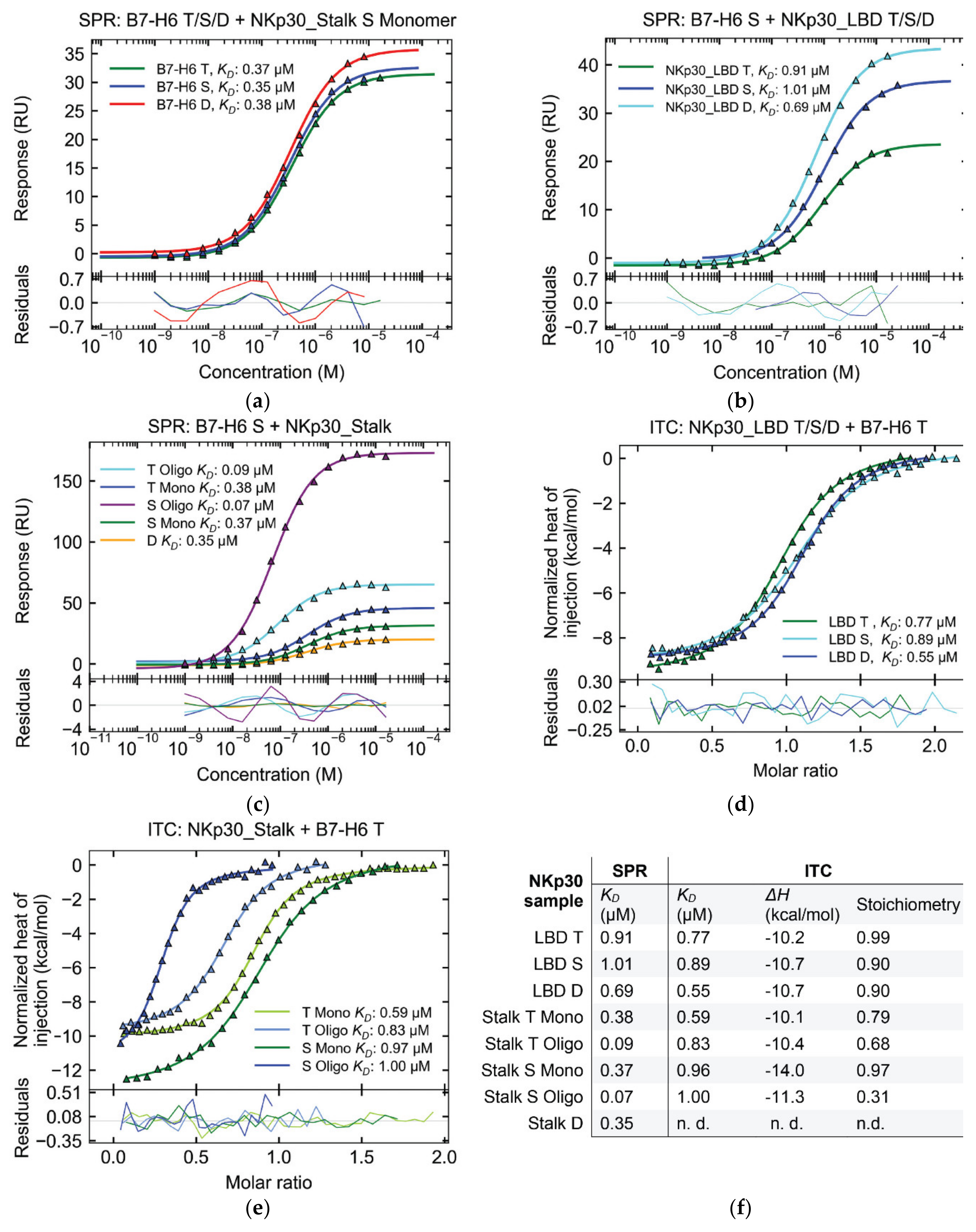
2.5. Affinity of NKp30:B7-H6 Interaction Differs between Surface and Solution
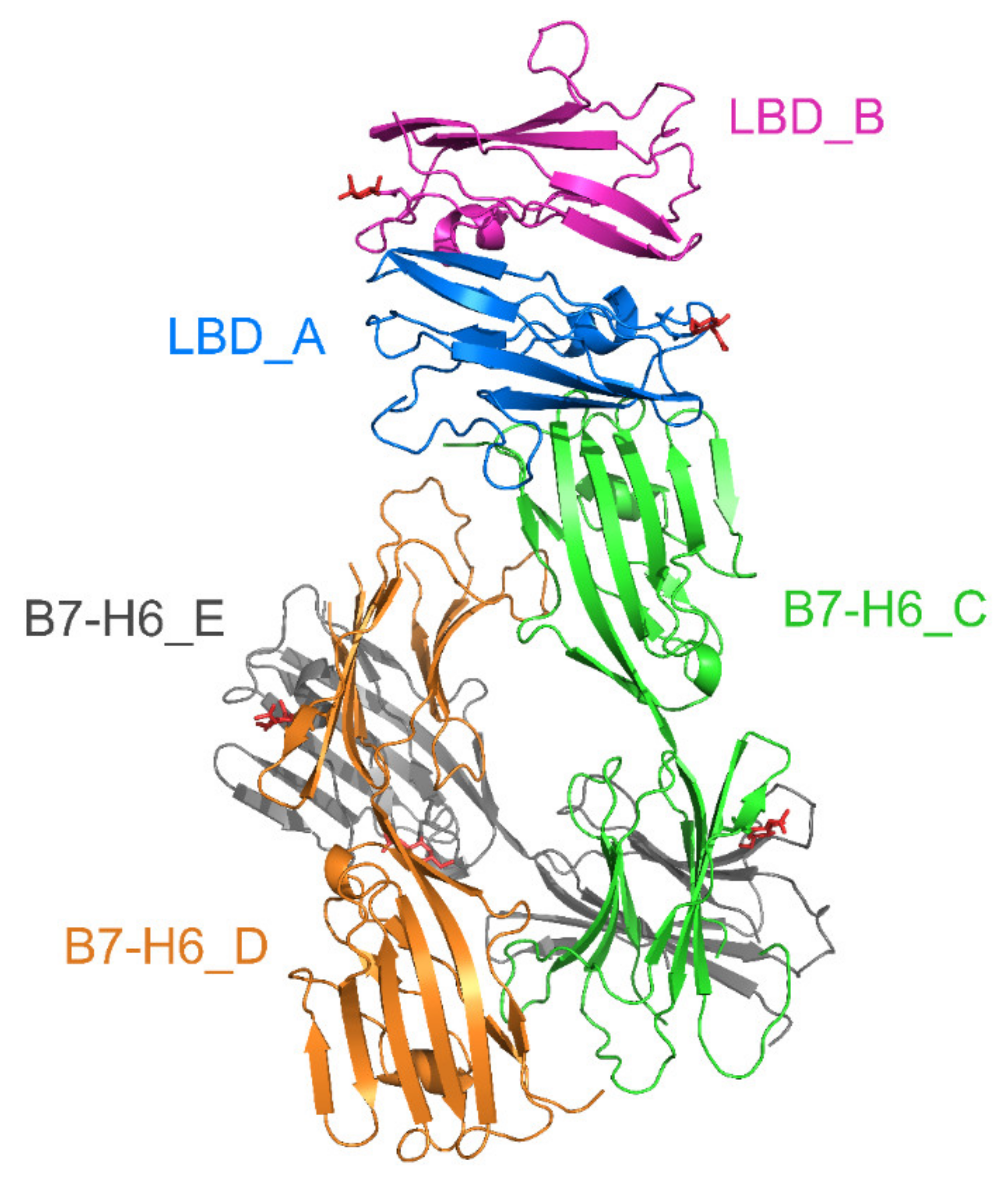
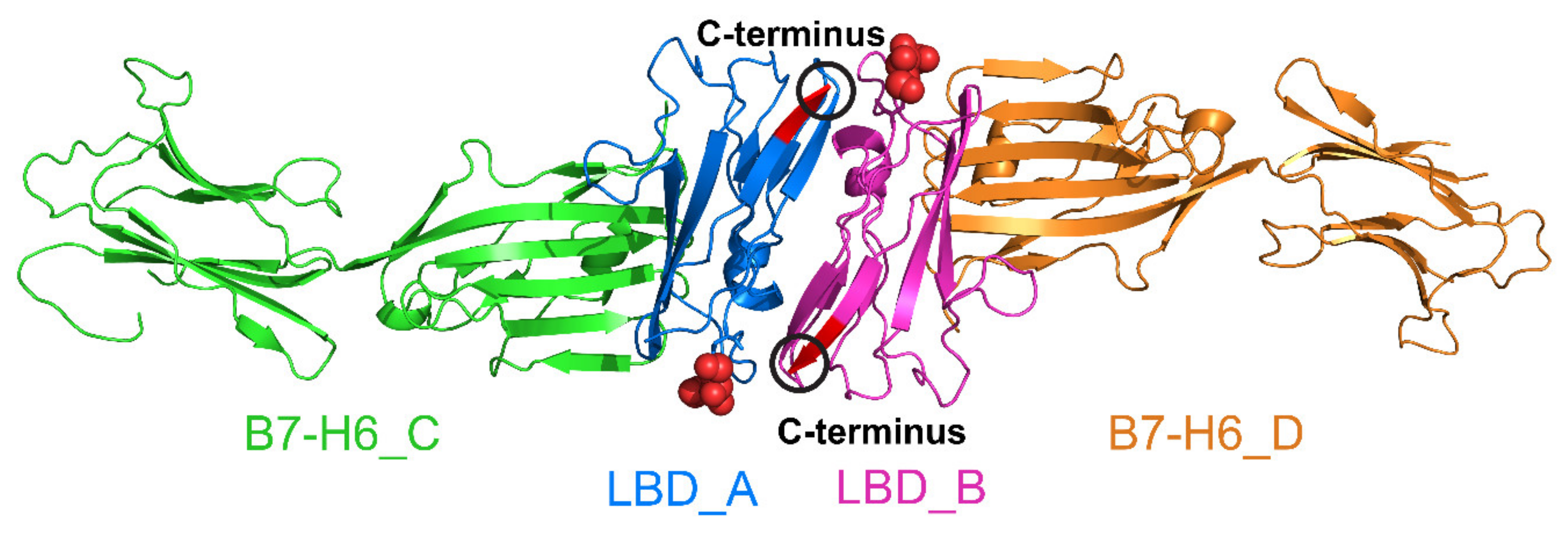
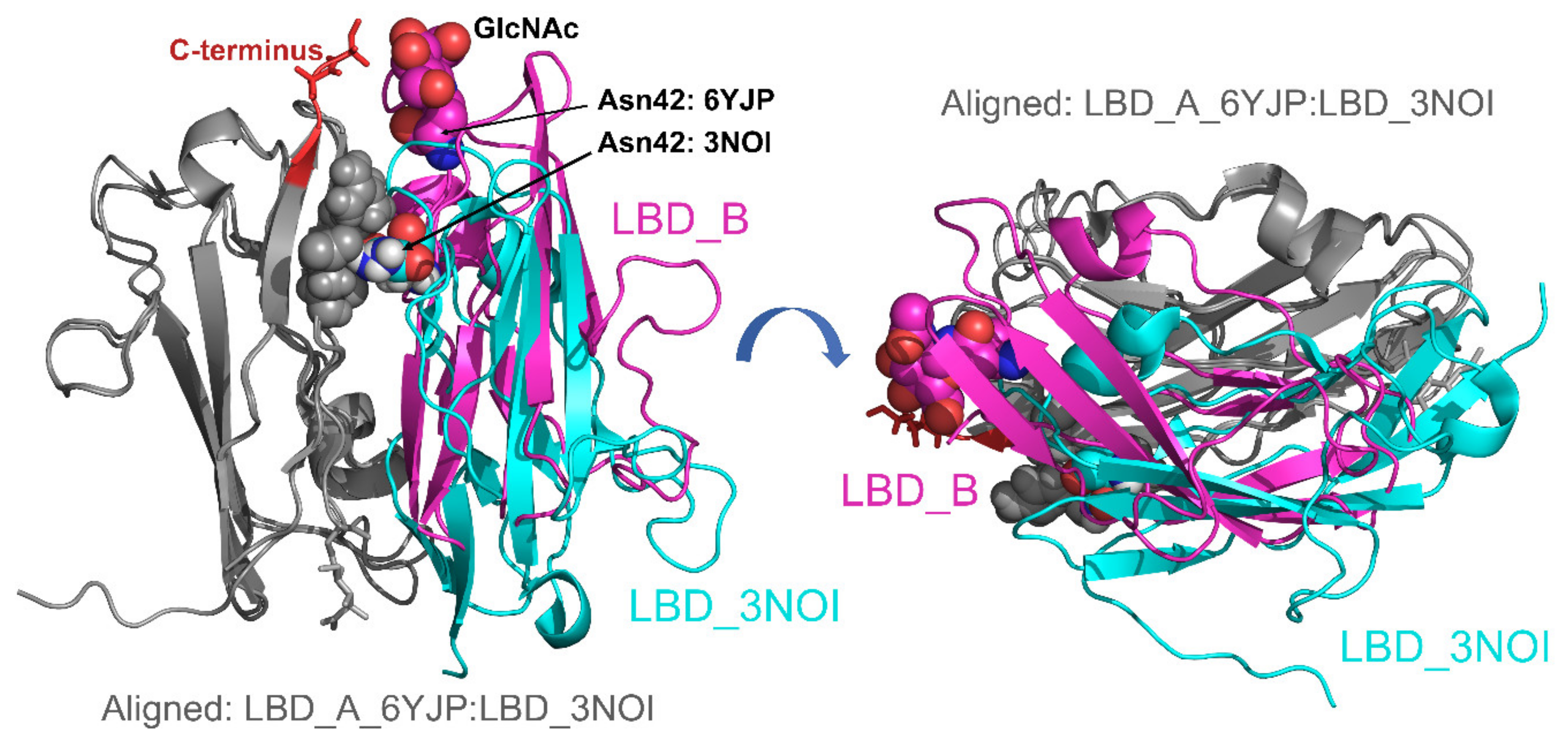
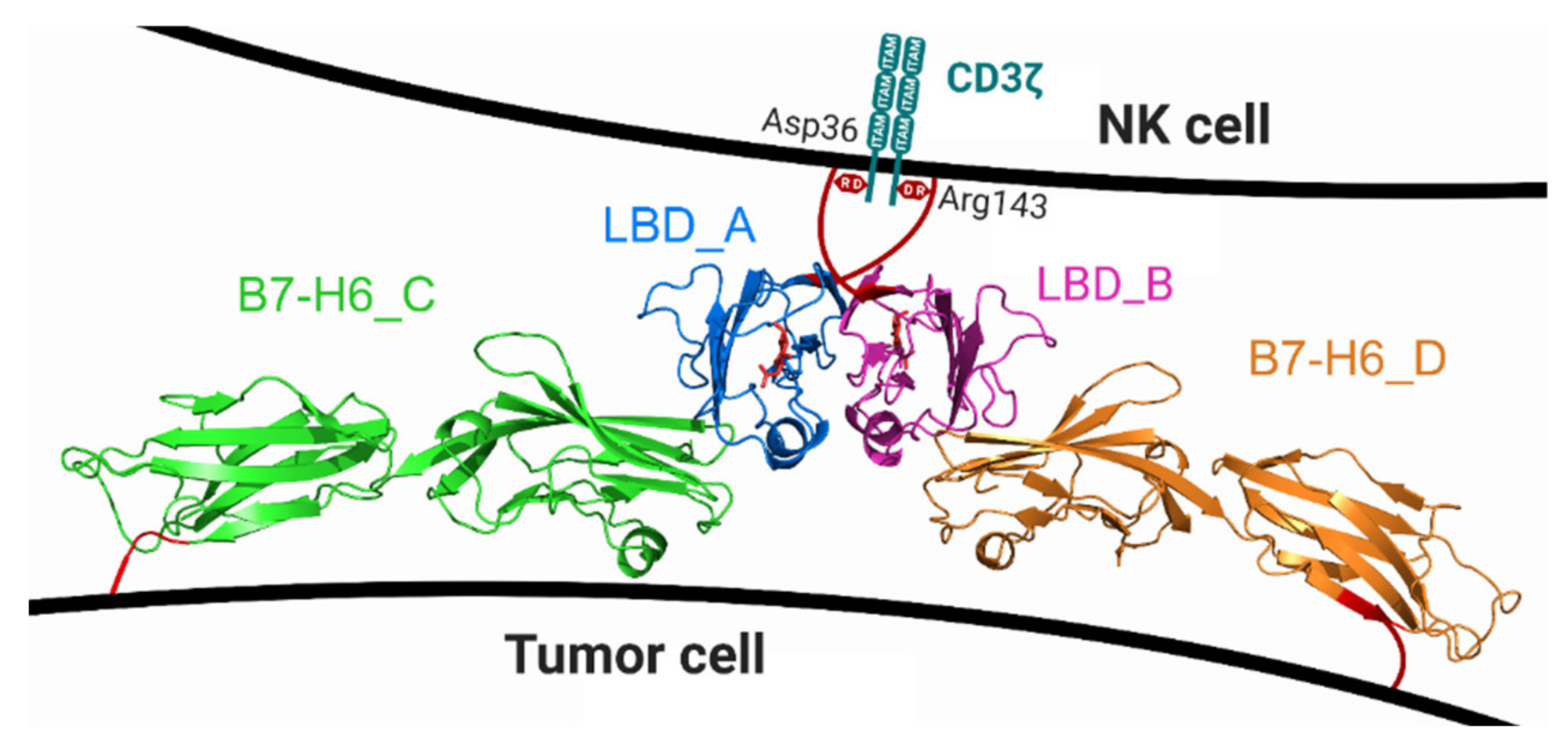
2.6. Crystal Structure of Glycosylated NKp30:B7-H6 Complex
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Vector Design
4.2. Protein Expression and Purification
4.3. Protein Labelling
4.4. Deglycosylation
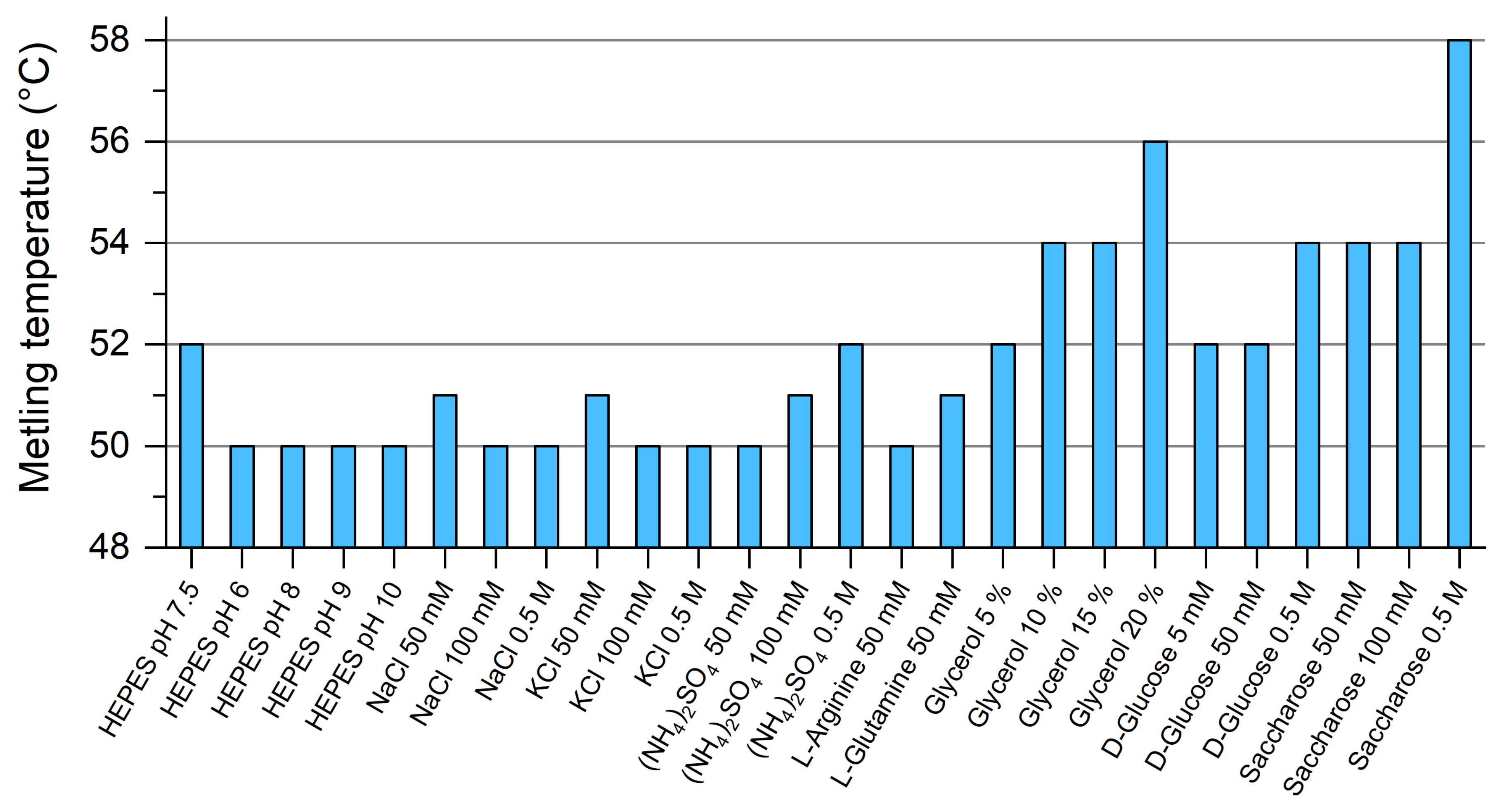
4.5. Differential Scanning Fluorimetry
4.6. Sedimentation Analysis
4.7. Size-Exclusion Chromatography with Multi-Angle Laser Light Scattering (SEC-MALS)
4.8. Isothermal Titration Calorimetry
4.9. Surface Plasmon Resonance
4.10. Protein Crystallization
4.11. Diffraction Data Collection
4.12. Structure Solution and Refinement
4.13. Data and Structure Deposition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kiessling, R.; Klein, E.; Wigzell, H. „Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Herberman, R.B.; Nunn, M.E.; Holden, H.T.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer 1975, 16, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Kruse, P.H.; Matta, J.; Ugolini, S.; Vivier, E. Natural cytotoxicity receptors and their ligands. Immunol. Cell Biol. 2014, 92, 221–229. [Google Scholar] [CrossRef]
- Biassoni, R. Human Natural Killer Receptors, Co-Receptors, and Their Ligands. Curr. Protoc. Immunol. 2009, 84. [Google Scholar] [CrossRef]
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and Molecular Characterization of Nkp30, a Novel Triggering Receptor Involved in Natural Cytotoxicity Mediated by Human Natural Killer Cells. J. Exp. Med. 1999, 190, 1505–1516. [Google Scholar] [CrossRef]
- Memmer, S.; Weil, S.; Beyer, S.; Zöller, T.; Peters, E.; Hartmann, J.; Steinle, A.; Koch, J. The Stalk Domain of NKp30 Contributes to Ligand Binding and Signaling of a Preassembled NKp30-CD3ζ Complex. J. Biol. Chem. 2016, 291, 25427–25438. [Google Scholar] [CrossRef]
- Kaifu, T.; Escalière, B.; Gastinel, L.N.; Vivier, E.; Baratin, M. B7-H6/NKp30 interaction: A mechanism of alerting NK cells against tumors. Cell. Mol. Life Sci. 2011, 68, 3531–3539. [Google Scholar] [CrossRef]
- Hershkovitz, O.; Jarahian, M.; Zilka, A.; Bar-Ilan, A.; Landau, G.; Jivov, S.; Tekoah, Y.; Glicklis, R.; Gallagher, J.T.; Hoffmann, S.C.; et al. Altered glycosylation of recombinant NKp30 hampers binding to heparan sulfate: A lesson for the use of recombinant immunoreceptors as an immunological tool. Glycobiology 2008, 18, 28–41. [Google Scholar] [CrossRef]
- Chisholm, S.E.; Reyburn, H.T. Recognition of Vaccinia Virus-Infected Cells by Human Natural Killer Cells Depends on Natural Cytotoxicity Receptors. J. Virol. 2006, 80, 2225–2233. [Google Scholar] [CrossRef]
- Arnon, T.I.; Achdout, H.; Levi, O.; Markel, G.; Saleh, N.; Katz, G.; Gazit, R.; Gonen-Gross, T.; Hanna, J.; Nahari, E.; et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat. Immunol. 2005, 6, 515–523. [Google Scholar] [CrossRef]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Binici, J.; Koch, J. BAG-6, a jack of all trades in health and disease. Cell. Mol. Life Sci. 2014, 71, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Schlecker, E.; Fiegler, N.; Arnold, A.; Altevogt, P.; Rose-John, S.; Moldenhauer, G.; Sucker, A.; Paschen, A.; Von Strandmann, E.P.; Textor, S.; et al. Metalloprotease-Mediated Tumor Cell Shedding of B7-H6, the Ligand of the Natural Killer Cell-Activating Receptor NKp30. Cancer Res. 2014, 74, 3429–3440. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, H.; Geng, J.; Zheng, X.; Wei, H.; Sun, R.; Tian, Z. Tumor-released Galectin-3, a Soluble Inhibitory Ligand of Human NKp30, Plays an Important Role in Tumor Escape from NK Cell Attack. J. Biol. Chem. 2014, 289, 33311–33319. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mo, J.; Jia, X.; He, Y. The B7 Family Member B7-H6: A New Bane of Tumor. Pathol. Oncol. Res. 2018, 24, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zeng, T.; Xiao, Z.; Hu, Q.; Li, Y.; Tan, X.; Yue, H.; Wang, W.; Tan, H.; Zou, J. Immunological role and underlying mechanisms of B7-H6 in tumorigenesis. Clin. Chim. Acta 2020, 502, 191–198. [Google Scholar] [CrossRef]
- Matta, J.; Baratin, M.; Chiche, L.; Forel, J.-M.; Cognet, C.; Thomas, G.; Farnarier, C.; Piperoglou, C.; Papazian, L.; Chaussabel, D.; et al. Induction of B7-H6, a ligand for the natural killer cell–activating receptor NKp30, in inflammatory conditions. Blood 2013, 122, 394–404. [Google Scholar] [CrossRef]
- Joyce, M.G.; Tran, P.; Zhuravleva, M.A.; Jaw, J.; Colonna, M.; Sun, P.D. Crystal structure of human natural cytotoxicity receptor NKp30 and identification of its ligand binding site. Proc. Natl. Acad. Sci. USA 2011, 108, 6223–6228. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Q.; Mariuzza, R.A. Structure of the human activating natural cytotoxicity receptor NKp30 bound to its tumor cell ligand B7-H6. J. Exp. Med. 2011, 208, 703–714. [Google Scholar] [CrossRef]
- Herrmann, J.; Berberich, H.; Hartmann, J.; Beyer, S.; Davies, K.E.; Koch, J. Homo-oligomerization of the Activating Natural Killer Cell Receptor NKp30 Ectodomain Increases Its Binding Affinity for Cellular Ligands. J. Biol. Chem. 2014, 289, 765–777. [Google Scholar] [CrossRef]
- Hartmann, J.; Tran, T.-V.; Kaudeer, J.; Oberle, K.; Herrmann, J.; Quagliano, I.; Abel, T.; Cohnen, A.; Gatterdam, V.; Jacobs, A.; et al. The Stalk Domain and the Glycosylation Status of the Activating Natural Killer Cell Receptor NKp30 Are Important for Ligand Binding. J. Biol. Chem. 2012, 287, 31527–31539. [Google Scholar] [CrossRef] [PubMed]
- Binici, J.; Hartmann, J.; Herrmann, J.; Schreiber, C.; Beyer, S.; Güler, G.; Vogel, V.; Tumulka, F.; Abele, R.; Mäntele, W.; et al. A Soluble Fragment of the Tumor Antigen BCL2-associated Athanogene 6 (BAG-6) Is Essential and Sufficient for Inhibition of NKp30 Receptor-dependent Cytotoxicity of Natural Killer Cells. J. Biol. Chem. 2013, 288, 34295–34303. [Google Scholar] [CrossRef] [PubMed]
- Aricescu, A.R.; Lu, W.; Jones, E.Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Reeves, P.J.; Callewaert, N.; Contreras, R.; Khorana, H.G. Structure and function in rhodopsin: High-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. USA 2002, 99, 13419–13424. [Google Scholar] [CrossRef]
- Bláha, J.; Maraun, M.; Novák, P.; Vaněk, O. Expression and purification of soluble and stable ectodomain of natural killer cell receptor LLT1 through high-density transfection of suspension adapted HEK293S GnTI− cells. Protein Expr. Purif. 2015, 109, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Dong, C. New B7 Family Checkpoints in Human Cancers. Mol. Cancer Ther. 2017, 16, 1203–1211. [Google Scholar] [CrossRef]
- Wu, M.-R.; Zhang, T.; Gacerez, A.T.; Coupet, T.A.; Demars, L.R.; Sentman, C.L. B7H6-Specific Bispecific T Cell Engagers Lead to Tumor Elimination and Host Antitumor Immunity. J. Immunol. 2015, 194, 5305–5311. [Google Scholar] [CrossRef]
- Gacerez, A.T.; Hua, C.K.; Ackerman, M.E.; Sentman, C.L. Chimeric antigen receptors with human scFvs preferentially induce T cell anti-tumor activity against tumors with high B7H6 expression. Cancer Immunol. Immunother. 2018, 67, 749–759. [Google Scholar] [CrossRef]
- Kellner, C.; Maurer, T.; Hallack, D.; Repp, R.; Van De Winkel, J.G.J.; Parren, P.W.H.I.; Valerius, T.; Humpe, A.; Gramatzki, M.; Peipp, M.; et al. Mimicking an Induced Self Phenotype by Coating Lymphomas with the NKp30 Ligand B7-H6 Promotes NK Cell Cytotoxicity. J. Immunol. 2012, 189, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Kellner, C.; Günther, A.; Humpe, A.; Repp, R.; Klausz, K.; Derer, S.; Valerius, T.; Ritgen, M.; Brüggemann, M.; Van De Winkel, J.G.; et al. Enhancing natural killer cell-mediated lysis of lymphoma cells by combining therapeutic antibodies with CD20-specific immunoligands engaging NKG2D or NKp30. OncoImmunology 2016, 5, e1058459. [Google Scholar] [CrossRef]
- Peipp, M.; Derer, S.; Lohse, S.; Staudinger, M.; Klausz, K.; Valerius, T.; Gramatzki, M.; Kellner, C. HER2-specific immunoligands engaging NKp30 or NKp80 trigger NK-cell-mediated lysis of tumor cells and enhance antibody-dependent cell-mediated cytotoxicity. Oncotarget 2015, 6, 32075–32088. [Google Scholar] [CrossRef] [PubMed]
- Jaron-Mendelson, M.; Yossef, R.; Appel, M.; Zilka, A.; Hadad, U.; Afergan, F.; Rosental, B.; Engel, S.; Nedvetzki, S.; Braiman, A.; et al. Dimerization of NKp46 Receptor Is Essential for NKp46-Mediated Lysis: Characterization of the Dimerization Site by Epitope Mapping. J. Immunol. 2012, 188, 6165–6174. [Google Scholar] [CrossRef] [PubMed]
- Hadad, U.; Thauland, T.J.; Martinez, O.M.; Butte, M.J.; Porgador, A.; Krams, S.M. NKp46 Clusters at the Immune Synapse and Regulates NK Cell Polarization. Front. Immunol. 2015, 6, 216. [Google Scholar] [CrossRef]
- Arnon, T.I.; Markel, G.; Bar-Ilan, A.; Hanna, J.H.; Fima, E.; Benchetrit, F.; Galili, R.; Cerwenka, A.; Benharroch, D.; Sion-Vardy, N.; et al. Harnessing Soluble NK Cell Killer Receptors for the Generation of Novel Cancer Immune Therapy. PLoS ONE 2008, 3, e2150. [Google Scholar] [CrossRef]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Xu, X.; Li, Y.; Gauthier, L.; Chen, Q.; Vivier, E.; Mariuzza, R.A. Expression, crystallization and X-ray diffraction analysis of a complex between B7-H6, a tumor cell ligand for the natural cytotoxicity receptor NKp30, and an inhibitory antibody. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 697–701. [Google Scholar] [CrossRef]
- Xu, X.; Narni-Mancinelli, E.; Cantoni, C.; Li, Y.; Guia, S.; Gauthier, L.; Chen, Q.; Moretta, A.; Vély, F.; Eisenstein, E.; et al. Structural Insights into the Inhibitory Mechanism of an Antibody against B7-H6, a Stress-Induced Cellular Ligand for the Natural Killer Cell Receptor NKp30. J. Mol. Biol. 2016, 428, 4457–4466. [Google Scholar] [CrossRef]
- Durocher, Y. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002, 30, 9e. [Google Scholar] [CrossRef]
- Backliwal, G.; Hildinger, M.; Kuettel, I.; Delegrange, F.; Hacker, D.L.; Wurm, F.M. Valproic acid: A viable alternative to sodium butyrate for enhancing protein expression in mammalian cell cultures. Biotechnol. Bioeng. 2008, 101, 182–189. [Google Scholar] [CrossRef]
- Pompach, P.; Man, P.; Kavan, D.; Hofbauerová, K.; Kumar, V.; Bezouska, K.; Havlicek, V.; Novák, P. Modified electrophoretic and digestion conditions allow a simplified mass spectrometric evaluation of disulfide bonds. J. Mass Spectrom. 2009, 44, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Young, M.M.; Tang, N.; Hempel, J.C.; Oshiro, C.M.; Taylor, E.W.; Kuntz, I.D.; Gibson, B.W.; Dollinger, G. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc. Natl. Acad. Sci. USA 2000, 97, 5802–5806. [Google Scholar] [CrossRef]
- Kukačka, Z.; Rosulek, M.; Strohalm, M.; Kavan, D.; Novák, P. Mapping protein structural changes by quantitative cross-linking. Methods 2015, 89, 112–120. [Google Scholar] [CrossRef]
- Grüninger, F.; D’Arcy, A.; D’Arcy, B.; Chène, C. Deglycosylation of proteins for crystallization using recombinant fusion protein glycosidases. Protein Sci. 1996, 5, 2617–2622. [Google Scholar] [CrossRef]
- Schuck, P. Size-Distribution Analysis of Macromolecules by Sedimentation Velocity Ultracentrifugation and Lamm Equation Modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Ma, J.; Brown, P.H.; Zhao, H.; Schuck, P. Measuring macromolecular size distributions and interactions at high concentrations by sedimentation velocity. Nat. Commun. 2018, 9, 4415. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, T.H.; Brautigam, C.A. High-precision, automated integration of multiple isothermal titration calorimetric thermograms: New features of NITPIC. Methods 2015, 76, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Piszczek, G.; Schuck, P. SEDPHAT—A platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 2015, 76, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, C.A. Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol. 2015, 562, 109–133. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Winn, M.; Ballard, C.C.; Cowtan, K.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Vagin, A.; Lebedev, A. MoRDa, an automatic molecular replacement pipeline. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, S19. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Kovalevskiy, O.; Nicholls, R.; Murshudov, G. Automated refinement of macromolecular structures at low resolution using prior information. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | 6YJP |
|---|---|
| Data processing statistics | |
| Space group | C2 |
| Unit-cell parameters a, b, c (Å); α, β, γ (°) | 166.0, 86.5, 111.3; 90, 97.6 90 |
| Resolution range (Å) | 48.95-3.1 (3.29-3.1) |
| No. of observations | 99061 (12123) |
| No. of unique reflections | 27102 (3968) |
| Data completeness (%) | 95 (87) |
| Average redundancy | 3.7 (3.1) |
| Mosaicity (°) | 0.09 |
| Average I/σ(I) | 5.7 (0.8) |
| Solvent content (%) | 65 |
| Matthews coefficient (Å3/Da) | 3.54 |
| Wilson B-factor (Å2) | 105.5 |
| Rmerge | 0.104 (0.971) |
| Rpim | 0.081 (0.804) |
| CC1/2 | 0.995 (0.645) |
| Structure refinement parameters | |
| Rwork | 0.272 |
| Rfree | 0.322 |
| Rall | 0.275 |
| Average B-factor (Å2) | 155.6 |
| RMSD bond lengths from ideal (Å) | 0.005 |
| RMSD bond angles from ideal (°) | 1.59 |
| Number of non-hydrogen atoms | 6907 |
| Number of water molecules | 0 |
| Ramachandran statistics: residues in allowed/favored region (%) | 99.5/92.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skořepa, O.; Pazicky, S.; Kalousková, B.; Bláha, J.; Abreu, C.; Ječmen, T.; Rosůlek, M.; Fish, A.; Sedivy, A.; Harlos, K.; et al. Natural Killer Cell Activation Receptor NKp30 Oligomerization Depends on Its N-Glycosylation. Cancers 2020, 12, 1998. https://doi.org/10.3390/cancers12071998
Skořepa O, Pazicky S, Kalousková B, Bláha J, Abreu C, Ječmen T, Rosůlek M, Fish A, Sedivy A, Harlos K, et al. Natural Killer Cell Activation Receptor NKp30 Oligomerization Depends on Its N-Glycosylation. Cancers. 2020; 12(7):1998. https://doi.org/10.3390/cancers12071998
Chicago/Turabian StyleSkořepa, Ondřej, Samuel Pazicky, Barbora Kalousková, Jan Bláha, Celeste Abreu, Tomáš Ječmen, Michal Rosůlek, Alexander Fish, Arthur Sedivy, Karl Harlos, and et al. 2020. "Natural Killer Cell Activation Receptor NKp30 Oligomerization Depends on Its N-Glycosylation" Cancers 12, no. 7: 1998. https://doi.org/10.3390/cancers12071998
APA StyleSkořepa, O., Pazicky, S., Kalousková, B., Bláha, J., Abreu, C., Ječmen, T., Rosůlek, M., Fish, A., Sedivy, A., Harlos, K., Dohnálek, J., Skálová, T., & Vaněk, O. (2020). Natural Killer Cell Activation Receptor NKp30 Oligomerization Depends on Its N-Glycosylation. Cancers, 12(7), 1998. https://doi.org/10.3390/cancers12071998