Sugar Alcohols Have a Key Role in Pathogenesis of Chronic Liver Disease and Hepatocellular Carcinoma in Whole Blood and Liver Tissues


 ,
,
Abstract
1. Introduction
2. Results
2.1. Characteristics of Subjects
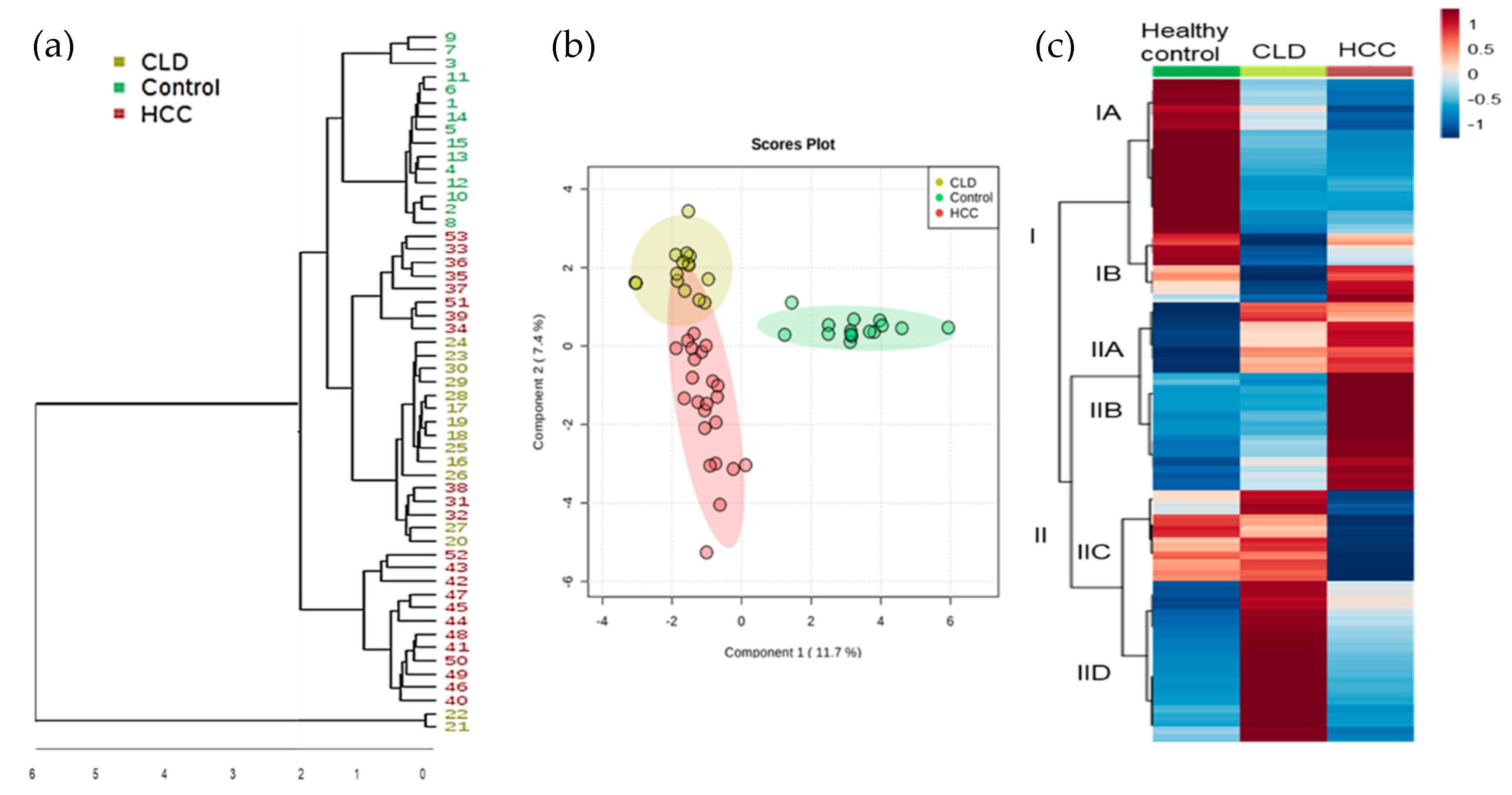
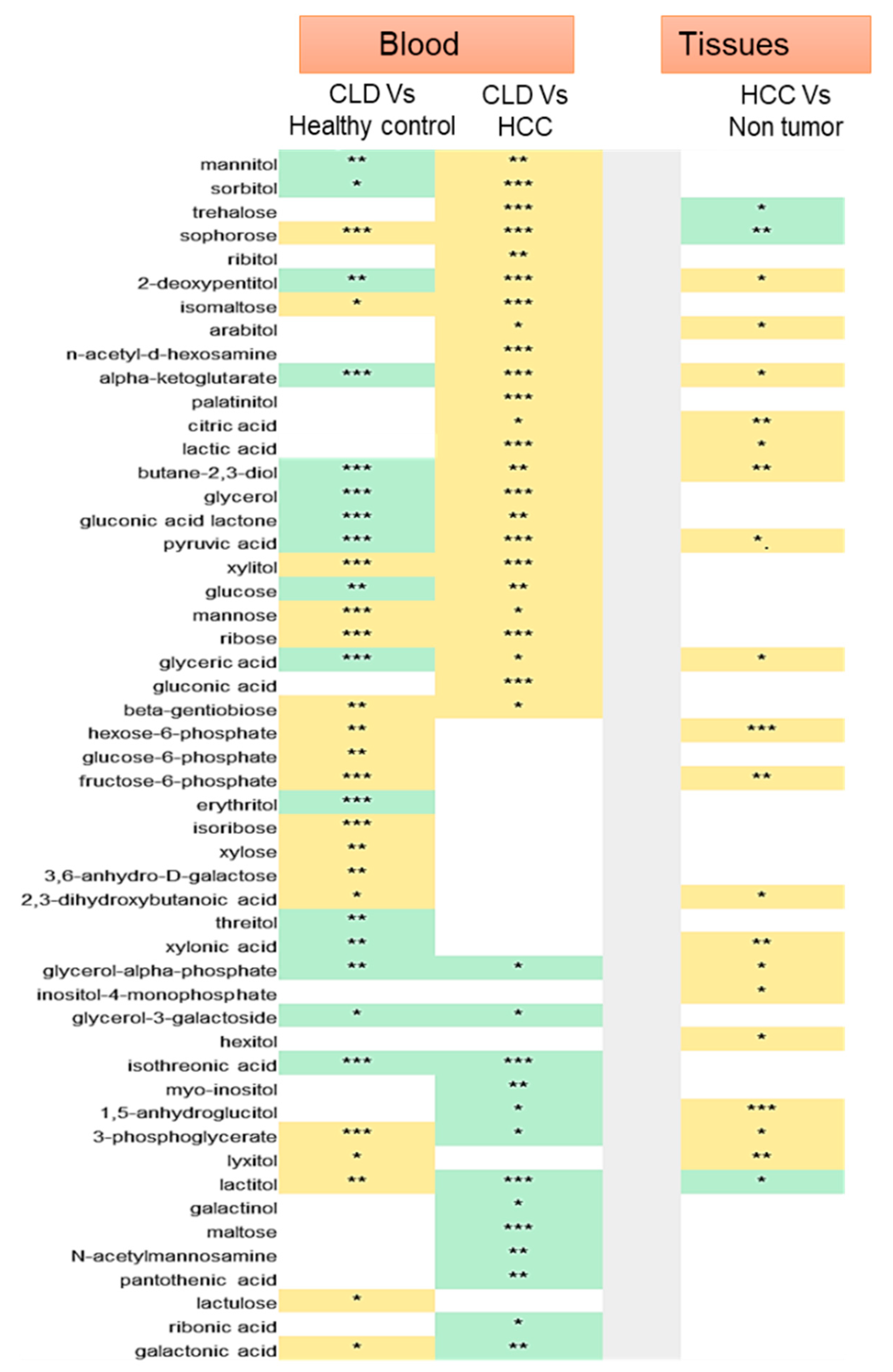
2.2. Statistical Metabolic Classes Differentiating HCC, CLD, and Healthy Control Blood Samples
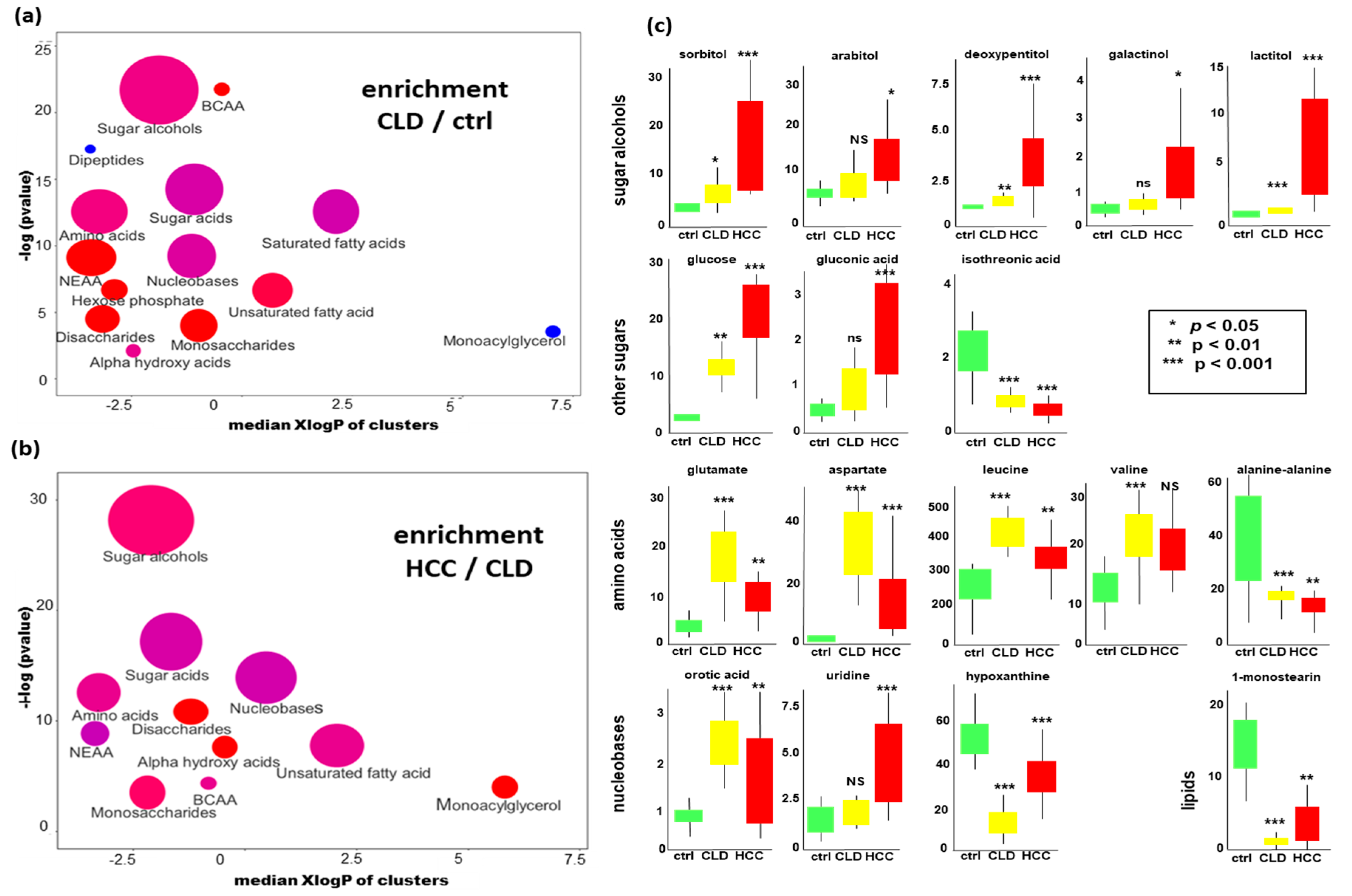
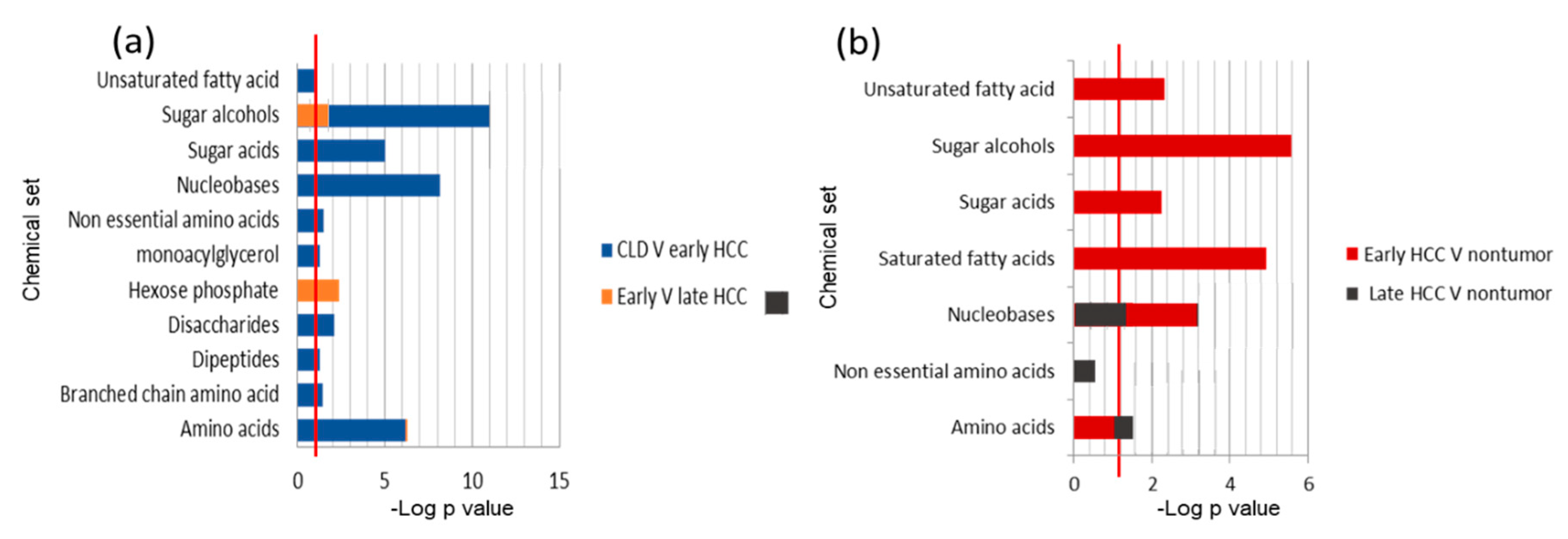
2.3. Chemical Set Enrichment Differences Between HCC, CLD, and Healthy Control Blood Samples
Metabolomics Data
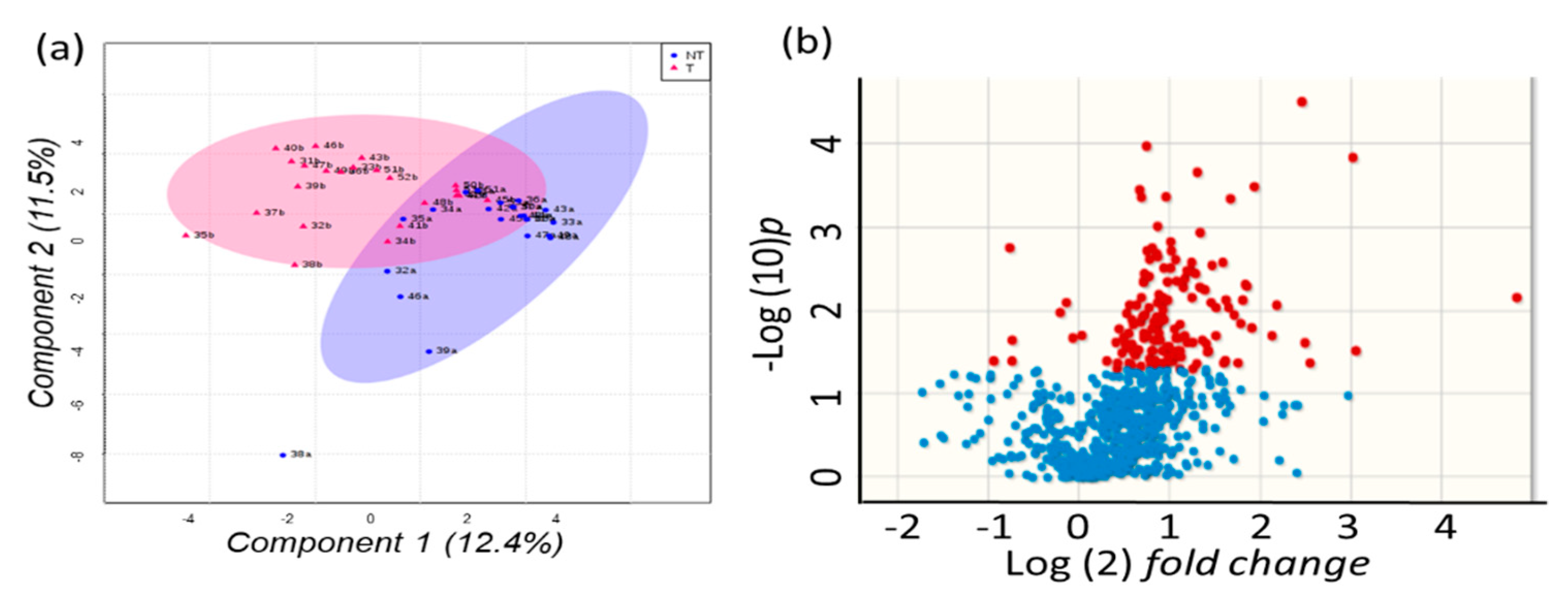
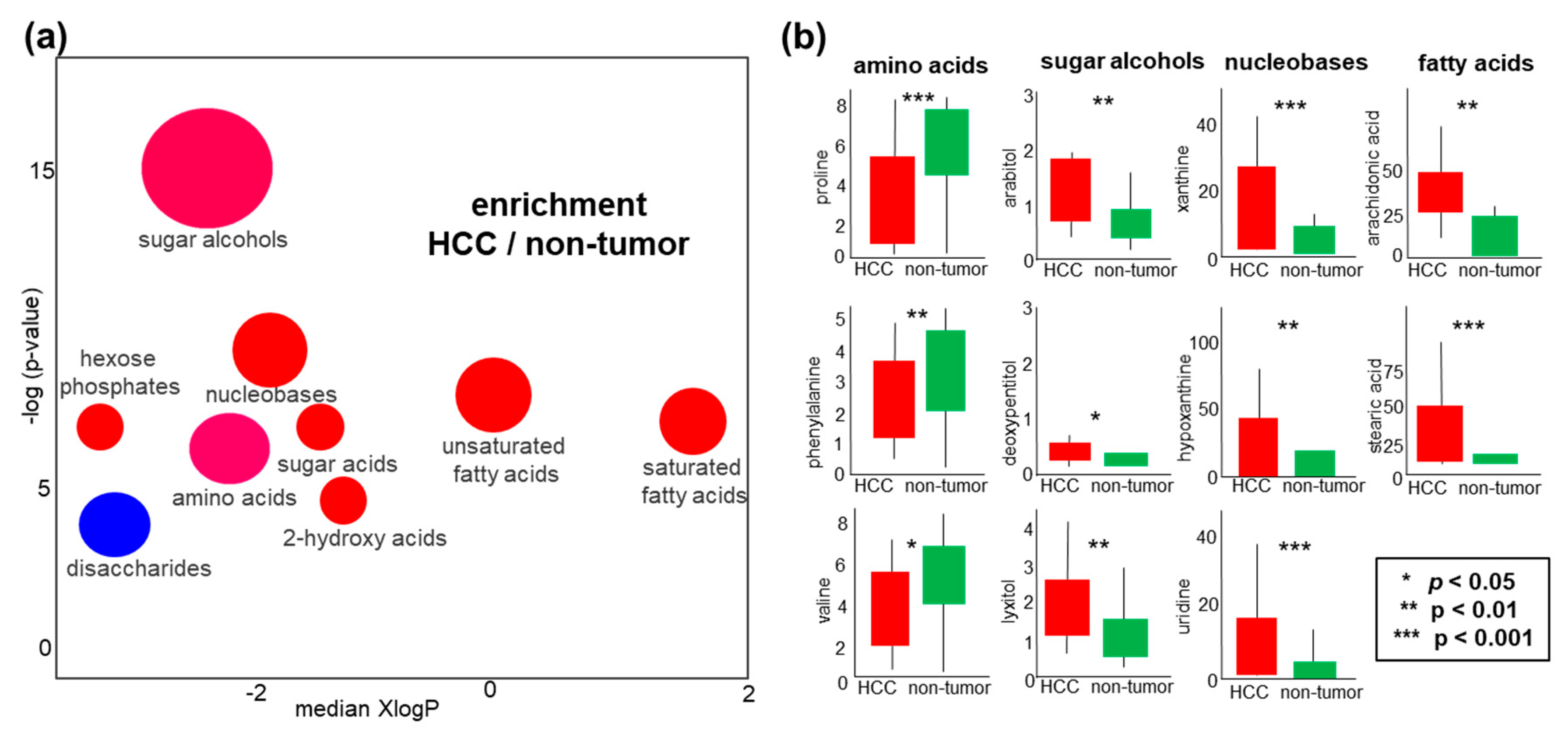
2.4. Metabolomics of Liver Tissues
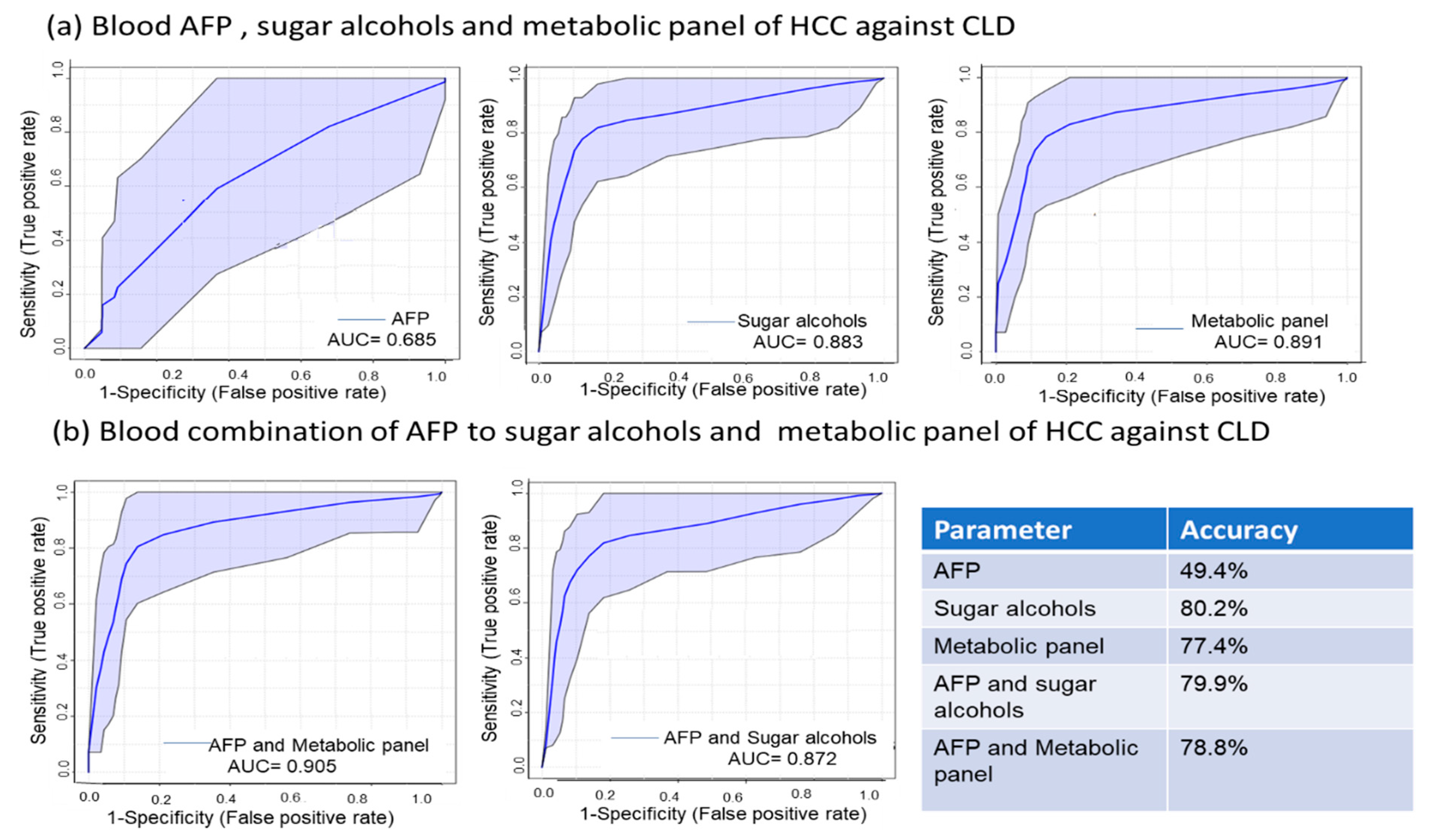
2.5. Sugar Alcohols are Highly Discriminant Metabolites Differentiating HCC from Non-HCC Subjects and HCC-Subjects from CLD Patients
2.6. Metabolite Clusters Enable Early Detection of HCC but not Differentiation of HCC Stage Subgroups
3. Discussion
4. Materials and Methods
4.1. Participants and Collection of Clinical Samples
4.2. Sample Pretreatment
4.3. Metabolomics Data Acquisition and Data Processing
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sayiner, M.; Golabi, P.; Younossi, Z.M. Disease Burden of Hepatocellular Carcinoma: A Global Perspective. Dig. Dis. Sci. 2019, 64, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 2013, 47, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elsalam, S.; Elwan, N.; Soliman, H.; Ziada, D.; Elkhalawany, W.; Salama, M.; Hawash, N.; Arafa, M.; Badawi, R.; Shehata, W.M.; et al. Epidemiology of liver cancer in Nile delta over a decade: A single-center study. South Asian J. Cancer 2018, 7, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Maucort-Boulch, D.; de Martel, C.; Franceschi, S.; Plummer, M. Fraction and incidence of liver cancer attributable to hepatitis B and C viruses worldwide. Int. J. Cancer 2018, 142, 2471–2477. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Yoon, S.K.; Lencioni, R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist 2010, 15 (Suppl. 4), 14–22. [Google Scholar] [CrossRef]
- Janevska, D.; Chaloska-Ivanova, V.; Janevski, V. Hepatocellular Carcinoma: Risk Factors, Diagnosis and Treatment. Open Access Maced. J. Med. Sci. 2015, 3, 732–736. [Google Scholar] [CrossRef]
- Shiels, M.S.; Engels, E.A.; Yanik, E.L.; McGlynn, K.A.; Pfeiffer, R.M.; O’Brien, T.R. Incidence of hepatocellular carcinoma among older Americans attributable to hepatitis C and hepatitis B: 2001 through 2013. Cancer 2019. [Google Scholar] [CrossRef]
- Petruzziello, A. Epidemiology of Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) Related Hepatocellular Carcinoma. Open Virol. J. 2018, 12, 26–32. [Google Scholar] [CrossRef]
- Kew, M.C. Epidemiology of hepatocellular carcinoma in sub-Saharan Africa. Ann. Hepatol. 2013, 12, 173–182. [Google Scholar] [CrossRef]
- Gomaa, A.; Allam, N.; Elsharkawy, A.; El Kassas, M.; Waked, I. Hepatitis C infection in Egypt: Prevalence, impact and management strategies. Hepatic Med. Evid. Res. 2017, 9, 17–25. [Google Scholar] [CrossRef]
- Poustchi, H.; Sepanlou, S.; Esmaili, S.; Mehrabi, N.; Ansarymoghadam, A. Hepatocellular carcinoma in the world and the middle East. Middle East J. Dig. Dis. 2010, 2, 31–41. [Google Scholar] [PubMed]
- Fateen, W.; Ryder, S.D. Screening for hepatocellular carcinoma: Patient selection and perspectives. J. Hepatocell. Carcinoma 2017, 4, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, G.P.; Abate, M.L.; Petrini, E.; Gaia, S.; Rizzetto, M.; Smedile, A. Highly sensitive alpha-fetoprotein, Lens culinaris agglutinin-reactive fraction of alpha-fetoprotein and des-gamma-carboxyprothrombin for hepatocellular carcinoma detection. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2016, 46, E130–E135. [Google Scholar] [CrossRef] [PubMed]
- Beyoglu, D.; Imbeaud, S.; Maurhofer, O.; Bioulac-Sage, P.; Zucman-Rossi, J.; Dufour, J.F.; Idle, J.R. Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology 2013, 58, 229–238. [Google Scholar] [CrossRef]
- Kajiura, D.; Yamanaka-Okumura, H.; Hirayama, A.; Tatano, H.; Endo, K.; Honma, M.; Igarashi, K.; Shoji, F.; Ikeda, S.; Yamaguchi, N.; et al. Perioperative serum and urine metabolome analyses in patients with hepatocellular carcinoma undergoing partial hepatectomy. Nutrition 2019, 58, 110–119. [Google Scholar] [CrossRef]
- Chettouh, H.; Lequoy, M.; Fartoux, L.; Vigouroux, C.; Desbois-Mouthon, C. Hyperinsulinaemia and insulin signalling in the pathogenesis and the clinical course of hepatocellular carcinoma. Liver Int. Off. J. Int. Assoc. Study Liver 2015, 35, 2203–2217. [Google Scholar] [CrossRef]
- Benfeitas, R.; Bidkhori, G.; Mukhopadhyay, B.; Klevstig, M.; Arif, M.; Zhang, C.; Lee, S.; Cinar, R.; Nielsen, J.; Uhlen, M.; et al. Characterization of heterogeneous redox responses in hepatocellular carcinoma patients using network analysis. EBioMedicine 2019, 40, 471–487. [Google Scholar] [CrossRef]
- Wang, X.W.; Hussain, S.P.; Huo, T.I.; Wu, C.G.; Forgues, M.; Hofseth, L.J.; Brechot, C.; Harris, C.C. Molecular pathogenesis of human hepatocellular carcinoma. Toxicology 2002, 181–182, 43–47. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Bandoh, S.; Roberts, L.R. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Garcia-Canaveras, J.C.; Donato, M.T.; Castell, J.V.; Lahoz, A. A comprehensive untargeted metabonomic analysis of human steatotic liver tissue by RP and HILIC chromatography coupled to mass spectrometry reveals important metabolic alterations. J. Proteome Res. 2011, 10, 4825–4834. [Google Scholar] [CrossRef]
- Patterson, A.D.; Maurhofer, O.; Beyoglu, D.; Lanz, C.; Krausz, K.W.; Pabst, T.; Gonzalez, F.J.; Dufour, J.F.; Idle, J.R. Aberrant lipid metabolism in hepatocellular carcinoma revealed by plasma metabolomics and lipid profiling. Cancer Res. 2011, 71, 6590–6600. [Google Scholar] [CrossRef] [PubMed]
- Osman, D.; Ali, O.; Obada, M.; El-Mezayen, H.; El-Said, H. Chromatographic determination of some biomarkers of liver cirrhosis and hepatocellular carcinoma in Egyptian patients. Biomed. Chromatogr. BMC 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Nahon, P.; Amathieu, R.; Triba, M.N.; Bouchemal, N.; Nault, J.C.; Ziol, M.; Seror, O.; Dhonneur, G.; Trinchet, J.C.; Beaugrand, M.; et al. Identification of serum proton NMR metabolomic fingerprints associated with hepatocellular carcinoma in patients with alcoholic cirrhosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 6714–6722. [Google Scholar] [CrossRef]
- Guo, W.; Tan, H.Y.; Wang, N.; Wang, X.; Feng, Y. Deciphering hepatocellular carcinoma through metabolomics: From biomarker discovery to therapy evaluation. Cancer Manag. Res. 2018, 10, 715–734. [Google Scholar] [CrossRef] [PubMed]
- Fitian, A.I.; Nelson, D.R.; Liu, C.; Xu, Y.; Ararat, M.; Cabrera, R. Integrated metabolomic profiling of hepatocellular carcinoma in hepatitis C cirrhosis through GC/MS and UPLC/MS-MS. Liver Int. Off. J. Int. Assoc. Study Liver 2014, 34, 1428–1444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Yan, G.; Han, Y.; Ye, Y.; Wang, X. Urinary metabolic profiling identifies a key role for glycocholic acid in human liver cancer by ultra-performance liquid-chromatography coupled with high-definition mass spectrometry. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 418, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Ressom, H.W.; Xiao, J.F.; Tuli, L.; Varghese, R.S.; Zhou, B.; Tsai, T.H.; Ranjbar, M.R.; Zhao, Y.; Wang, J.; Di Poto, C.; et al. Utilization of metabolomics to identify serum biomarkers for hepatocellular carcinoma in patients with liver cirrhosis. Anal. Chim. Acta 2012, 743, 90–100. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.; Nie, X.; Feng, X.; Chen, W.; Yue, Y.; Tang, H.; Deng, F. Metabonomic studies of human hepatocellular carcinoma using high-resolution magic-angle spinning 1H NMR spectroscopy in conjunction with multivariate data analysis. J. Proteome Res. 2007, 6, 2605–2614. [Google Scholar] [CrossRef]
- Le Moyec, L.; Triba, M.N.; Nahon, P.; Bouchemal, N.; Hantz, E.; Goossens, C.; Amathieu, R.; Savarin, P. Nuclear magnetic resonance metabolomics and human liver diseases: The principles and evidence associated with protein and carbohydrate metabolism. Biomed. Rep. 2017, 6, 387–395. [Google Scholar] [CrossRef]
- Ferrarini, A.; Di Poto, C.; He, S.; Tu, C.; Varghese, R.S.; Kara Balla, A.; Jayatilake, M.; Li, Z.; Ghaffari, K.; Fan, Z.; et al. Metabolomic Analysis of Liver Tissues for Characterization of Hepatocellular Carcinoma. J. Proteome Res. 2019, 18, 3067–3076. [Google Scholar] [CrossRef]
- Budhu, A.; Roessler, S.; Zhao, X.; Yu, Z.; Forgues, M.; Ji, J.; Karoly, E.; Qin, L.X.; Ye, Q.H.; Jia, H.L.; et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 2013, 144, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Xie, G.; Wang, X.; Fan, J.; Qiu, Y.; Zheng, X.; Qi, X.; Cao, Y.; Su, M.; Wang, X.; et al. Serum and urine metabolite profiling reveals potential biomarkers of human hepatocellular carcinoma. Mol. Cell. Proteom. MCP 2011, 10, M110.004945. [Google Scholar] [CrossRef]
- Nezami Ranjbar, M.R.; Luo, Y.; Di Poto, C.; Varghese, R.S.; Ferrarini, A.; Zhang, C.; Sarhan, N.I.; Soliman, H.; Tadesse, M.G.; Ziada, D.H.; et al. GC-MS Based Plasma Metabolomics for Identification of Candidate Biomarkers for Hepatocellular Carcinoma in Egyptian Cohort. PLoS ONE 2015, 10, e0127299. [Google Scholar] [CrossRef]
- Gao, R.; Cheng, J.; Fan, C.; Shi, X.; Cao, Y.; Sun, B.; Ding, H.; Hu, C.; Dong, F.; Yan, X. Serum Metabolomics to Identify the Liver Disease-Specific Biomarkers for the Progression of Hepatitis to Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 18175. [Google Scholar] [CrossRef]
- Gong, Z.G.; Zhao, W.; Zhang, J.; Wu, X.; Hu, J.; Yin, G.C.; Xu, Y.J. Metabolomics and eicosanoid analysis identified serum biomarkers for distinguishing hepatocellular carcinoma from hepatitis B virus-related cirrhosis. Oncotarget 2017, 8, 63890–63900. [Google Scholar] [CrossRef]
- Di Poto, C.; Ferrarini, A.; Zhao, Y.; Varghese, R.S.; Tu, C.; Zuo, Y.; Wang, M.; Nezami Ranjbar, M.R.; Luo, Y.; Zhang, C.; et al. Metabolomic Characterization of Hepatocellular Carcinoma in Patients with Liver Cirrhosis for Biomarker Discovery. Cancer Epidemiol. Biomark. Prev. 2017, 26, 675–683. [Google Scholar] [CrossRef]
- Wu, E.M.; Wong, L.L.; Hernandez, B.Y.; Ji, J.F.; Jia, W.; Kwee, S.A.; Kalathil, S. Gender differences in hepatocellular cancer: Disparities in nonalcoholic fatty liver disease/steatohepatitis and liver transplantation. Hepatoma Res. 2018, 4. [Google Scholar] [CrossRef]
- Yu, M.W.; Chen, C.J. Elevated serum testosterone levels and risk of hepatocellular carcinoma. Cancer Res. 1993, 53, 790–794. [Google Scholar]
- Xiao, J.; Zhao, Y.; Varghese, R.S.; Zhou, B.; Di Poto, C.; Zhang, L.; Tadesse, M.G.; Ziada, D.H.; Shetty, K.; Ressom, H.W. Evaluation of metabolite biomarkers for hepatocellular carcinoma through stratified analysis by gender, race, and alcoholic cirrhosis. Cancer Epidemiol. Biomark. Prev. 2014, 23, 64–72. [Google Scholar] [CrossRef]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef]
- Lai, Z.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K.; et al. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 2018, 15, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Barupal, D.K.; Fiehn, O. Chemical Similarity Enrichment Analysis (ChemRICH) as alternative to biochemical pathway mapping for metabolomic datasets. Sci. Rep. 2017, 7, 14567. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Yamada, T.; Hamajima, M.; Itoh, M.; Katayama, T.; Bork, P.; Goto, S.; Kanehisa, M. KEGG Atlas mapping for global analysis of metabolic pathways. Nucleic Acids Res. 2008, 36, W423–W426. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Wang, N.; Qin, W. Gut Microbiota and Hepatocellular Carcinoma. Gastrointest. Tumors 2015, 2, 33–40. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.M.; Niu, Y.; Zhang, X.; Yin, J.B.; Zhao, C.J.; Wang, R.T. Serum Zonulin in HBV-Associated Chronic Hepatitis, Liver Cirrhosis, and Hepatocellular Carcinoma. Dis. Markers 2019, 2019, 5945721. [Google Scholar] [CrossRef]
- Kim, H.S.; El-Serag, H.B. The Epidemiology of Hepatocellular Carcinoma in the USA. Curr. Gastroenterol. Rep. 2019, 21, 17. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Muralidharan, P.; Raj, J.P. Update in global trends and aetiology of hepatocellular carcinoma. Contemp. Oncol. 2018, 22, 141–150. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Attwa, M.H.; El-Etreby, S.A. Guide for diagnosis and treatment of hepatocellular carcinoma. World J. Hepatol. 2015, 7, 1632–1651. [Google Scholar] [CrossRef]
- Patel, N.; Yopp, A.C.; Singal, A.G. Diagnostic delays are common among patients with hepatocellular carcinoma. J. Natl. Compr. Cancer Netw. JNCCN 2015, 13, 543–549. [Google Scholar] [CrossRef]
- Bai, D.S.; Zhang, C.; Chen, P.; Jin, S.J.; Jiang, G.Q. The prognostic correlation of AFP level at diagnosis with pathological grade, progression, and survival of patients with hepatocellular carcinoma. Sci. Rep. 2017, 7, 12870. [Google Scholar] [CrossRef] [PubMed]
- Bialecki, E.S.; Di Bisceglie, A.M. Diagnosis of hepatocellular carcinoma. HPB Off. J. Int. Hepato Pancreato Biliary Assoc. 2005, 7, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Mehinovic, L.; Islamagic, E.; Husic-Selimovic, A.; Kurtovic-Kozaric, A.; Vukobrat-Bijedic, Z.; Suljevic, D. Evaluation of Diagnostic Efficiency of Alpha-Fetoprotein in Patients with Liver Cirrhosis and Hepatocellular Carcinoma: Single-Center Experience. Open Access Maced. J. Med. Sci. 2018, 6, 1668–1673. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.E.; Dvorin, E.; Drugan, A.; Johnson, M.P.; Yaron, Y.; Evans, M.I. Race-ethnicity-specific variation in multiple-marker biochemical screening: Alpha-fetoprotein, hCG, and estriol. Obstet. Gynecol. 1997, 89, 355–358. [Google Scholar] [CrossRef]
- Gopal, P.; Yopp, A.C.; Waljee, A.K.; Chiang, J.; Nehra, M.; Kandunoori, P.; Singal, A.G. Factors that affect accuracy of alpha-fetoprotein test in detection of hepatocellular carcinoma in patients with cirrhosis. Clin. Gastroenterol. Hepatol. 2014, 12, 870–877. [Google Scholar] [CrossRef]
- Stringer, K.A.; Younger, J.G.; McHugh, C.; Yeomans, L.; Finkel, M.A.; Puskarich, M.A.; Jones, A.E.; Trexel, J.; Karnovsky, A. Whole Blood Reveals More Metabolic Detail of the Human Metabolome than Serum as Measured by 1H-NMR Spectroscopy: Implications for Sepsis Metabolomics. Shock 2015, 44, 200–208. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers 2018, 10, 447. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Li, Z.; Guan, M.; Lin, Y.; Cui, X.; Zhang, Y.; Zhao, Z.; Zhu, J. Aberrant Lipid Metabolism in Hepatocellular Carcinoma Revealed by Liver Lipidomics. Int. J. Mol. Sci. 2017, 18, 2550. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.Y.; Ann, D.K. When fats commit crimes: Fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun. 2018, 38, 47. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.H.; Hu, W.J.; Chen, B.C.; Grahn, T.H.; Zhao, Y.R.; Bao, H.L.; Zhu, Y.F.; Zhang, Q.Y. BCAT1, a key prognostic predictor of hepatocellular carcinoma, promotes cell proliferation and induces chemoresistance to cisplatin. Liver Int. Off. J. Int. Assoc. Study Liver 2016, 36, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64–70. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788. [Google Scholar] [CrossRef]
- Choi, B.H.; Coloff, J.L. The Diverse Functions of Non-Essential Amino Acids in Cancer. Cancers 2019, 11, 675. [Google Scholar] [CrossRef]
- Reyes, R.K.; Motiwala, T.; Jacob, S.T. Regulation of glucose metabolism in hepatocarcinogenesis by microRNAs. Gene Expr. 2014, 16, 85–92. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef]
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef]
- Amann, T.; Maegdefrau, U.; Hartmann, A.; Agaimy, A.; Marienhagen, J.; Weiss, T.S.; Stoeltzing, O.; Warnecke, C.; Scholmerich, J.; Oefner, P.J.; et al. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am. J. Pathol. 2009, 174, 1544–1552. [Google Scholar] [CrossRef]
- Mano, Y.; Aishima, S.; Kubo, Y.; Tanaka, Y.; Motomura, T.; Toshima, T.; Shirabe, K.; Baba, S.; Maehara, Y.; Oda, Y. Correlation between biological marker expression and fluorine-18 fluorodeoxyglucose uptake in hepatocellular carcinoma. Am. J. Clin. Pathol. 2014, 142, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.Z.; Qu, S.B.; Wang, D.S. Reprogramming of glucose metabolism in hepatocellular carcinoma: Progress and prospects. World J. Gastroenterol. 2016, 22, 9933–9943. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pan, L.; Gao, C.; Xu, H.; Li, Y.; Zhang, L.; Ma, L.; Meng, L.; Sun, X.; Qin, H. Quercetin Inhibits the Proliferation of Glycolysis-Addicted HCC Cells by Reducing Hexokinase 2 and Akt-mTOR Pathway. Molecules 2019, 24, 1993. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef]
- Schwab, A.; Siddiqui, A.; Vazakidou, M.E.; Napoli, F.; Bottcher, M.; Menchicchi, B.; Raza, U.; Saatci, O.; Krebs, A.M.; Ferrazzi, F.; et al. Polyol Pathway Links Glucose Metabolism to the Aggressiveness of Cancer Cells. Cancer Res. 2018, 78, 1604–1618. [Google Scholar] [CrossRef]
- Yan, L.J. Redox imbalance strmeess in diabetes mellitus: Role of the polyol pathway. Anim. Models Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef]
- Chung, S.S.; Ho, E.C.; Lam, K.S.; Chung, S.K. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. JASN 2003, 14, S233–S236. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Obrosova, I.G.; Chung, S.S.; Kador, P.F. Diabetic cataracts: Mechanisms and management. Diabetes Metab. Res. Rev. 2010, 26, 172–180. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D.; Stasinopoulos, M.; Zelenko, Z.; Shiloach, J. Polyol accumulation in muscle and liver in a mouse model of type 2 diabetes. J. Diabetes Complicat. 2016, 30, 999–1007. [Google Scholar] [CrossRef]
- Sarode, G.V.; Kim, K.; Kieffer, D.A.; Shibata, N.M.; Litwin, T.; Czlonkowska, A.; Medici, V. Metabolomics profiles of patients with Wilson disease reveal a distinct metabolic signature. Metabolomics 2019, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, A.; D’Ascenzo, S.; Knijn, A.; Iorio, E.; Dolo, V.; Pavan, A.; Podo, F. Detection of polyol accumulation in a new ovarian carcinoma cell line, CABA I: A(1)H NMR study. Br. J. Cancer 2002, 86, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Regenold, W.T.; Shapiro, P. Inhibition of aldose reductase enhances HeLa cell sensitivity to chemotherapeutic drugs and involves activation of extracellular signal-regulated kinases. Anti Cancer Drugs 2002, 13, 859–868. [Google Scholar] [CrossRef]
- Uzozie, A.; Nanni, P.; Staiano, T.; Grossmann, J.; Barkow-Oesterreicher, S.; Shay, J.W.; Tiwari, A.; Buffoli, F.; Laczko, E.; Marra, G. Sorbitol dehydrogenase overexpression and other aspects of dysregulated protein expression in human precancerous colorectal neoplasms: A quantitative proteomics study. Mol. Cell. Proteom. MCP 2014, 13, 1198–1218. [Google Scholar] [CrossRef]
- Saraswat, M.; Mrudula, T.; Kumar, P.U.; Suneetha, A.; Rao, T.S.; Srinivasulu, M.; Reddy, B. Overexpression of aldose reductase in human cancer tissues. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2006, 12, CR525–CR529. [Google Scholar]
- Bagga, P.; Wilson, N.; Rich, L.; Marincola, F.M.; Schnall, M.D.; Hariharan, H.; Haris, M.; Reddy, R. Sugar alcohol provides imaging contrast in cancer detection. Sci. Rep. 2019, 9, 11092. [Google Scholar] [CrossRef]
- Sanchez-Lozada, L.G.; Andres-Hernando, A.; Garcia-Arroyo, F.E.; Cicerchi, C.; Li, N.; Kuwabara, M.; Roncal-Jimenez, C.A.; Johnson, R.J.; Lanaspa, M.A. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J. Biol. Chem. 2019, 294, 4272–4281. [Google Scholar] [CrossRef]
- Takahashi, M.; Fujii, J.; Miyoshi, E.; Hoshi, A.; Taniguchi, N. Elevation of aldose reductase gene expression in rat primary hepatoma and hepatoma cell lines: Implication in detoxification of cytotoxic aldehydes. Int. J. Cancer 1995, 62, 749–754. [Google Scholar] [CrossRef]
- Zeindl-Eberhart, E.; Jungblut, P.R.; Otto, A.; Rabes, H.M. Identification of tumor-associated protein variants during rat hepatocarcinogenesis. Aldose reductase. J. Biol. Chem. 1994, 269, 14589–14594. [Google Scholar]
- Cao, D.; Fan, S.T.; Chung, S.S. Identification and characterization of a novel human aldose reductase-like gene. J. Biol. Chem. 1998, 273, 11429–11435. [Google Scholar] [CrossRef]
- Lee, K.W.; Ko, B.C.; Jiang, Z.; Cao, D.; Chung, S.S. Overexpression of aldose reductase in liver cancers may contribute to drug resistance. Anti Cancer Drugs 2001, 12, 129–132. [Google Scholar] [CrossRef]
- Petrash, J.M. All in the family: Aldose reductase and closely related aldo-keto reductases. Cell Mol. Life Sci. 2004, 61, 737–749. [Google Scholar] [CrossRef]
- Samanta, B.K.; Chandra, N.C.; Ghosh, S.; Mukherjee, K.L. Aldose metabolism in developing human fetal brain and liver. Experientia 1984, 40, 1420–1422. [Google Scholar] [CrossRef]
- Takahashi, M.; Hoshi, A.; Fujii, J.; Miyoshi, E.; Kasahara, T.; Suzuki, K.; Aozasa, K.; Taniguchi, N. Induction of aldose reductase gene expression in LEC rats during the development of the hereditary hepatitis and hepatoma. Jpn. J. Cancer Res. Gann 1996, 87, 337–341. [Google Scholar] [CrossRef]
- Scuric, Z.; Stain, S.C.; Anderson, W.F.; Hwang, J.J. New member of aldose reductase family proteins overexpressed in human hepatocellular carcinoma. Hepatology 1998, 27, 943–950. [Google Scholar] [CrossRef]
- Tammali, R.; Srivastava, S.K.; Ramana, K.V. Targeting aldose reductase for the treatment of cancer. Curr. Cancer Drug Targets 2011, 11, 560–571. [Google Scholar] [CrossRef]
- Brown, K.E.; Broadhurst, K.A.; Mathahs, M.M.; Kladney, R.D.; Fimmel, C.J.; Srivastava, S.K.; Brunt, E.M. Immunodetection of aldose reductase in normal and diseased human liver. Histol. Histopathol. 2005, 20, 429–436. [Google Scholar] [CrossRef]
- Qiu, L.; Wu, X.; Chau, J.F.; Szeto, I.Y.; Tam, W.Y.; Guo, Z.; Chung, S.K.; Oates, P.J.; Chung, S.S.; Yang, J.Y. Aldose reductase regulates hepatic peroxisome proliferator-activated receptor alpha phosphorylation and activity to impact lipid homeostasis. J. Biol. Chem. 2008, 283, 17175–17183. [Google Scholar] [CrossRef]
- Zhao, J.X.; Yuan, Y.W.; Cai, C.F.; Shen, D.Y.; Chen, M.L.; Ye, F.; Mi, Y.J.; Luo, Q.C.; Cai, W.Y.; Zhang, W.; et al. Aldose reductase interacts with AKT1 to augment hepatic AKT/mTOR signaling and promote hepatocarcinogenesis. Oncotarget 2017, 8, 66987–67000. [Google Scholar] [CrossRef]
- Guo, C.; Chen, L.; Huang, J.; Wang, Y.; Shi, C.; Gao, J.; Hong, Y.; Chen, T.; Qiu, L. Aldose reductase inhibitor protects mice from alcoholic steatosis by repressing saturated fatty acid biosynthesis. Chem. Biol. Interact. 2018, 287, 41–48. [Google Scholar] [CrossRef]
- Castellvi, A.; Crespo, I.; Crosas, E.; Camara-Artigas, A.; Gavira, J.A.; Aranda, M.A.G.; Pares, X.; Farres, J.; Juanhuix, J. Efficacy of aldose reductase inhibitors is affected by oxidative stress induced under X-ray irradiation. Sci. Rep. 2019, 9, 3177. [Google Scholar] [CrossRef] [PubMed]
- Ramana, K.V.; Srivastava, S.K. Aldose reductase: A novel therapeutic target for inflammatory pathologies. Int. J. Biochem. Cell Biol. 2010, 42, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Tammali, R.; Reddy, A.B.; Saxena, A.; Rychahou, P.G.; Evers, B.M.; Qiu, S.; Awasthi, S.; Ramana, K.V.; Srivastava, S.K. Inhibition of aldose reductase prevents colon cancer metastasis. Carcinogenesis 2011, 32, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.C.W.; Carella, M.A.; Papa, S.; Bubici, C. High Expression of Glycolytic Genes in Cirrhosis Correlates With the Risk of Developing Liver Cancer. Front. Cell Dev. Biol. 2018, 6, 138. [Google Scholar] [CrossRef]
- Gupta, H.; Youn, G.S.; Shin, M.J.; Suk, K.T. Role of Gut Microbiota in Hepatocarcinogenesis. Microorganisms 2019, 7, 121. [Google Scholar] [CrossRef]
- El-Sokkary, R.H.; Tash, R.M.E.; Meawed, T.E.; El Seifi, O.S.; Mortada, E.M. Detection of hepatitis C virus (HCV) among health care providers in an Egyptian university hospital: Different diagnostic modalities. Infect. Drug Resist. 2017, 10, 357–364. [Google Scholar] [CrossRef]
- Motawi, T.K.; Mady, A.E.; Shaheen, S.; Elshenawy, S.Z.; Talaat, R.M.; Rizk, S.M. Genetic variation in microRNA-100 (miR-100) rs1834306 T/C associated with Hepatitis B virus (HBV) infection: Correlation with expression level. Infect. Genet. Evol. 2019, 73, 444–449. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics by Gas Chromatography-Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr. Protoc. Mol. Biol. 2016, 114, 30.4.1–30.4.32. [Google Scholar] [CrossRef]
- Chong, J.; Yamamoto, M.; Xia, J. MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites 2019, 9, 57. [Google Scholar] [CrossRef]
- Le Cao, K.A.; Boitard, S.; Besse, P. Sparse PLS discriminant analysis: Biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinform. 2011, 12, 253. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | HCC | Chronic Liver Disease (CLD) | Healthy Control | p-Value |
|---|---|---|---|---|
| Number | 23 | 15 | 15 | |
| Gender (M/F) | 17/6 | 11/4 | 10/5 | 0.877 (1) |
| Age (Years) Mean (range) | 53 (23–73) | 46 (24–67) | 50 (23–64) | 0.311 (2) |
| Alanine transaminase (IU/L) Mean (range) | 56 (18–160) | 58 (20–134) | 27 (16–37) | 0.878 (a) 0.003 (2) |
| Aspartate transaminase (IU/L) Mean (range) | 65 (21–195) | 48 (20–119) | 26 (18–35) | 0.115 (a) 0.001 (2) |
| Hemoglobin (g/L) Mean (range) | 12.9 (9.3–16.7) | 14 (11.9–16.6) | 13 (11.8–14.3) | 0.011 (a) 0.02 (2) |
| Platelet count (103/L) Mean (range) | 193 (53–686) | 240 (172–355) | 217 (166–260) | 0.158 (a) 0.370 (2) |
| International normalized ratio Mean (range) | 1.34 (1–2.8) | 1.03 (0.9–1.14) | 0.97 (0.9–1.1) | 0.005 (a) 0.001 (2) |
| current HCV infection (Y/N) (c) | 13/10 | 12/3 | 0/15 | 3.2 × 10−5 (1) 0.13 (b) |
| current HBV infection (Y/N) (c) | 2/21 | 3/12 | 0/15 | 0.18 (1) 0.25 (b) |
| Alfa fetoprotein (AFP) (ng/mL) Mean (range) | 268.3 (2.6–2000) | 8.9 (1.53–60) | 2.6 (1.8–3.5) | 0.021 (a) 0.016 (2) |
| Age of the Patients (years) Mean (range) | 53 (23–73) |
|---|---|
| Gender | |
| Male | 17 (74%) |
| Female | 6 (26%) |
| Tumor stage (TNM) | |
| I (Early) | 7 (30%) |
| II (Early) | 9 (39%) |
| III (Late) | 5 (22%) |
| IV (Late) | 2 (9%) |
| Focal Lesion | |
| Single | 15 (65%) |
| Multiple | 8 (35%) |
| Cirrhosis | |
| Yes | 21 (91%) |
| No | 2 (9%) |
| Tumor Size | |
| <5 cm | 19 (83%) |
| >5 cm | 4 (17%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, I.T.; Fiehn, O.; Elfert, A.; Helal, M.; Salama, I.; El-Said, H. Sugar Alcohols Have a Key Role in Pathogenesis of Chronic Liver Disease and Hepatocellular Carcinoma in Whole Blood and Liver Tissues. Cancers 2020, 12, 484. https://doi.org/10.3390/cancers12020484
Ismail IT, Fiehn O, Elfert A, Helal M, Salama I, El-Said H. Sugar Alcohols Have a Key Role in Pathogenesis of Chronic Liver Disease and Hepatocellular Carcinoma in Whole Blood and Liver Tissues. Cancers. 2020; 12(2):484. https://doi.org/10.3390/cancers12020484
Chicago/Turabian StyleIsmail, Israa T., Oliver Fiehn, Ashraf Elfert, Marwa Helal, Ibrahim Salama, and Hala El-Said. 2020. "Sugar Alcohols Have a Key Role in Pathogenesis of Chronic Liver Disease and Hepatocellular Carcinoma in Whole Blood and Liver Tissues" Cancers 12, no. 2: 484. https://doi.org/10.3390/cancers12020484
APA StyleIsmail, I. T., Fiehn, O., Elfert, A., Helal, M., Salama, I., & El-Said, H. (2020). Sugar Alcohols Have a Key Role in Pathogenesis of Chronic Liver Disease and Hepatocellular Carcinoma in Whole Blood and Liver Tissues. Cancers, 12(2), 484. https://doi.org/10.3390/cancers12020484