Therapeutic Targeting of Autophagy for Renal Cell Carcinoma Therapy


Abstract
1. Introduction
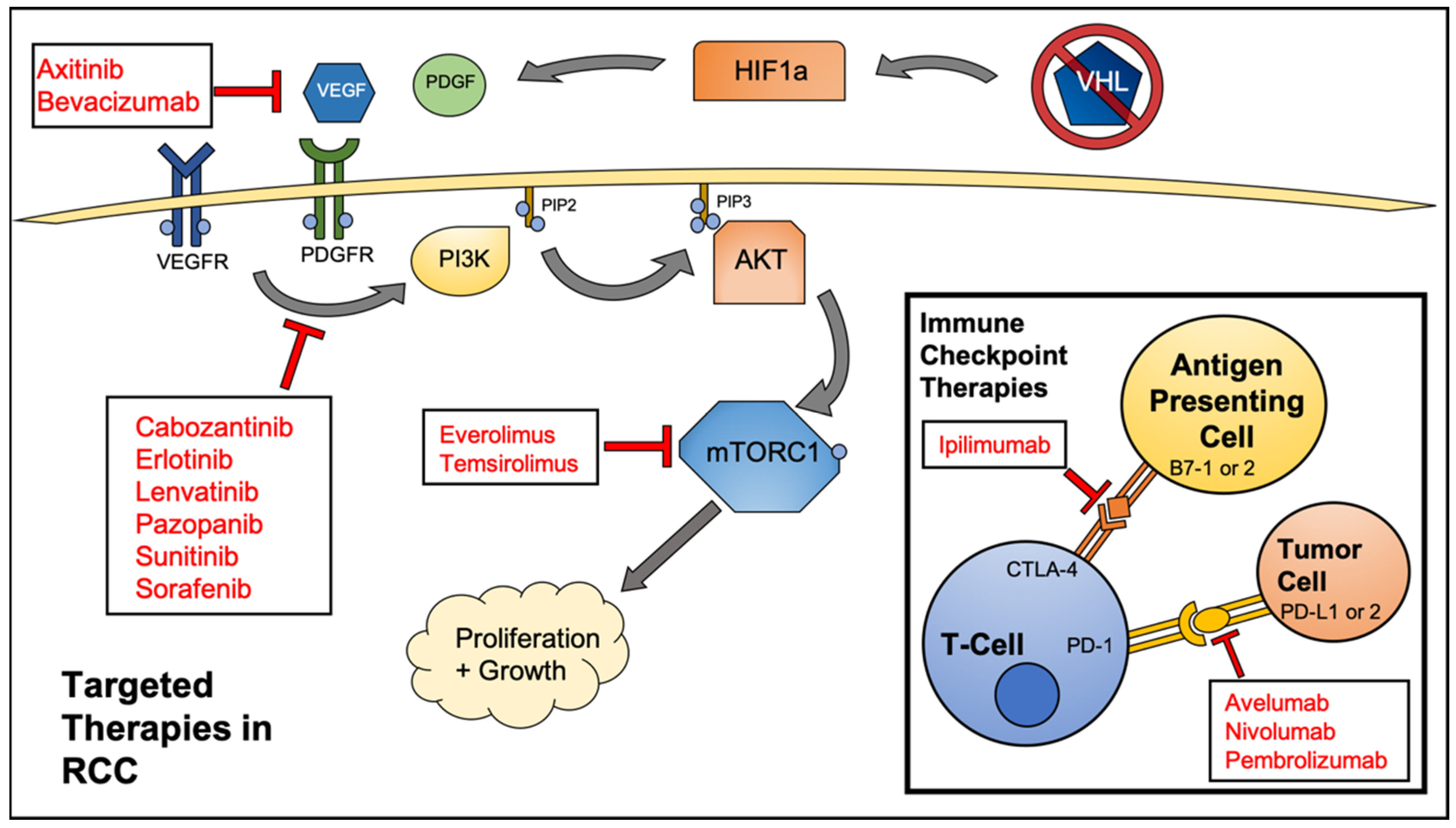
2. Targeted Therapeutics for RCC
3. Autophagy
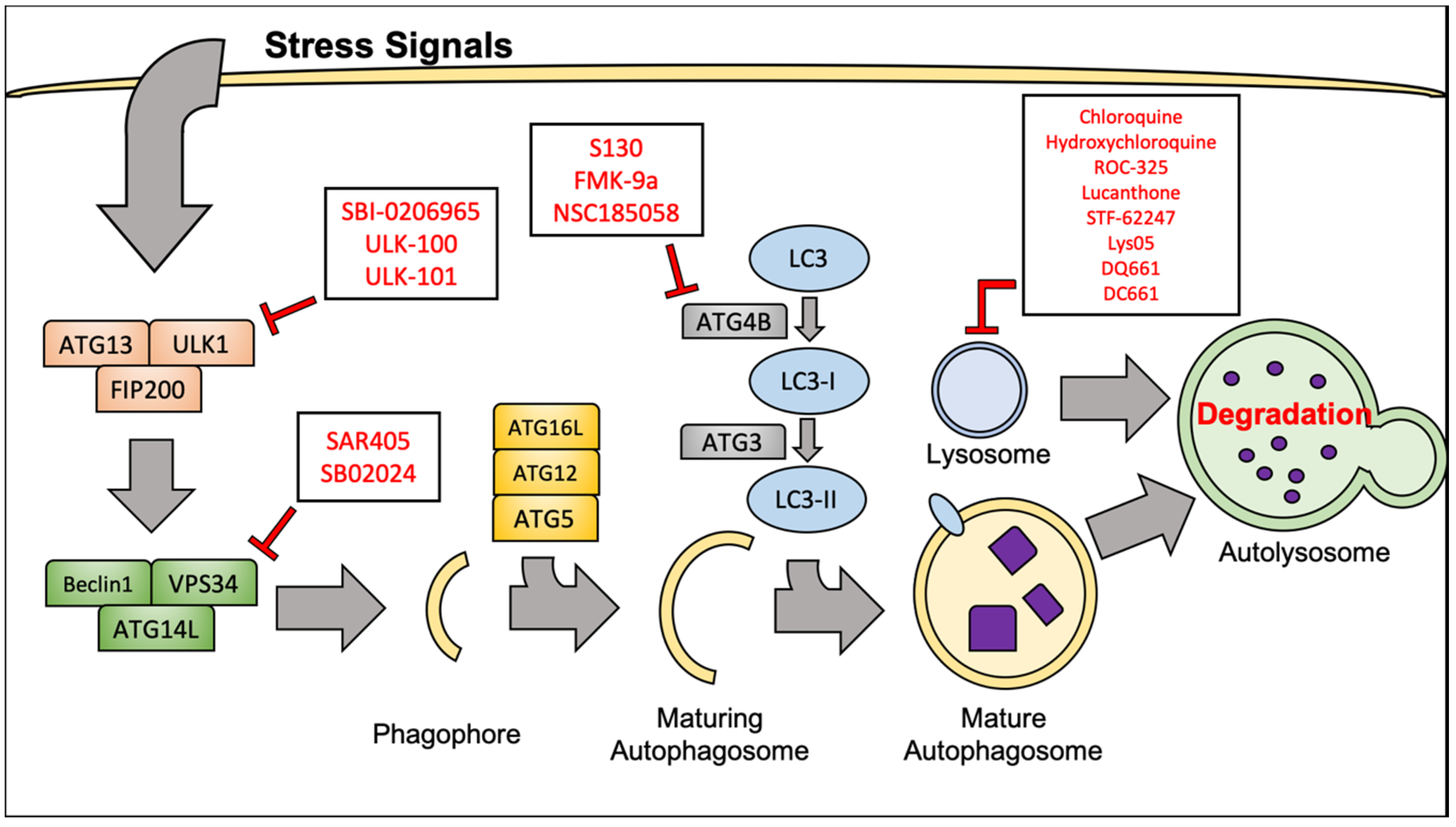
3.1. Molecular Mechanisms of Autophagy
3.2. Targeting Autophagy to Improve RCC Therapeutic Outcome
4. Inhibition of Autophagy for Therapy of RCC
4.1. Chloroquine and Hydroxychloroquine
4.2. Lucanthone
4.3. ROC-325
4.4. STF-62247
4.5. Lys05, DQ661, and DC661
4.6. VPS34 Inhibitors
4.7. ULK1 Inhibitors
4.8. ATG4B Inhibitors
5. Immune Checkpoint Inhibitor Therapy and Autophagy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RCC | renal cell carcinoma |
| VHL | Von-Hippel Lindau tumor suppressor |
| VEGF | vascular endothelial growth factor |
| PDGF | platelet-derived growth factor |
| ATG | autophagy-related gene |
| PFS | progression-free survival |
| PR | partial response |
| DLT | dose-limiting toxicity |
| SD | stable disease |
| CR | complete response |
| HDAC | histone deacetylase |
| AE | adverse event |
| HCQ | hydroxychloroquine |
| CQ | chloroquine |
| IHC | immunohistochemistry |
| ULK | UNC-51 like kinase |
| CTLA-4 | cytotoxic t-lymphocyte associated protein 4 |
| PD-L1 | Programmed cell death ligand 1 |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.C.; Rini, B.I. Treatment of renal cell carcinoma: Current status and future directions. CA A Cancer J. Clin. 2017, 67, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Noone, A.; Howlander, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.; et al. Cancer Statistics Review; SEER: Bethesda, MD, USA, 2018. [Google Scholar]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Kidney Cancer; Version 2.2020. Available online: https://www.nccn.org/professionals/physician_gls/pdf/kidney.pdf (accessed on 1 November 2019).
- Eggener, S.E.; Yossepowitch, O.; Pettus, J.A.; Snyder, M.E.; Motzer, R.J.; Russo, P. Renal cell carcinoma recurrence after nephrectomy for localized disease: Predicting survival from time of recurrence. J. Clin. Oncol. 2006, 24, 3101–3106. [Google Scholar] [CrossRef]
- Kim, W.Y.; Kaelin, W.G. Role of VHL gene mutation in human cancer. J. Clin. Oncol. 2004, 22, 4991–5004. [Google Scholar] [CrossRef]
- Creighton, C.J.; Morgan, M.; Gunaratne, P.H.; Wheeler, D.A.; Gibbs, R.A.; Robertson, G.; Chu, A.; Beroukhim, R.; Cibulskis, K.; Signoretti, S.; et al. Comprehensivemolecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- Duran, I.; Lambea, J.; Maroto, P.; González-Larriba, J.L.; Flores, L.; Granados-Principal, S.; Graupera, M.; Sáez, B.; Vivancos, A.; Casanovas, O. Resistance to Targeted Therapies in Renal Cancer: The Importance of Changing the Mechanism of Action. Target. Oncol. 2017, 12, 19–35. [Google Scholar] [CrossRef]
- Rini, B.I.; Atkins, M.B. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009, 10, 992–1000. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Negrier, S.; Escudier, B.; Lasset, C.; Douillard, J.-Y.; Savary, J.; Chevreau, C.; Ravaud, A.; Mercatello, A.; Peny, J.; Mousseau, M.; et al. Recombinant Human Interleukin-2, Recombinant Human Interferon Alfa-2a, or Both in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2002, 338, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomised phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef]
- Powles, T.; Motzer, R.J.; Escudier, B.; Pal, S.; Kollmannsberger, C.; Pikiel, J.; Gurney, H.; Rha, S.Y.; Park, S.H.; Geertsen, P.F.; et al. Outcomes based on prior therapy in the phase 3 METEOR trial of cabozantinib versus everolimus in advanced renal cell carcinoma. Br. J. Cancer 2018, 119, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, R.M.; Kabbinavar, F.F.; Figlin, R.A.; Flaherty, K.; Srinivas, S.; Vaishampayan, U.; Drabkin, H.A.; Dutcher, J.; Ryba, S.; Xia, Q.; et al. Randomized phase II study of erlotinib combined with bevacizumab compared with bevacizumab alone in metastatic renal cell cancer. J. Clin. Oncol. 2007, 25, 4536–4541. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Hudes, G.; O’Toole, T.; Tomczak, P.; Bodrogi, I.; Sosman, J.; Kapoor, A.; Staroslawska, E.; Kovacevic, Z.; McDermott, D.; Dutcher, J.; et al. Temsirolimus, Interferon Alfa, or Both for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Bellmunt, J.; Négrier, S.; Bajetta, E.; Melichar, B.; Bracarda, S.; Ravaud, A.; Golding, S.; Jethwa, S.; Sneller, V. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Minasian, L.M.; Motzer, R.J.; Gluck, L.; Mazumdar, M.; Vlamis, V.; Krown, S.E. Interferon alfa-2a in advanced renal cell carcinoma: Treatment results and survival in 159 patients with long-term follow-up. J. Clin. Oncol. 1993, 11, 1368–1375. [Google Scholar] [CrossRef]
- Fyfe, G.; Fisher, R.I.; Rosenberg, S.A.; Sznol, M.; Parkinson, D.R.; Louie, A.C. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J. Clin. Oncol. 1995, 13, 688–696. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy*[S]. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Ann. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 2004, 36, 2445–2462. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401. [Google Scholar] [CrossRef]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Autophagy as a target for cancer therapy: New developments. Cancer Manag. Res. 2012, 4, 357–365. [Google Scholar] [CrossRef]
- Carew, J.S.; Nawrocki, S.T.; Cleveland, J.L. Modulating autophagy for therapeutic benefit. Autophagy 2007, 3, 464–467. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, L.; Zhang, Y.; Leng, Y.; Pei, X.Y.; Lin, H.; Jones, R.; Orlowski, R.Z.; Dai, Y.; Grant, S. Targeting SQSTM1/p62 induces cargo loading failure and converts autophagy to apoptosis via NBK/Bik. Mol. Cell Biol. 2014, 34, 3435–3449. [Google Scholar] [CrossRef]
- Bouhamdani, N.; Comeau, D.; Cormier, K.; Turcotte, S. STF-62247 accumulates in lysosomes and blocks late stages of autophagy to selectively target von Hippel-Lindau-inactivated cells. Am. J. Physiol. Cell Physiol. 2019, 316, C605–C620. [Google Scholar] [CrossRef]
- Turcotte, S.; Chan, D.A.; Sutphin, P.D.; Hay, M.P.; Denny, W.A.; Giaccia, A.J. A Molecule Targeting VHL-Deficient Renal Cell Carcinoma that Induces Autophagy. Cancer Cell 2008, 14, 90–102. [Google Scholar] [CrossRef]
- Bray, K.; Mathew, R.; Lau, A.; Kamphorst, J.J.; Fan, J.; Chen, J.; Chen, H.Y.; Ghavami, A.; Stein, M.; DiPaola, R.S.; et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef]
- Zheng, B.; Zhu, H.; Gu, D.; Pan, X.; Qian, L.; Xue, B.; Yang, D.; Zhou, J.; Shan, Y. MiRNA-30a-mediated autophagy inhibition sensitizes renal cell carcinoma cells to sorafenib. Biochem. Biophys. Res. Comun. 2015, 459, 234–239. [Google Scholar] [CrossRef]
- Li, H.; Jin, X.; Zhang, Z.; Xing, Y.; Kong, X. Inhibition of autophagy enhances apoptosis induced by the PI3K/AKT/mTor inhibitor NVP-BEZ235 in renal cell carcinoma cells. Cell Biochem. Funct. 2013, 31, 427–433. [Google Scholar] [CrossRef]
- Singla, M.; Bhattacharyya, S. Autophagy as a potential therapeutic target during epithelial to mesenchymal transition in renal cell carcinoma: An in vitro study. Biomed. Pharmacother. 2017, 94, 332–340. [Google Scholar] [CrossRef]
- Homewood, C.A.; Warhurst, D.C.; Peters, W.; Baggaley, V.C. Lysosomes, pH and the anti-malarial action of chloroquine. Nature 1972, 235, 50–52. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef]
- Haas, N.B.; Appleman, L.J.; Stein, M.; Redlinger, M.; Wilks, M.; Xu, X.; Onorati, A.; Kalavacharla, A.; Kim, T.; Zhen, C.J.; et al. Autophagy inhibition to augment mTOR inhibition: A phase I/II trial of everolimus and hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin. Cancer Res. 2019, 25, 2080–2087. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, J.M.; Kaveney, A.D.; Malhotra, J.; Spencer, K.; Portal, D.; Goodin, S.; Tan, A.R.; Aisner, J.; Moss, R.A.; Lin, H.; et al. A phase I trial of MK-2206 and hydroxychloroquine in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Carew, J.S.; Espitia, C.M.; Esquivel, J.A., 2nd; Mahalingam, D.; Kelly, K.R.; Reddy, G.; Giles, F.J.; Nawrocki, S.T. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J. Biol. Chem. 2011, 286, 6602–6613. [Google Scholar] [CrossRef]
- Carew, J.S.; Nawrocki, S.T. Drain the lysosome: Development of the novel orally available autophagy inhibitor ROC-325. Autophagy 2017, 13, 765–766. [Google Scholar] [CrossRef]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Han, Y.; Visconte, V.; Phillips, J.; Nawrocki, S.T. Disruption of autophagic degradation with ROC-325 antagonizes renal cell carcinoma pathogenesis. Clin. Cancer Res. 2017, 23, 2869–2879. [Google Scholar] [CrossRef]
- Jones, T.M.; Espitia, C.; Wang, W.; Nawrocki, S.T.; Carew, J.S. Moving beyond hydroxychloroquine: The novel lysosomal autophagy inhibitor ROC-325 shows significant potential in preclinical studies. Cancer Commun. 2019, 39, 72. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Han, Y.; Visconte, V.; Przychodzen, B.; Espitia, C.M.; Phillips, J.; Anwer, F.; Advani, A.; Carraway, H.E.; Kelly, K.R.; et al. The novel autophagy inhibitor ROC-325 augments the antileukemic activity of azacitidine. Leukemia 2019, 33, 2971–2974. [Google Scholar] [CrossRef]
- Zielke, S.; Meyer, N.; Mari, M.; Abou-El-Ardat, K.; Reggiori, F.; van Wijk, S.J.L.; Kögel, D.; Fulda, S. Loperamide, pimozide, and STF-62247 trigger autophagy-dependent cell death in glioblastoma cells. Cell Death Dis. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Anbalagan, S.; Pires, I.M.; Blick, C.; Hill, M.A.; Ferguson, D.J.P.; Chan, D.A.; Hammond, E.M. Radiosensitization of renal cell carcinoma in vitro through the induction of autophagy. Radiother. Oncol. 2012, 103, 388–393. [Google Scholar] [CrossRef]
- Kozako, T.; Sato, K.; Uchida, Y.; Kato, N.; Aikawa, A.; Ogata, K.; Kamimura, H.; Uemura, H.; Yoshimitsu, M.; Ishitsuka, K.; et al. The small molecule STF-62247 induces apoptotic and autophagic cell death in leukemic cells. Oncotarget 2018, 9, 27645–27655. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Winkler, J.D. Lys05: A new lysosomal autophagy inhibitor. Autophagy 2012, 8, 1383–1384. [Google Scholar] [CrossRef]
- McAfee, Q.; Zhang, Z.; Samanta, A.; Levi, S.M.; Ma, X.H.; Piao, S.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E.; et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef]
- Rebecca, V.W.; Nicastri, M.C.; McLaughlin, N.; Fennelly, C.; McAfee, Q.; Ronghe, A.; Nofal, M.; Lim, C.Y.; Witze, E.; Chude, C.I.; et al. A Unified Approach to Targeting the Lysosome’s Degradative and Growth Signaling Roles. Cancer Discov. 2017, 7, 1266–1283. [Google Scholar] [CrossRef]
- Rebecca, V.W.; Nicastri, M.C.; Fennelly, C.; Chude, C.I.; Barber-Rotenberg, J.S.; Ronghe, A.; McAfee, Q.; McLaughlin, N.P.; Zhang, G.; Goldman, A.R.; et al. PPT1 Promotes Tumor Growth and Is the Molecular Target of Chloroquine Derivatives in Cancer. Cancer Discov. 2019, 9, 220–229. [Google Scholar] [CrossRef]
- Pasquier, B. SAR405, a PIK3C3/VPS34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy 2015, 11, 725–726. [Google Scholar] [CrossRef]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef]
- Dyczynski, M.; Yu, Y.; Otrocka, M.; Parpal, S.; Braga, T.; Henley, A.B.; Zazzi, H.; Lerner, M.; Wennerberg, K.; Viklund, J.; et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018, 435, 32–43. [Google Scholar] [CrossRef]
- Egan, D.F.; Chun, M.G.H.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef]
- Martin, K.R.; Celano, S.L.; Solitro, A.R.; Gunaydin, H.; Scott, M.; O’Hagan, R.C.; Shumway, S.D.; Fuller, P.; MacKeigan, J.P. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. Iscience 2018, 8, 74–84. [Google Scholar] [CrossRef]
- Chu, J.; Fu, Y.; Xu, J.; Zheng, X.; Gu, Q.; Luo, X.; Dai, Q.; Zhang, S.; Liu, P.; Hong, L.; et al. ATG4B inhibitor FMK-9a induces autophagy independent on its enzyme inhibition. Arch. Biochem. Biophys. 2018, 644, 29–36. [Google Scholar] [CrossRef]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2019, 15, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.M.; Kim, J.S.; Horenstein, N.; Dunn, W.A., Jr. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Larkin, J.; Oya, M.; Thistlethwaite, F.; Martignoni, M.; Nathan, P.; Powles, T.; McDermott, D.; Robbins, P.B.; Chism, D.D.; et al. Preliminary results for avelumab plus axitinib as first-line therapy in patients with advanced clear-cell renal-cell carcinoma (JAVELIN Renal 100): An open-label, dose-finding and dose-expansion, phase 1b trial. Lancet Oncol. 2018, 19, 451–460. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: Phase I results from a multicenter, open-label phase I/II trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef]
- Phadwal, K.; Alegre-Abarrategui, J.; Watson, A.S.; Pike, L.; Anbalagan, S.; Hammond, E.M.; Wade-Martins, R.; McMichael, A.; Klenerman, P.; Simon, A.K. A novel method for autophagy detection in primary cells: Impaired levels of macroautophagy in immunosenescent T cells. Autophagy 2012, 8, 677–689. [Google Scholar] [CrossRef]
- Clarke, A.J.; Riffelmacher, T.; Braas, D.; Cornall, R.J.; Simon, A.K. B1a B cells require autophagy for metabolic homeostasis and self-renewal. J. Exp. Med. 2018, 215, 399–413. [Google Scholar] [CrossRef]
- Xu, X.; Araki, K.; Li, S.; Han, J.H.; Ye, L.; Tan, W.G.; Konieczny, B.T.; Bruinsma, M.W.; Martinez, J.; Pearce, E.L.; et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat. Immunol. 2014, 15, 1152–1161. [Google Scholar] [CrossRef]
- Starobinets, H.; Ye, J.; Broz, M.; Barry, K.; Goldsmith, J.; Marsh, T.; Rostker, F.; Krummel, M.; Debnath, J. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J. Clin. Investig. 2016, 126, 4417–4429. [Google Scholar] [CrossRef]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef] [PubMed]
- Khazen, R.; Muller, S.; Gaudenzio, N.; Espinosa, E.; Puissegur, M.P.; Valitutti, S. Melanoma cell lysosome secretory burst neutralizes the CTL-mediated cytotoxicity at the lytic synapse. Nat. Commun. 2016, 7, 10823. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T.; et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Liu, J.; Liu, X.; Han, Y.; Zhang, J.; Liu, D.; Ma, G.; Li, C.; Liu, L.; Kong, D. Nanovaccine Incorporated with Hydroxychloroquine Enhances Antigen Cross-Presentation and Promotes Antitumor Immune Responses. ACS Appl. Mater. Interfaces 2018, 10, 30983–30993. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Category | Therapeutic Name | Target(s) | Comparator | PFS (in Months) vs. Comparator |
|---|---|---|---|---|
| Small Molecule Kinase Inhibitors | Axitinib [14] | VEGF, PDGF | Sorafenib | 6–7 vs. 4–7 |
| Cabozantinib [15] | VEGFR-1,2,3, MET, FLT3, TIE-2, AXL, TRKB | Everolimus | 7.4–9.1 vs. 3.7–5.1 | |
| Erlotinib [16] | EGFR | Bevacizumab | 9.9 vs. 8.5 | |
| Lenvatinib [17] | VEGFR2 | Everolimus | 7.4 vs. 5.5 | |
| Pazopanib [18] | VEGFR-1,2,3, PDGFR, c-kit | Placebo | 9.2 vs. 4.2 | |
| Sorafenib [19] | RAF, VEGFR, PDGFR | Placebo | 5.5 vs. 2.8 | |
| Sunitinib [20] | VEGFR2, PDGFRb, c-kit, FLT3 | Interferon-alpha | 11 vs. 5 | |
| mTOR Inhibitors | Everolimus [21] | FKBP-12 | Placebo | 4 vs. 1.9 |
| Temsirolimus [22] | mTOR | Interferon-alpha | 5.5 vs. 3.1 | |
| Monoclonal Antibodies | Avelumab [23] | PD-L1 | Sunitinib | 13.8 vs. 8.4 |
| Bevacizumab [24] | VEGF | Interferon-alpha | 10.2 vs. 5.4 | |
| Ipilimumab [25] | CTLA4 | Sunitinib | 11.6 vs. 8.4 | |
| Nivolumab [26] | PD-1 | Everolimus | 4.6 vs. 4.4 | |
| Pembrolizumab [27] | PD-1 | Sunitinib | 15.1 vs. 11.1 | |
| Cytokine Therapy | Interferon alfa-2a [28] | Immunostimulatory | N/A | 10% Response Rate |
| Interleukin-2 [29] | Immunostimulatory | N/A | 14% Response Rate |
| Clinical Trial Identifier | Autophagy-Modulating Compound | Interventions | Phase | Neoplasm | DLTs | Response Rate |
|---|---|---|---|---|---|---|
| NCT01510119 [53] | HCQ | Everolimus | I/II | Previously Treated RCC | None in Phase I; Grades 3–4 AE’s <10% | SD or PR: 67%; Median PFS 6.3 Months |
| NCT01144169 | HCQ | Surgery | I | Primary RCC | N/A | N/A |
| NCT01480154 | HCQ | MK2206 | I | Advanced Solid Tumors | N/A | N/A |
| NCT01550367 | HCQ | IL-2 | I/II | Metastatic RCC | Grades 3–5 AE’s 96.6% | SD/PR/CR: 69%; Median PFS 5.5 Months |
| NCT01023737 [54] | HCQ | Vorinostat | I | Advanced Solid Tumors | Grades 3–4 AE’s 18.5% | RCC Patient: PR for >50 cycles |
| Inhibitor | Autophagy Target | Cancer Type | References |
|---|---|---|---|
| Hydroxychloroquine | Lysosome | RCC, etc. | [21,53,54,55] |
| Chloroquine | Lysosome | RCC, etc. | [51,52] |
| ROC-325 | Lysosome | RCC, AML | [57,58,59,60] |
| Lucanthone | Lysosome | Breast | [41,56] |
| STF-62247 | Lysosome | RCC, Glioblastoma, T-cell Leukemia | [45,61,62,63] |
| Lys05, DQ661, DC661 | Lysosome, PPT1 | Melanoma, Colon, Glioma | [64,65,66,67] |
| SAR405, SB02024 | VPS34 | RCC, Cervical | [68,69,70] |
| SBI-0206965, ULK-100, ULK-101 | ULK1 | Lung | [71,72] |
| S130, FMK-9a, NSC185058 | ATG4B | Cervical, Colon, Osteosarcoma, GBM | [73,74,75,76] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, T.M.; Carew, J.S.; Nawrocki, S.T. Therapeutic Targeting of Autophagy for Renal Cell Carcinoma Therapy. Cancers 2020, 12, 1185. https://doi.org/10.3390/cancers12051185
Jones TM, Carew JS, Nawrocki ST. Therapeutic Targeting of Autophagy for Renal Cell Carcinoma Therapy. Cancers. 2020; 12(5):1185. https://doi.org/10.3390/cancers12051185
Chicago/Turabian StyleJones, Trace M., Jennifer S. Carew, and Steffan T. Nawrocki. 2020. "Therapeutic Targeting of Autophagy for Renal Cell Carcinoma Therapy" Cancers 12, no. 5: 1185. https://doi.org/10.3390/cancers12051185
APA StyleJones, T. M., Carew, J. S., & Nawrocki, S. T. (2020). Therapeutic Targeting of Autophagy for Renal Cell Carcinoma Therapy. Cancers, 12(5), 1185. https://doi.org/10.3390/cancers12051185