Inhibition of Alternative Cancer Cell Metabolism of EGFR Mutated Non-Small Cell Lung Cancer Serves as a Potential Therapeutic Strategy

 , ,
, ,
Abstract
1. Introduction
2. Results
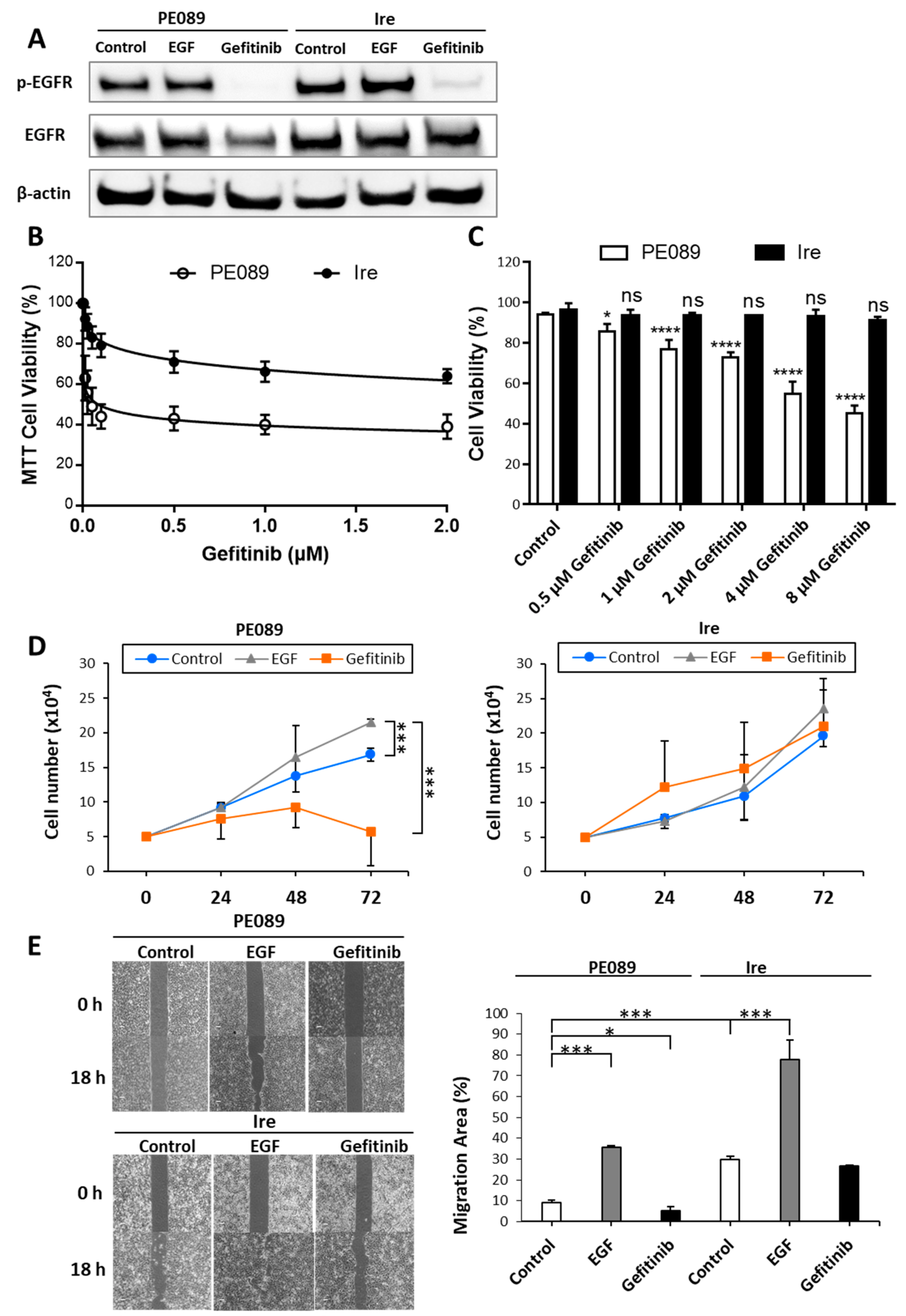
2.1. Ire Cells Are Gefitinib-Resistant Lung Adenocarcinomas
2.2. Enhanced Mitochondrial Translocation of EGFR and Mitochondrial Bioenergetics in TKI-Resistant Ire Cells
2.3. Higher Activity and Higher mRNA and Protein Expression Levels in Mitochondrial Complexes in TKI-Resistant Ire Cells
2.4. Gefitinib-Resistant Ire Cells Relied on the OXPHOS Pathway to Generate ATP
2.5. Enhanced Expression of OXPHOS Pathway-Related Genes in Ire Cells
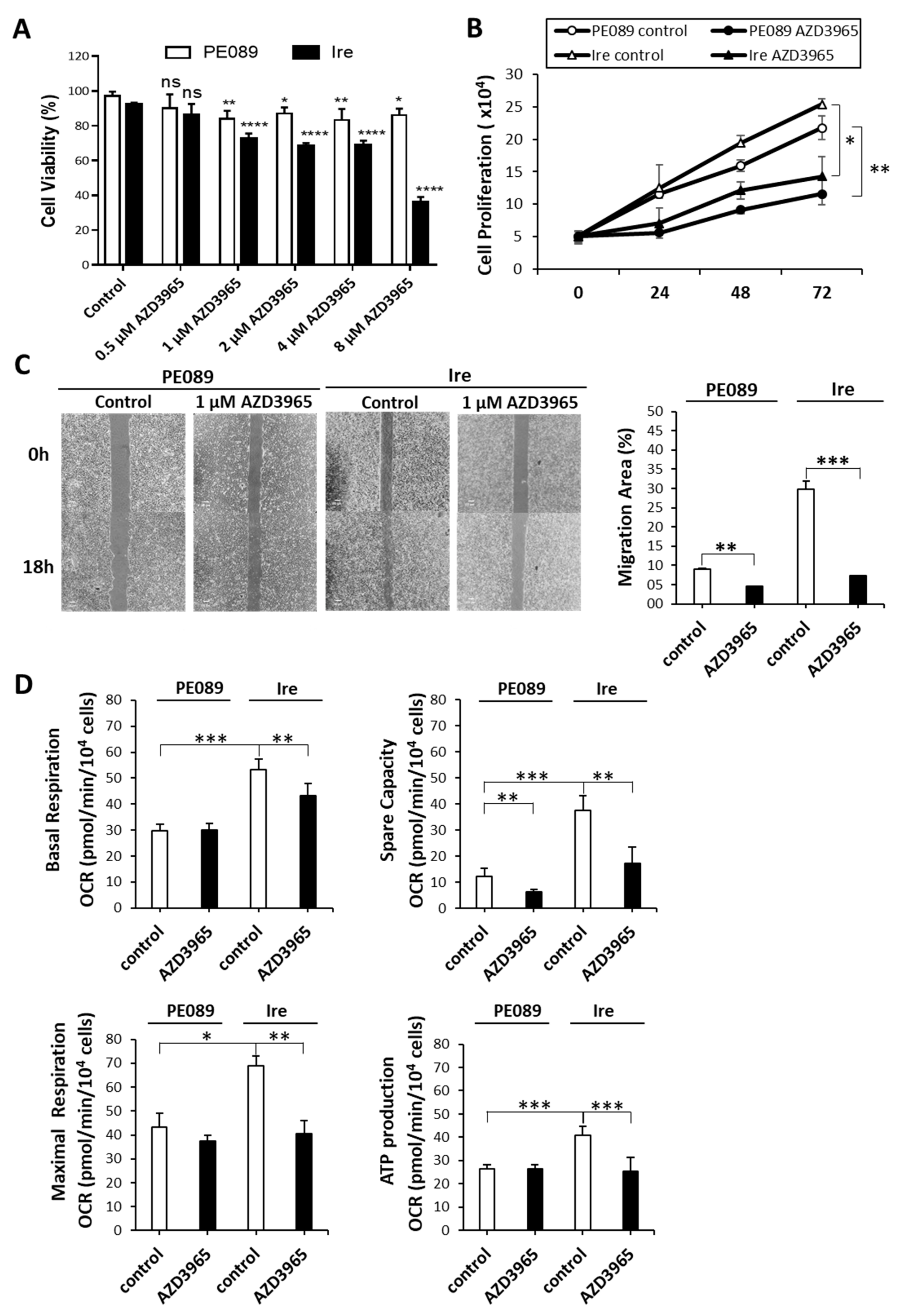
2.6. MCT-1 Can Serve as a Therapeutic Target for Gefitinib-Resistant Ire Cells In Vitro
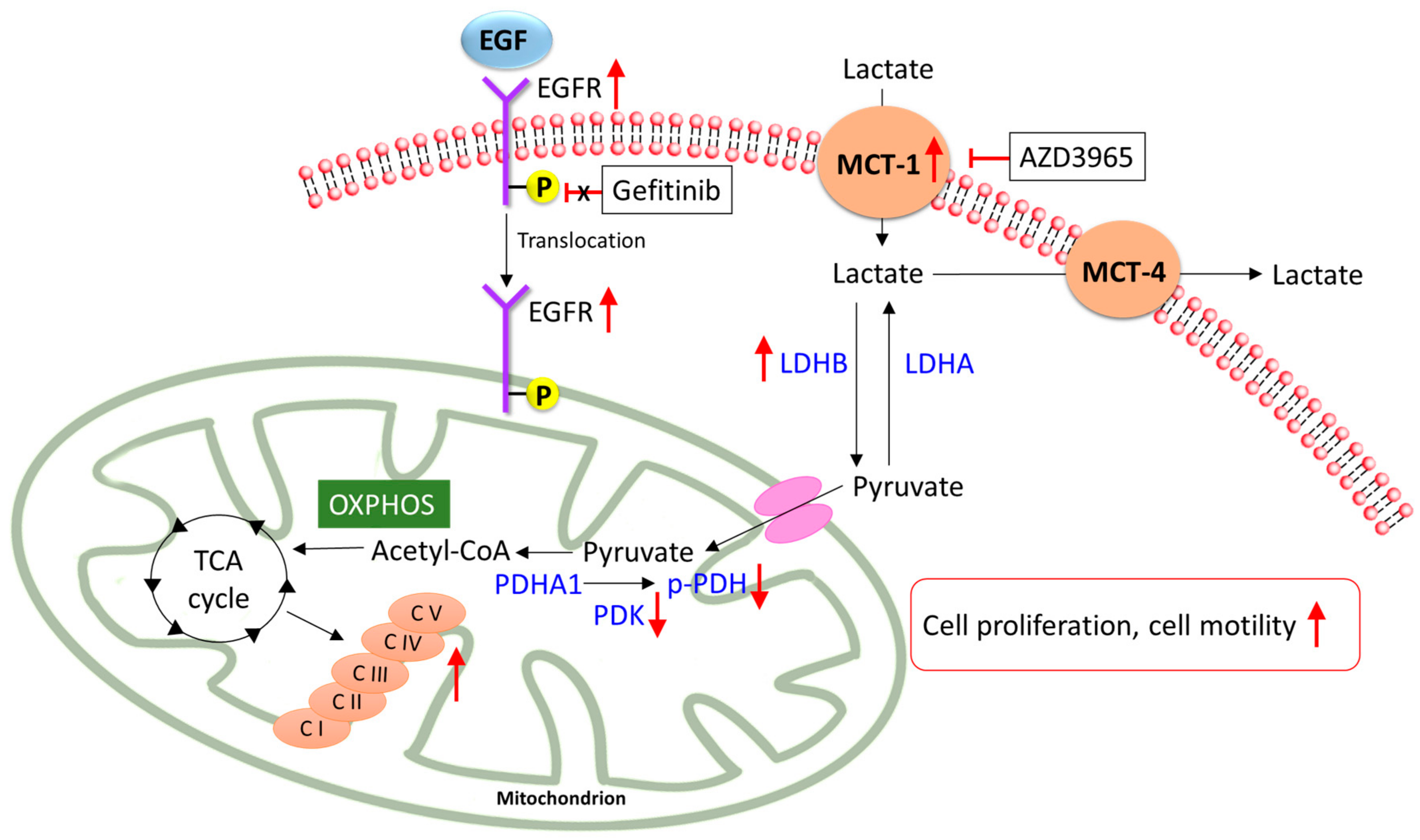
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Immunoblotting
4.4. Cell Proliferation
4.5. Cell Viability
4.6. Wound Healing Assay
4.7. Mitochondrial Extraction
4.8. Immunofluorescence Analysis
4.9. Mitochondrial Bioenergetics Assay
4.10. Mitochondrial Complex Activity Assay
4.11. Real-Time Quantitative RT-PCR
4.12. Statistical analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef]
- Wang, B.Y.; Huang, J.Y.; Cheng, C.Y.; Lin, C.H.; Ko, J.; Liaw, Y.P. Lung cancer and prognosis in taiwan: A population-based cancer registry. J. Thorac. Oncol. 2013, 8, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Peled, N.; Wynes, M.W.; Yoshida, K.; Pardo, M.; Mascaux, C.; Ohira, T.; Tsuboi, M.; Matsubayashi, J.; Nagao, T.; et al. Novel epidermal growth factor receptor mutation-specific antibodies for non-small cell lung cancer: Immunohistochemistry as a possible screening method for epidermal growth factor receptor mutations. J. Thorac. Oncol. 2010, 5, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H.; Chiang, C.J.; Tseng, J.S.; Yang, T.Y.; Hsu, K.H.; Chen, K.C.; Wang, C.L.; Chen, C.Y.; Yen, S.H.; Tsai, C.M.; et al. EGFR mutation, smoking, and gender in advanced lung adenocarcinoma. Oncotarget 2017, 8, 98384–98393. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Chariot, A. EGFR and NF-kappaB: Partners in cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Red Brewer, M.; Yun, C.H.; Lai, D.; Lemmon, M.A.; Eck, M.J.; Pao, W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E3595–E3604. [Google Scholar] [CrossRef]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef]
- Nukaga, S.; Yasuda, H.; Tsuchihara, K.; Hamamoto, J.; Masuzawa, K.; Kawada, I.; Naoki, K.; Matsumoto, S.; Mimaki, S.; Ikemura, S.; et al. Amplification of EGFR Wild-Type Alleles in Non-Small Cell Lung Cancer Cells Confers Acquired Resistance to Mutation-Selective EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2017, 77, 2078–2089. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Wang, S.; Tsui, S.T.; Liu, C.; Song, Y.; Liu, D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 59. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Fujiwara, S.; Kawano, Y.; Yuki, H.; Okuno, Y.; Nosaka, K.; Mitsuya, H.; Hata, H. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br. J. Cancer 2013, 108, 170–178. [Google Scholar] [CrossRef]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Weinhouse, S. On respiratory impairment in cancer cells. Science 1956, 124, 267–269. [Google Scholar] [CrossRef]
- Newsholme, E.A.; Crabtree, B.; Ardawi, M.S. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci. Rep. 1985, 5, 393–400. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Xu, X.D.; Shao, S.X.; Jiang, H.P.; Cao, Y.W.; Wang, Y.H.; Yang, X.C.; Wang, Y.L.; Wang, X.S.; Niu, H.T. Warburg effect or reverse Warburg effect? A review of cancer metabolism. Oncol. Res. Treat. 2015, 38, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371. [Google Scholar] [CrossRef]
- San-Millan, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Mahieu, N.G.; Huang, X.; Singh, M.; Crawford, P.A.; Johnson, S.L.; Gross, R.W.; Schaefer, J.; Patti, G.J. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 2016, 12, 937. [Google Scholar] [CrossRef]
- Cao, X.; Zhu, H.; Ali-Osman, F.; Lo, H.W. EGFR and EGFRvIII undergo stress- and EGFR kinase inhibitor-induced mitochondrial translocalization: A potential mechanism of EGFR-driven antagonism of apoptosis. Mol. Cancer 2011, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Che, T.F.; Lin, C.W.; Wu, Y.Y.; Chen, Y.J.; Han, C.L.; Chang, Y.L.; Wu, C.T.; Hsiao, T.H.; Hong, T.M.; Yang, P.C. Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes metastasis in NSCLC. Oncotarget 2015, 6, 37349–37366. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.S.; Huang, C.Y.; Kuo, Y.Z.; Kang, W.Y.; Liu, G.Y.; Huang, A.M.; Yu, H.J.; Lai, M.K.; Huang, S.P.; Wu, W.J.; et al. Characterization of membranous and cytoplasmic EGFR expression in human normal renal cortex and renal cell carcinoma. J. Biomed. Sci. 2009, 16, 82. [Google Scholar] [CrossRef] [PubMed]
- Boerner, J.L.; Demory, M.L.; Silva, C.; Parsons, S.J. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol. Cell Biol. 2004, 24, 7059–7071. [Google Scholar] [CrossRef] [PubMed]
- Demory, M.L.; Boerner, J.L.; Davidson, R.; Faust, W.; Miyake, T.; Lee, I.; Huttemann, M.; Douglas, R.; Haddad, G.; Parsons, S.J. Epidermal growth factor receptor translocation to the mitochondria: Regulation and effect. J. Biol. Chem. 2009, 284, 36592–36604. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Chen, Y.J.; Li, L.Y.; Wei, Y.L.; Hsu, S.C.; Tsai, S.L.; Chiu, P.C.; Huang, W.P.; Wang, Y.N.; Chen, C.H.; et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J. Biol. Chem. 2011, 286, 20558–20568. [Google Scholar] [CrossRef]
- Beloueche-Babari, M.; Wantuch, S.; Casals Galobart, T.; Koniordou, M.; Parkes, H.G.; Arunan, V.; Chung, Y.L.; Eykyn, T.R.; Smith, P.D.; Leach, M.O. MCT1 inhibitor AZD3965 increases mitochondrial metabolism, facilitating combination therapy and noninvasive magnetic resonance spectroscopy. Cancer Res. 2017, 77, 5913–5924. [Google Scholar] [CrossRef]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef]
- Li, C.; Iida, M.; Dunn, E.F.; Ghia, A.J.; Wheeler, D.L. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009, 28, 3801–3813. [Google Scholar] [CrossRef]
- Takenaka, T.; Katayama, M.; Sugiyama, A.; Hagiwara, M.; Fujii, I.; Takatani-Nakase, T.; Kobayashi, S.S.; Nakase, I. Gefitinib enhances mitochondrial biological functions in NSCLCs with EGFR mutations at a high cell density. Anticancer Res. 2017, 37, 4779–4788. [Google Scholar] [CrossRef]
- Dasgupta, S.; Soudry, E.; Mukhopadhyay, N.; Shao, C.; Yee, J.; Lam, S.; Lam, W.; Zhang, W.; Gazdar, A.F.; Fisher, P.B.; et al. Mitochondrial DNA mutations in respiratory complex-I in never-smoker lung cancer patients contribute to lung cancer progression and associated with EGFR gene mutation. J. Cell Physiol. 2012, 227, 2451–2460. [Google Scholar] [CrossRef]
- Urra, F.A.; Munoz, F.; Lovy, A.; Cardenas, C. The Mitochondrial Complex(I)ty of Cancer. Front. Oncol. 2017, 7, 118. [Google Scholar] [CrossRef]
- He, X.; Zhou, A.; Lu, H.; Chen, Y.; Huang, G.; Yue, X.; Zhao, P.; Wu, Y. Suppression of mitochondrial complex I influences cell metastatic properties. PLoS ONE 2013, 8, e61677. [Google Scholar] [CrossRef]
- Jeon, J.H.; Kim, D.K.; Shin, Y.; Kim, H.Y.; Song, B.; Lee, E.Y.; Kim, J.K.; You, H.J.; Cheong, H.; Shin, D.H.; et al. Migration and invasion of drug-resistant lung adenocarcinoma cells are dependent on mitochondrial activity. Exp. Mol. Med. 2016, 48, e277. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, X.; Zhao, M.; Wang, Y.; Cheng, X.; Wang, D.; Xu, Y.; Du, Z.; Yu, X. Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells. BMC Cancer 2011, 11, 170. [Google Scholar] [CrossRef]
- Sotgia, F.; Lisanti, M.P. Mitochondrial markers predict survival and progression in non-small cell lung cancer (NSCLC) patients: Use as companion diagnostics. Oncotarget 2017, 8, 68095–68107. [Google Scholar] [CrossRef] [PubMed]
- Mandujano-Tinoco, E.A.; Gallardo-Perez, J.C.; Marin-Hernandez, A.; Moreno-Sanchez, R.; Rodriguez-Enriquez, S. Anti-mitochondrial therapy in human breast cancer multi-cellular spheroids. Biochim. Biophys. Acta 2013, 1833, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Chang, C.P.; Tsao, C.C.; Xu, J. Oligomycin-induced bioenergetic adaptation in cancer cells with heterogeneous bioenergetic organization. J. Biol. Chem. 2010, 285, 12647–12654. [Google Scholar] [CrossRef]
- Jeoung, N.H. Pyruvate dehydrogenase kinases: Therapeutic targets for diabetes and cancers. Diabetes Metab. J. 2015, 39, 188–197. [Google Scholar] [CrossRef]
- Hong, C.S.; Graham, N.A.; Gu, W.; Espindola Camacho, C.; Mah, V.; Maresh, E.L.; Alavi, M.; Bagryanova, L.; Krotee, P.A.L.; Gardner, B.K.; et al. MCT1 Modulates Cancer Cell Pyruvate Export and Growth of Tumors that Co-express MCT1 and MCT4. Cell Rep. 2016, 14, 1590–1601. [Google Scholar] [CrossRef]
- Pinheiro, C.; Penna, V.; Morais-Santos, F.; Abrahao-Machado, L.F.; Ribeiro, G.; Curcelli, E.C.; Olivieri, M.V.; Morini, S.; Valenca, I.; Ribeiro, D.; et al. Characterization of monocarboxylate transporters (MCTs) expression in soft tissue sarcomas: Distinct prognostic impact of MCT1 sub-cellular localization. J. Transl. Med. 2014, 12. [Google Scholar] [CrossRef]
- Polanski, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.A.; Bell, N.; Blair, H.; Sikka, A.; Thomas, H.; Phillips, N.; Nakjang, S.; Miwa, S.; Crossland, R.; Rand, V.; et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica 2017, 102, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Salabei, J.K.; Gibb, A.A.; Hill, B.G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014, 9, 421–438. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Catalogue Number | Vendor |
|---|---|---|
| EGFR | sc-03 | Santa Cruz Biotech |
| p-EGFR | #3777 | Cell Signaling Technology |
| MCT-1 | AB3538P | EMD Millipore |
| MCT-4 | AB3316P | EMD Millipore |
| PDK-1α | ADI-KAP-PK112 | Enzo Life Sciences |
| p-PDH | AP1062 | EMD Millipore |
| LDHA | #2012 | Cell Signaling Technology |
| LDHB | GTX101747 | GeneTex |
| β-actin | GTX110564 | GeneTex |
| COX IV | NB110-39115 | Novus biologicals |
| MS604 | ab110413 | Abcam |
| HSP60 | 611562 | BD Biosciences |
| Gene | Sequence (5′→3′) | |
|---|---|---|
| SCL16A1 | Forward | TGGATGGAGAGGAAGCTTTCTAAT |
| Reverse | CACACCAGATTTTCCAGCTTTC | |
| SCL16A3 | Forward | GAGTTTGGGATCGGCTACAG |
| Reverse | CGGTTCACGCACACACTG | |
| ACTB | Forward | CCAACCGCGAGAAGATGA |
| Reverse | CCAGAGGCGTACAGGGATAG | |
| LDHA | Forward | AGCCCGATTCCGTTACC |
| Reverse | CACCAGCAACATTCATTCCA | |
| LDHB | Forward | CTAGATTTCGCTACCTTAT |
| Reverse | TCATTGTCAGTTCCCATT | |
| PDHA1 | Forward | TGTGGAAGAACTAAAGGAAATTGATGT |
| Reverse | TTCCAAAGGTGGCTCAGGAT | |
| PDK1 | Forward | CCGCTCTCCATGAAGCAGTT |
| Reverse | TTGCCGCAGAAACATAAATGAG | |
| NDUFB8 | Forward | AGCCAGGTATTGACTGAATGTA |
| Reverse | CACAGCACTGAGTTTTATTAGGGA | |
| SDHB | Forward | GACACCAACCTCAATAAGGTCTC |
| Reverse | GGCTCAATGGATTTGTACTGTGC | |
| UQCRC2 | Forward | CAAAGTTGCCCCCAAACTTA |
| Reverse | AGCCATGTTTTCCCTTGTTG | |
| ATP5A | Forward | GGTCAGCCGTCTCAGTCCATT |
| Reverse | AACTAGCATCAACAGGTCCTC | |
| ND1 | L3441 | ACTACAACCCTTCGCTGACG |
| H3557 | AGAAGAGCGATGGTGAGAGC | |
| CYTB | L15260 | AGTCCCACCCTCACACGATTC |
| H15396 | TTATCGGAATGGGAGGTGATTC | |
| COI | L7075 | GAGGCTTCATTCACTGATTTCC |
| H7255 | TTTCATGTGGTGTATGCATCG | |
| ATP6 | L8903 | CCCACTTCTTACCACAAGGC |
| H9059 | GTGGCGCTTCCAATTAGGTG | |
| B2M | Forward | CCAGCAGAGAATGGAAAGTCAA |
| Reverse | CTCTCTCCATTCTTCAGTAAGTCAACT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.-Y.; Hsu, L.-H.; Chen, C.-Y.; Chang, G.-C.; Chang, H.-W.; Hung, Y.-M.; Liu, K.-J.; Kao, S.-H. Inhibition of Alternative Cancer Cell Metabolism of EGFR Mutated Non-Small Cell Lung Cancer Serves as a Potential Therapeutic Strategy. Cancers 2020, 12, 181. https://doi.org/10.3390/cancers12010181
Huang C-Y, Hsu L-H, Chen C-Y, Chang G-C, Chang H-W, Hung Y-M, Liu K-J, Kao S-H. Inhibition of Alternative Cancer Cell Metabolism of EGFR Mutated Non-Small Cell Lung Cancer Serves as a Potential Therapeutic Strategy. Cancers. 2020; 12(1):181. https://doi.org/10.3390/cancers12010181
Chicago/Turabian StyleHuang, Chung-Yu, Li-Han Hsu, Chung-Yeh Chen, Gee-Chen Chang, Hui-Wen Chang, Yi-Mei Hung, Ko-Jiunn Liu, and Shu-Huei Kao. 2020. "Inhibition of Alternative Cancer Cell Metabolism of EGFR Mutated Non-Small Cell Lung Cancer Serves as a Potential Therapeutic Strategy" Cancers 12, no. 1: 181. https://doi.org/10.3390/cancers12010181
APA StyleHuang, C.-Y., Hsu, L.-H., Chen, C.-Y., Chang, G.-C., Chang, H.-W., Hung, Y.-M., Liu, K.-J., & Kao, S.-H. (2020). Inhibition of Alternative Cancer Cell Metabolism of EGFR Mutated Non-Small Cell Lung Cancer Serves as a Potential Therapeutic Strategy. Cancers, 12(1), 181. https://doi.org/10.3390/cancers12010181