Immunotherapy Associated Pulmonary Toxicity: Biology Behind Clinical and Radiological Features

 , ,
, , 


Abstract
1. Introduction
2. Immune Checkpoint Blockade: Biological Bases for its Use in Cancer Immunotherapy
3. Immune Related Adverse Events in Lung Due to Immune Checkpoint Blockade
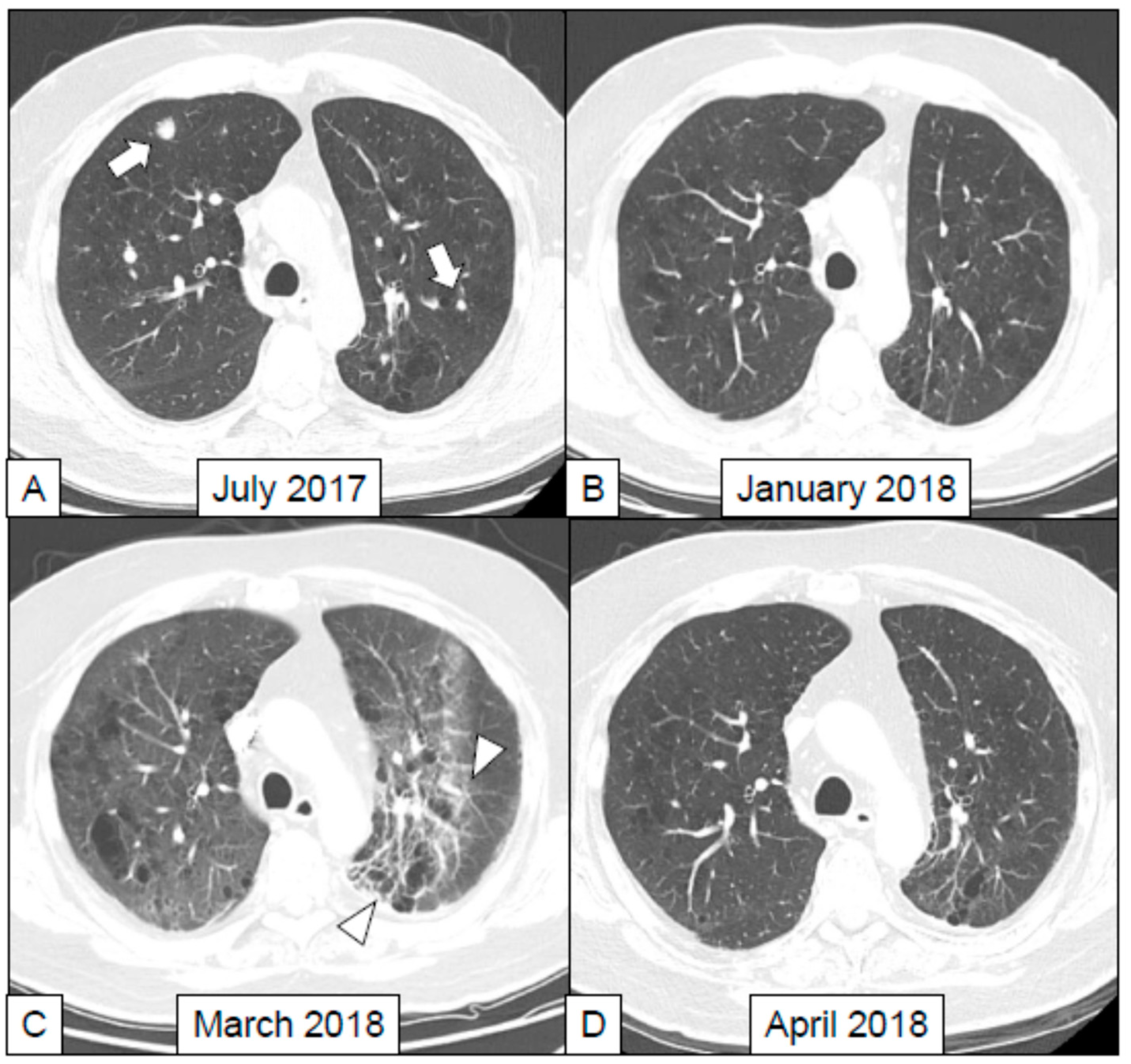
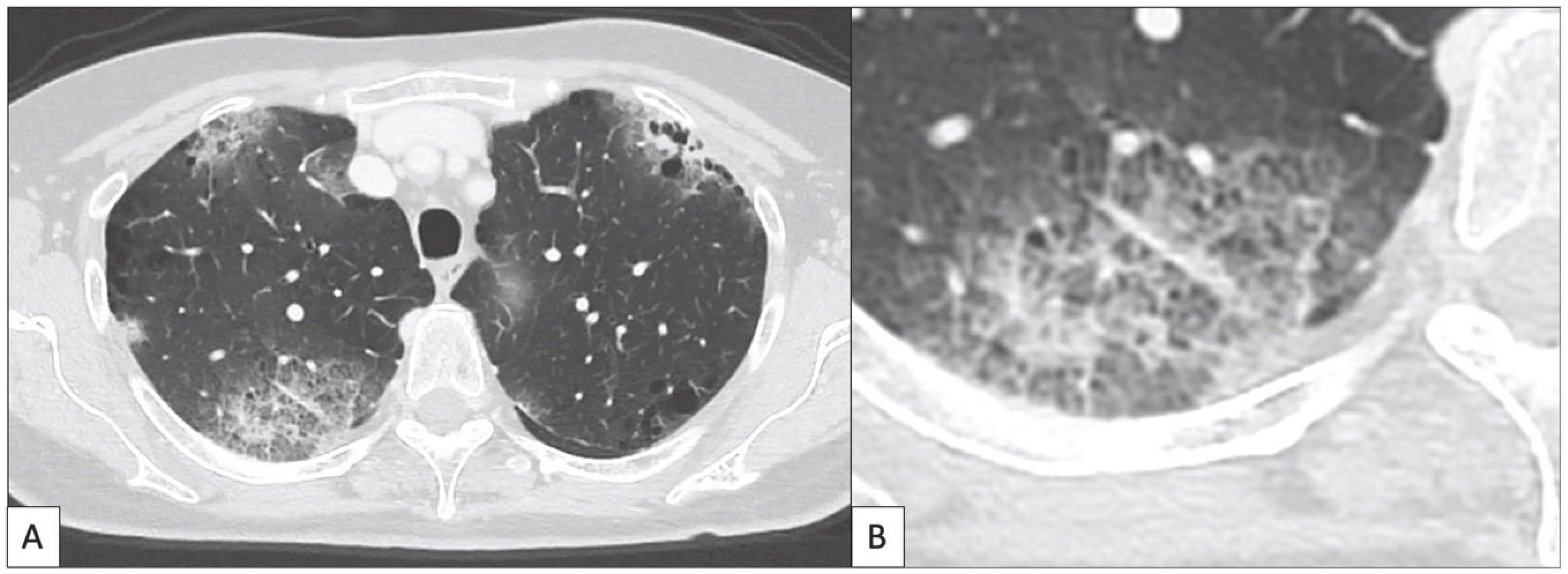
4. Immune Related Adverse Events in Lung: Findings at Imaging
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? Bmc Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.J.; Tirumani, S.H.; Van den Abbeele, A.D.; Koo, P.J.; Jacene, H.A. Cancer immunotherapy: Imaging assessment of novel treatment response patterns and immune-related adverse events. Radiographics 2015, 35, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Mechanism of action of immunotherapy. Semin. Oncol. 2014, 41 (Suppl. 5), S3–S13. [Google Scholar] [CrossRef]
- Morse, M.A.; Mosca, P.J.; Clay, T.M.; Lyerly, H.K. Dendritic cell maturation in active immunotherapy strategies. Expert Opin. Biol. Ther. 2002, 2, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kokate, R. A systematic overview of cancer immunotherapy: an emerging therapy. Pharm. Pharmacol. Int. J. 2017, 5, 31–35. [Google Scholar] [CrossRef]
- Hwang, W.L.; Pike, L.R.G.; Royce, T.J.; Mahal, B.A.; Loeffler, J.S. Safety of combining radiotherapy with immune-checkpoint inhibition. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Braschi-Amirfarzan, M.; Tirumani, S.H.; Hodi, F.S., Jr.; Nishino, M. Immune-Checkpoint Inhibitors in the Era of Precision Medicine: What Radiologists Should Know. Korean J. Radiol. 2017, 18, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. KEYNOTE-024 Investigators. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Napolitano, S.; Brancaccio, G.; Argenziano, G.; Martinelli, E.; Morgillo, F.; Ciardiello, F.; Troiani, T. It is finally time for adjuvant therapy in melanoma. Cancer Treat. Rev. 2018, 69, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. KEYNOTE-407 Investigators. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. KEYNOTE-189 Investigators. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C. The benefits of immunotherapy combinations. Nature 2017, 552, S67–S69. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Crittenden, M.; Wargo, J. Combination Immunotherapy Development in Melanoma. In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology Annual Meeting Faculty: Chicago, IL, USA, 2018; pp. 197–207. [Google Scholar]
- Brahmer, J.R.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Health-related quality-of-life results for pembrolizumab versus chemotherapy in advanced, PD-L1-positive NSCLC (KEYNOTE-024): A multicentre, international, randomised, open-label phase 3 trial. Lancet Oncol. 2017, 18, 1600–1609. [Google Scholar] [CrossRef]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K.; ESMO Guidelines Committee. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv264–iv266. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chaudhary, N.; Garg, M.; Floudas, C.S.; Soni, P.; Chandra, A.B. Current Diagnosis and Management of Immune Related Adverse Events (irAEs) Induced by Immune Checkpoint Inhibitor Therapy. Front. Pharmacol. 2017, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Sosa, A.; Lopez Cadena, E.; Simon Olive, C.; Karachaliou, N.; Rosell, R. Clinical assessment of immune-related adverse events. Ther. Adv. Med Oncol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.; Lee, R.; Bowyer, S.; Fusi, A.; Lorigan, P. Optimal management of immune-related toxicities associated with checkpoint inhibitors in lung cancer. Lung Cancer 2015, 88, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Golstein, P. Lymphocyte activation: T-cell regulation by CTLA-4. Curr. Biol. CB 1996, 6, 398–400. [Google Scholar] [CrossRef]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Nguyen, H.; Chambers, C.; Kang, J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl. Acad. Sci. USA 2010, 107, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.S. Treg and CTLA-4: Two intertwining pathways to immune tolerance. J. Autoimmun. 2013, 45, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A. Clinical development of the anti-CTLA-4 antibody tremelimumab. Semin. Oncol. 2010, 37, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Tian, J.; Liu, H.; Li, F.; Niu, H.; Zhu, B. Efficacy and safety of anti-PD-1 and anti-PD-1 combined with anti-CTLA-4 immunotherapy to advanced melanoma: A systematic review and meta-analysis of randomized controlled trials. Medicine 2017, 96, e7325. [Google Scholar] [CrossRef] [PubMed]
- Guazzelli, A.; Bakker, E.; Krstic-Demonacos, M.; Lisanti, M.P.; Sotgia, F.; Mutti, L. Anti-CTLA-4 therapy for malignant mesothelioma. Immunotherapy 2017, 9, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Flaherty, K.; Goff, S. Emerging Strategies in Systemic Therapy for the Treatment of Melanoma. In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology Annual Meeting Faculty: Chicago, IL, USA, 2018; pp. 751–758. [Google Scholar]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Podojil, J.R.; Miller, S.D. Molecular mechanisms of T-cell receptor and costimulatory molecule ligation/blockade in autoimmune disease therapy. Immunol. Rev. 2009, 229, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A.; Chatterjee, P.; Li, L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014, 20, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.S.; Chew, V.; Sieow, J.L.; Goh, S.; Yeong, J.P.; Soon, A.L.; Ricciardi-Castagnoli, P. PD-1 expression on dendritic cells suppresses CD8(+) T cell function and antitumor immunity. Oncoimmunology 2015, 5, e1085146. [Google Scholar] [CrossRef] [PubMed]
- Kinter, A.L.; Godbout, E.J.; McNally, J.P.; Sereti, I.; Roby, G.A.; O’Shea, M.A.; Fauci, A.S. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J. Immunol. 2008, 181, 6738–6746. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Lee, S.W.; Seo, S.K.; Choi, I.W.; Choi, I.; Lee, S.W. Interferon-sensitive response element (ISRE) is mainly responsible for IFN-alpha-induced upregulation of programmed death-1 (PD-1) in macrophages. Biochim. Et Biophys. Acta 2008, 1779, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Wang, S.; Zhu, Y.; Luo, L.; Zhu, G.; Flies, S.; Xu, H.; Ruff, W.; Broadwater, M.; Choi, I.H.; et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood 2009, 113, 5811–5818. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Arasanz, H.; Gato-Cañas, M.; Zuazo, M.; Ibañez-Vea, M.; Breckpot, K.; Kochan, G.; Escors, D. PD1 signal transduction pathways in T cells. Oncotarget 2017, 8, 51936–51945. [Google Scholar] [CrossRef] [PubMed]
- Abdin, S.M.; Zaher, D.M.; Arafa, E.A.; Omar, H.A. Tackling Cancer Resistance by Immunotherapy: Updated Clinical Impact and Safety of PD-1/PD-L1 Inhibitors. Cancers (Basel) 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Zheng, P. Tumor cells versus host immune cells: Whose PD-L1 contributes to PD-1/PD-L1 blockade mediated cancer immunotherapy? Cell Biosci. 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Kowanetz, M.; Zou, W.; Gettinger, S.N.; Koeppen, H.; Kockx, M.; Schmid, P.; Kadel, E.E., 3rd; Wistuba, I.; Chaft, J.; Rizvi, N.A.; et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti-PD-L1). Proc. Natl. Acad. Sci. USA 2018, 115, E10119–E10126. [Google Scholar] [CrossRef] [PubMed]
- Champiat, S.; Lambotte, O.; Barreau, E.; Belkhir, R.; Berdelou, A.; Carbonnel, F.; Cauquil, C.; Chanson, P.; Collins, M.; Durrbach, A.; et al. Management of immune checkpoint blockade dysimmune toxicities: A collaborative position paper. Ann. Oncol. 2016, 27, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Rozali, E.N.; Hato, S.V.; Robinson, B.W.; Lake, R.A.; Lesterhuis, W.J. Programmed death ligand 2 in cancer-induced immune suppression. Clin. Dev. Immunol. 2012, 2012, 656340. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Yao, H.; Li, C.; Fang, J.Y.; Xu, J. Regulation of PD-L1: Emerging Routes for Targeting Tumor Immune Evasion. Front. Pharmacol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Masugi, Y.; Nishihara, R.; Hamada, T.; Song, M.; da Silva, A.; Kosumi, K.; Gu, M.; Shi, Y.; Li, W.; Liu, L.; et al. Tumor PDCD1LG2 (PD-L2) Expression and the Lymphocytic Reaction to Colorectal Cancer. Cancer Immunol. Res. 2017, 5, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Eigentler, T.K.; Hassel, J.C.; Berking, C.; Aberle, J.; Bachmann, O.; Grünwald, V.; Kähler, K.C.; Loquai, C.; Reinmuth, N.; Steins, M.; et al. Diagnosis, monitoring and management of immune-related adverse drug reactions of anti-PD-1 antibody therapy. Cancer Treat. Rev. 2016, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Wang, X.; Woo, K.M.; Iyriboz, T.; Halpenny, D.; Cunningham, J.; Chaft, J.E.; Segal, N.H.; Callahan, M.K.; Lesokhin, A.M.; et al. Pneumonitis in Patients Treated With Anti-Programmed Death-1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. 2017, 35, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Hassel, J.C.; Heinzerling, L.; Aberle, J.; Bähr, O.; Eigentler, T.K.; Grimm, M.O.; Grünwald, V.; Leipe, J.; Reinmuth, N.; Tietze, J.K.; et al. Combined immune checkpoint blockade (anti-PD-1/anti-CTLA-4): Evaluation and management of adverse drug reactions. Cancer Treat. Rev. 2017, 57, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Common Terminology Criteria for Adverse Events (CTCAE). Available online: https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_50 (accessed on 1 July 2018).
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef] [PubMed]
- Possick, J.D. Pulmonary Toxicities from Checkpoint Immunotherapy for Malignancy. Clin. Chest. Med. 2017, 38, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Sholl, L.M.; Hodi, F.S. Anti–PD-1–Related Pneumonitis during Cancer Immunotherapy. N. Engl. J. Med. 2015, 373, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Barjaktarevic, I.Z.; Qadir, N.; Suri, A.; Santamauro, J.T.; Stover, D. Organizing pneumonia as a side effect of ipilimumab treatment of melanoma. Chest 2013, 143, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Kanai, O.; Nakatani, K.; Fujita, K.; Okamura, M.; Mio, T. Concurrence of nivolumab-induced interstitial lung disease and cancer invasion. Respirol. Case Rep. 2017, 5, e00257. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.; Velcheti, V.; Mukhopadhyay, S.; Stoller, J.K. Focal lung infiltrate complicating PD-1 inhibitor use: A new pattern of drug-associated lung toxicity? Respir. Med. Case Rep. 2016, 19, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Grenier, P.; Valeyre, D.; Cluzel, P.; Brauner, M.W.; Lenoir, S.; Chastang, C. Chronic diffuse interstitial lung disease: Diagnostic value of chest radiography and high-resolution CT. Radiology 1991, 179, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Eguchi, K.; Ohmatsu, H.; Kakinuma, R.; Naruke, T.; Suemasu, K.; Moriyama, N. Peripheral lung cancer: Screening and detection with low-dose spiral CT versus radiography. Radiology 1996, 201, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King TEJr Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Ramaiya, N.H.; Awad, M.M.; Sholl, L.M.; Maattala, J.A.; Taibi, M.; Hatabu, H.; Ott, P.A.; Armand, P.F.; Hodi, F.S. PD-1 Inhibitor-Related Pneumonitis in Advanced Cancer Patients: Radiographic Patterns and Clinical Course. Clin. Cancer Res. 2016, 22, 6051–6060. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Mang, C.; Grosse, C.; Schmid, K.; Stiebellehner, L.; Bankier, A.A. What every radiologist should know about idiopathic interstitial pneumonias. Radiographics 2007, 27, 595–615. [Google Scholar] [CrossRef] [PubMed]
- Palmucci, S.; Roccasalva, F.; Puglisi, S.; Torrisi, S.E.; Vindigni, V.; Mauro, L.A.; Ettorre, G.C.; Piccoli, M.; Vancheri, C. Clinical and radiological features of idiopathic interstitial pneumonias (IIPs): A pictorial review. Insights Imaging 2014, 5, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Johkoh, T.; Fukuoka, J.; Tanaka, T. Rare idiopathic intestinal pneumonias (IIPs) and histologic patterns in new ATS/ERS multidisciplinary classification of the IIPs. Eur. J. Radiol. 2015, 84, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, M.A.; Cham, M.D.; Beasley, M.B.; Ward, T.J.; Jacobi, A.H.; Eber, C.D.; Padilla, M.L. Idiopathic interstitial pneumonias: A radiology-pathology correlation based on the revised 2013 American Thoracic Society-European Respiratory Society classification system. Curr. Probl. Diagn. Radiol. 2015, 44, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Berthod, G.; Lazor, R.; Letovanec, I.; Romano, E.; Noirez, L.; Mazza Stalder, J.; Speiser, D.E.; Peters, S.; Michielin, O. Pulmonary sarcoid-like granulomatosis induced by ipilimumab. J. Clin. Oncol. 2012, 30, e156–e159. [Google Scholar] [CrossRef] [PubMed]
- Dimitriou, F.; Frauchiger, A.L.; Urosevic-Maiwald, M.; Naegeli, M.C.; Goldinger, S.M.; Barysch, M.; Franzen, D.; Kamarachev, J.; Braun, R.; Dummer, R.; et al. Sarcoid-like reactions in patients receiving modern melanoma treatment. Melanoma Res. 2018, 28, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Firwana, B.; Ravilla, R.; Raval, M.; Hutchins, L.; Mahmoud, F. Sarcoidosis-like syndrome and lymphadenopathy due to checkpoint inhibitors. J. Oncol. Pharm. Pract. 2017, 23, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Depeursinge, A.; Chin, A.S.; Leung, A.N.; Terrone, D.; Bristow, M.; Rosen, G.; Rubin, D.L. Automated classification of usual interstitial pneumonia using regional volumetric texture analysis in high-resolution computed tomography. Investig. Radiol. 2015, 50, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Jung, K.H.; Lee, Y.; Kim, H.J.; Kim, N.; Jun, S.; Seo, J.B.; Lynch, D.A. Comparison of Shallow and Deep Learning Methods on Classifying the Regional Pattern of Diffuse Lung Disease. J. Digit. Imaging. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Christodoulidis, S.; Anthimopoulos, M.; Ebner, L.; Christe, A.; Mougiakakou, S. Multisource Transfer Learning With Convolutional Neural Networks for Lung Pattern Analysis. IEEE J. Biomed. Health Inform. 2017, 21, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Anthimopoulos, M.; Christodoulidis, S.; Ebner, L.; Christe, A.; Mougiakakou, S. Lung Pattern Classification for Interstitial Lung Diseases Using a Deep Convolutional Neural Network. IEEE Trans. Med. Imaging 2016, 35, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Than, J.C.M.; Saba, L.; Noor, N.M.; Rijal, O.M.; Kassim, R.M.; Yunus, A.; Suri, H.S.; Porcu, M.; Suri, J.S. Lung disease stratification using amalgamation of Riesz and Gabor transforms in machine learning framework. Comput. Biol. Med. 2017, 89, 197–211. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Immune Checkpoint Blockade | |
|---|---|
| European Medicine Agency | Food and Drug Administration |
| Ipilimumab (anti-CTLA-4) | |
| Melanoma Unresectable or metastatic disease in adults and adolescents (12 years and older) Unresectable or metastatic disease in adults in combination with nivolumab | Melanoma Unresectable or metastatic disease in adults and pediatric (12 years and older) patients Adjuvant treatment of patients with involvement of regional LN (>1 mm) after complete resection, including total lymphadenectomy |
| RCC 1st line treatment of adult patients with intermediate or poor risk disease in combination with nivolumab | RCC Intermediate or poor risk, previously untreated patients, in combination with nivolumab |
| MSI-H or dMMR CRC Metastatic patients (adult and pediatric 12 years and older patients) that has progressed after fluoropyrimidine, oxaliplatin, and irinotecan, in combination with nivolumab | |
| Pembrolizumab (anti-PD-1) | |
| Melanoma Unresectable or metastatic disease in adults Adjuvant treatment of adults with stage III disease and LN involvement who have undergone complete resection | Melanoma Unresectable or metastatic disease |
| NSCLC 1st line treatment of metastatic adult patients whose tumors express PD-L1 with a ≥ 50% TPS with no EGFR or ALK mutations In combination with pemetrexed and platinum chemotherapy, for the 1st line treatment of metastatic non-squamous adult patients whose tumors have no EGFR or ALK mutations Locally advanced or metastatic adult patients whose tumors express PD-L1 with a ≥ 1% TPS and who have received at least one prior chemotherapy regimen. Patients with EGFR or ALK positive mutations should also have received targeted therapy | NSCLC In combination with pemetrexed and platinum chemotherapy, as 1st line treatment of patients with metastatic nonsquamous disease, with no EGFR or ALK mutations In combination with carboplatin and either paclitaxel or nabpaclitaxel, as 1st line treatment of patients with metastatic squamous disease As a single agent for the 1st line treatment of patients with metastatic disease whose tumors have high PD-L1 expression (TPS ≥50%), with no EGFR or ALK mutations As a single agent for the treatment of patients with metastatic disease whose tumors express PD-L1 (TPS ≥1%) with disease progression on or after platinum-containing chemotherapy; patients with EGFR or ALK mutations should have disease progression on FDA-approved therapy for these mutations |
| cHL Adult patients with relapsed or refractory disease who have failed ASCT and BV, or who are transplant-ineligible and have failed BV | HNSCC Current or metastatic disease with progression on or after platinum containing chemotherapy |
| UC Locally advanced or metastatic disease in adults who have received prior platinum-containing chemotherapy Locally advanced or metastatic UC in adults not eligible for cisplatin-containing chemotherapy and whose tumors express PD-L1 with a CPS ≥ 10 Recurrent or metastatic HNSCC in adults whose tumors express PD-L1 with a ≥ 50% TPS and progressing on or after platinum-containing chemotherapy | cHL Adult and pediatric patients with refractory disease, or who have relapsed after 3 or more prior lines of therapy |
| PMBCL Adult and pediatric patients with refractory disease, or who have relapsed after 2 or more prior lines of therapy | |
| UC Locally advanced or metastatic disease not eligible for cisplatin-containing chemotherapy and whose tumors express PD-L1 (CPS ≥10), or in patients who are not eligible for any platinum-containing chemotherapy regardless of PD-L1 status Locally advanced or metastatic UC who have disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum containing chemotherapy | |
| MSI-H cancer Adult and pediatric patients with unresectable or metastatic MSI-H or dMMR solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options MSI-H or dMMR CRC that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan | |
| Gastric or gastroesophageal junction adenocarcinoma Recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (CPS ≥1), with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy | |
| Cervical cancer Recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) | |
| HCC Previous treatment with sorafenib. | |
| MCC Adult and pediatric patients with recurrent locally advanced or metastatic disease | |
| NSCLC 1st line treatment of metastatic NSCLC in adults whose tumors express PD-L1 with a ≥ 50% TPS with no EGFR or ALK mutations In combination with pemetrexed and platinum chemotherapy, for the 1st line treatment of metastatic non-squamous disease in adults whose tumors have no EGFR or ALK mutations Locally advanced or metastatic disease in adults whose tumors express PD-L1 with a ≥ 1% TPS and who have received at least one prior chemotherapy regimen. Patients with EGFR or ALK positive mutations should also have received targeted therapy | NSCLC In combination with pemetrexed and platinum chemotherapy, as 1st line treatment of patients with metastatic nonsquamous NSCLC, with no EGFR or ALK mutations In combination with carboplatin and either paclitaxel or nabpaclitaxel, as 1st line treatment of patients with metastatic squamous NSCLC As a single agent for the 1st line treatment of patients with metastatic NSCLC whose tumors have high PD-L1 expression (TPS ≥50%), with no EGFR or ALK mutations As a single agent for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) with disease progression on or after platinum-containing chemotherapy; patients with EGFR or ALK mutations should have disease progression on FDA-approved therapy for these mutations |
| cHL Adult patients with relapsed or refractory disease who have failed ASCT and BV, or who are transplant-ineligible and have failed BV | HNSCC Current or metastatic disease with progression on or after platinum containing chemotherapy |
| UC Locally advanced or metastatic disease in adults who have received prior platinum-containing chemotherapy Locally advanced or metastatic disease in adults not eligible for cisplatin-containing chemotherapy and whose tumors express PD-L1 with a CPS ≥ 10 | cHL Adult and pediatric patients with refractory disease, or who have relapsed after 3 or more prior lines of therapy |
| PMBCL Adult and pediatric patients with refractory disease, or who have relapsed after 2 or more prior lines of therapy | |
| UC Locally advanced or metastatic patients not eligible for cisplatin-containing chemotherapy and whose tumors express PD-L1 (CPS ≥10), or not eligible for any platinum-containing chemotherapy regardless of PD-L1 status Locally advanced or metastatic disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum containing chemotherapy | |
| MSI-H cancer Adult and pediatric patients with unresectable or metastatic MSI-H or dMMR solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options MSI-H or dMMR CRC that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan | |
| HNSCC Recurrent or metastatic HNSCC in adults whose tumors express PD-L1 with a ≥ 50% TPS and progressing on or after platinum-containing chemotherapy | Gastric or gastroesophageal junction adenocarcinoma Recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (CPS ≥1), with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy |
| Cervical cancer Recurrent or metastatic disease with progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) | |
| HCC Previous treatment with sorafenib. | |
| MCC Adult and pediatric patients with recurrent locally advanced or metastatic disease | |
| Nivolumab (anti-PD-1) | |
| Melanoma Advanced or metastatic disease in adults, alone or in combination with ipilimumab Adjuvant treatment of adults with involvement of LN or metastatic disease who have undergone complete resection | Melanoma Unresectable or metastatic disease as single agent or in combination with ipilimumab Adjuvant treatment of melanoma with LN involvement or metastatic disease who have undergone complete resection |
| NSCLC Locally advanced or metastatic disease after prior chemotherapy in adults | NSCLC Metastatic disease with progression on or after platinum-based chemotherapy; patients with EGFR or ALK mutations should have disease progression on FDA-approved therapy for these mutations |
| SCLC Metastatic disease with progression after platinum-based chemotherapy and at least one other line of therapy | |
| RCC After prior therapy in adult patients in combination with ipilimumab for the 1st line treatment in adults with in intermediate or poor risk advanced disease | RCC Advanced RCC who have received prior antiangiogenic therapy Intermediate or poor risk, previously untreated advanced RCC, in combination with ipilimumab |
| cHL Relapsed or refractory disease after ASCT and treatment with BV | cHL Adult patients that relapsed or progressed after HSCT and BV, or after 3 or more lines of systemic therapy that includes autologous HSCT |
| SCCHN Recurrent or metastatic disease progressing after platinum-based treatment | SCCHN Recurrent or metastatic disease with progression on or after a platinum-based therapy |
| UC Locally advanced unresectable or metastatic disease after failure of platinum-based treatment | UC Locally advanced or metastatic disease who have progression during or following platinum-containing chemotherapy, or have progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy |
| MSI-H or dMMR CRC Adult and pediatric (12 years and older) patients with MSI-H or dMMR metastatic CRC progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan, as a single agent or in combination with ipilimumab | |
| HCC Previous treatment with sorafenib | |
| Atezolizumab (anti-PD-L1) | |
| NSCLC Locally advanced or metastatic disease in adults previously treated with chemotherapy. Patients with EGFR or ALK mutations targeted treatments should also have received targeted therapy | UC Locally advanced or metastatic disease not eligible for cisplatin-containing chemotherapy and whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells covering ≥ 5% of the tumor area), or Locally advanced or metastatic disease not eligible for any platinum—containing chemotherapy regardless of PD-L1 status, or Locally advanced or metastatic disease that have disease progression during or following any platinum-containing chemotherapy, or within 12 months of neoadjuvant or adjuvant chemotherapy |
| UC Locally advanced or metastatic disease after platinum chemotherapy Locally advanced or metastatic disease ineligible for treatment with cisplatin and whose tumours have a PD-L1 expression ≥ 5% | NSCLC In combination with bevacizumab, paclitaxel, and carboplatin, for the 1st line treatment, of patients with metastatic non-squamous NSCLC with no EGFR or ALK genomic tumor aberrations metastatic disease progressing during or following platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations |
| Durvalumab (anti-PD-L1) | |
| NSCLC Locally advanced, unresectable disease in adults, whose tumors express PD-L1 on ≥ 1% of tumor cells and whose disease has not progressed following platinum-based chemoradiation therapy | UC Locally advanced or metastatic disease progressng during or following platinum-containing chemotherapy Locally advanced or metastatic disease progressing within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy |
| NSCLC Unresectable, stage III disease not progressing following concurrent platinum-based chemotherapy and radiation therapy | |
| Avelumab (anti-PD-L1) | |
| MCC Adult patients with metastatic disease | MCC Adult and pediatric (12 years and older) patients with metastatic disease |
| UC Locally advanced or metastatic disease progressing during or following platinum-containing chemotherapy Locally advanced or metastatic disease progressing within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy | |
| Cemiplimab-rwlc (anti-PD-1) | |
| Not approved | CSCC Patients with metastatic or locally advanced disease who are not candidates for curative surgery or curative radiation |
| Common Terminology Criteria for Adverse Events (CTCAE) Grading System | ||||
|---|---|---|---|---|
| Grade | General Criteria | Criteria for Pneumonitis | Criteria for Pulmunary Fibrosis | |
| 1 | Mild | Asymptomatic or mild symptoms that do not require intervention | Asymptomatic; clinical or diagnostic observations only; intervention not indicated | Radiologic pulmonary fibrosis <25% of lung volume associated with hypoxia |
| 2 | Moderate | It requires minimal, local or non invasive intervention | Symptomatic; medical intervention indicated; limiting instrumental activity of daily living (ADL) | Evidence of pulmonary hypertension; radiographic pulmonary fibrosis 25–50% associated with hypoxia |
| 3 | Severe or medically significant but not immediately life-threatening | It requires hospitalization or prolongation of hospitalization | Severe symptoms; limiting self care activity of daily living (ADL); oxygen indicated | Severe hypoxia; evidence of right-sided heart failure; radiographic pulmonary fibrosis > 50–75% |
| 4 | Life-threatening consequences | It requires urgent intervention | Life-threatening respiratory compromise; urgent intervention indicated (i.e., tracheotomy or intubation) | Life-threatening consequences (i.e., hemodynamic/pulmonary complications); intubation with ventilatory support indicated; radiographic pulmonary fibrosis >75% with severe honeycombing |
| 5 | Death | Death related to adverse event (AE) | Death | Death |
| Suggested Criteria for Diagnosis of ir-Pneumonitis | |
|---|---|
| Clinical criteria | History of immune checkpoint blockade (ICB) treatment |
| Symptoms and/or radiological evidence of pneumonitis | |
| Resistance to antibiotic treatment and absence of microrganisms in the bronchoalveolar lavage and sputum | |
| Exclusion of other possible etiologies | |
| Radiological criteria | Computed tomography (CT) findings of interstitial pneumonia, particularly in presence of: |
| cryptogenic organizing pneumonia (COP) -like pattern | |
| ground glass opacities (GGO) | |
| “sarcoid-like” pattern | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porcu, M.; De Silva, P.; Solinas, C.; Battaglia, A.; Schena, M.; Scartozzi, M.; Bron, D.; Suri, J.S.; Willard-Gallo, K.; Sangiolo, D.; et al. Immunotherapy Associated Pulmonary Toxicity: Biology Behind Clinical and Radiological Features. Cancers 2019, 11, 305. https://doi.org/10.3390/cancers11030305
Porcu M, De Silva P, Solinas C, Battaglia A, Schena M, Scartozzi M, Bron D, Suri JS, Willard-Gallo K, Sangiolo D, et al. Immunotherapy Associated Pulmonary Toxicity: Biology Behind Clinical and Radiological Features. Cancers. 2019; 11(3):305. https://doi.org/10.3390/cancers11030305
Chicago/Turabian StylePorcu, Michele, Pushpamali De Silva, Cinzia Solinas, Angelo Battaglia, Marina Schena, Mario Scartozzi, Dominique Bron, Jasjit S. Suri, Karen Willard-Gallo, Dario Sangiolo, and et al. 2019. "Immunotherapy Associated Pulmonary Toxicity: Biology Behind Clinical and Radiological Features" Cancers 11, no. 3: 305. https://doi.org/10.3390/cancers11030305
APA StylePorcu, M., De Silva, P., Solinas, C., Battaglia, A., Schena, M., Scartozzi, M., Bron, D., Suri, J. S., Willard-Gallo, K., Sangiolo, D., & Saba, L. (2019). Immunotherapy Associated Pulmonary Toxicity: Biology Behind Clinical and Radiological Features. Cancers, 11(3), 305. https://doi.org/10.3390/cancers11030305