
Microfluidic Single-Cell Proteomics Assay Chip: Lung Cancer Cell Line Case Study

 ,
, 
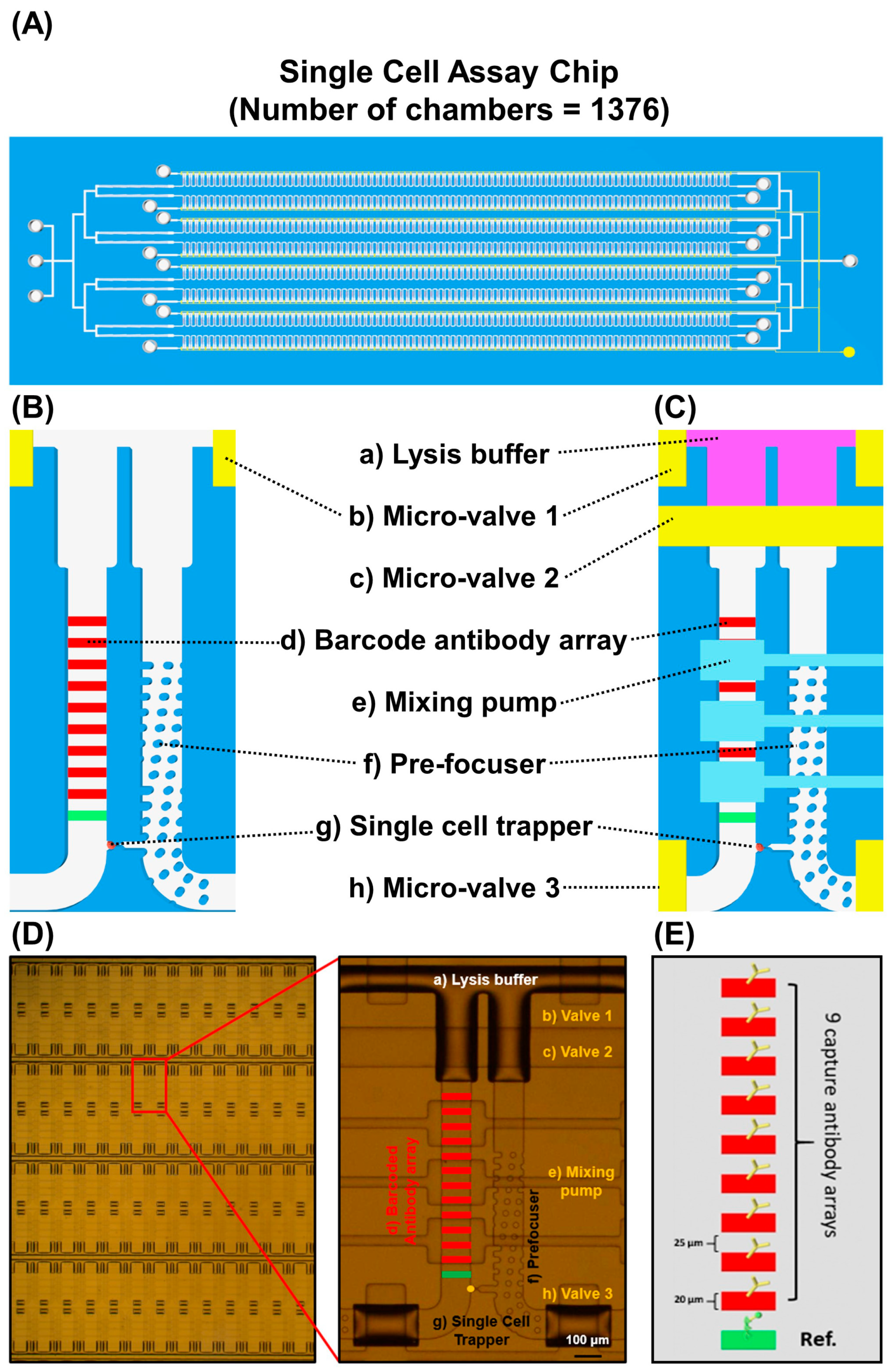{kind=link}
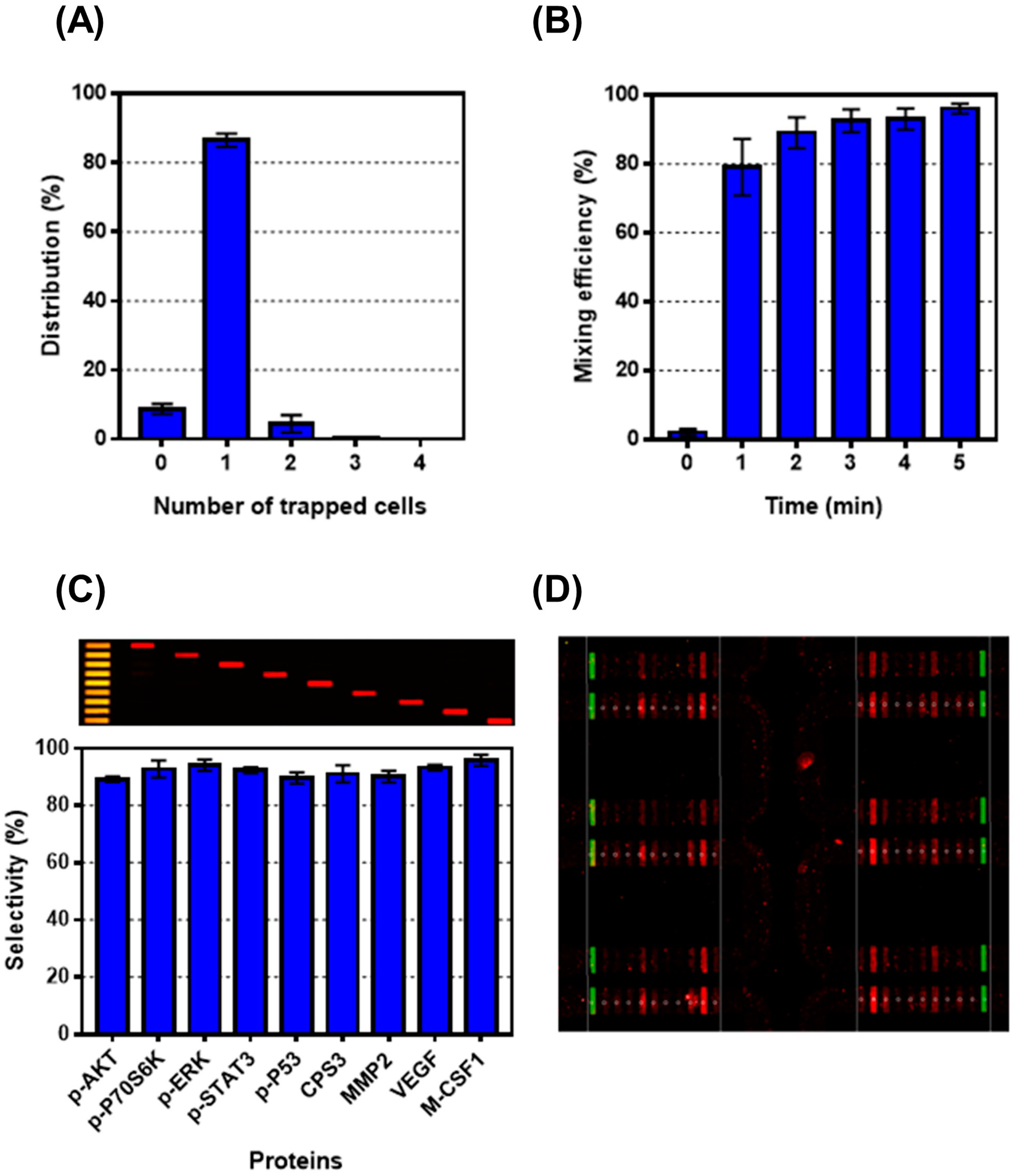{kind=link}
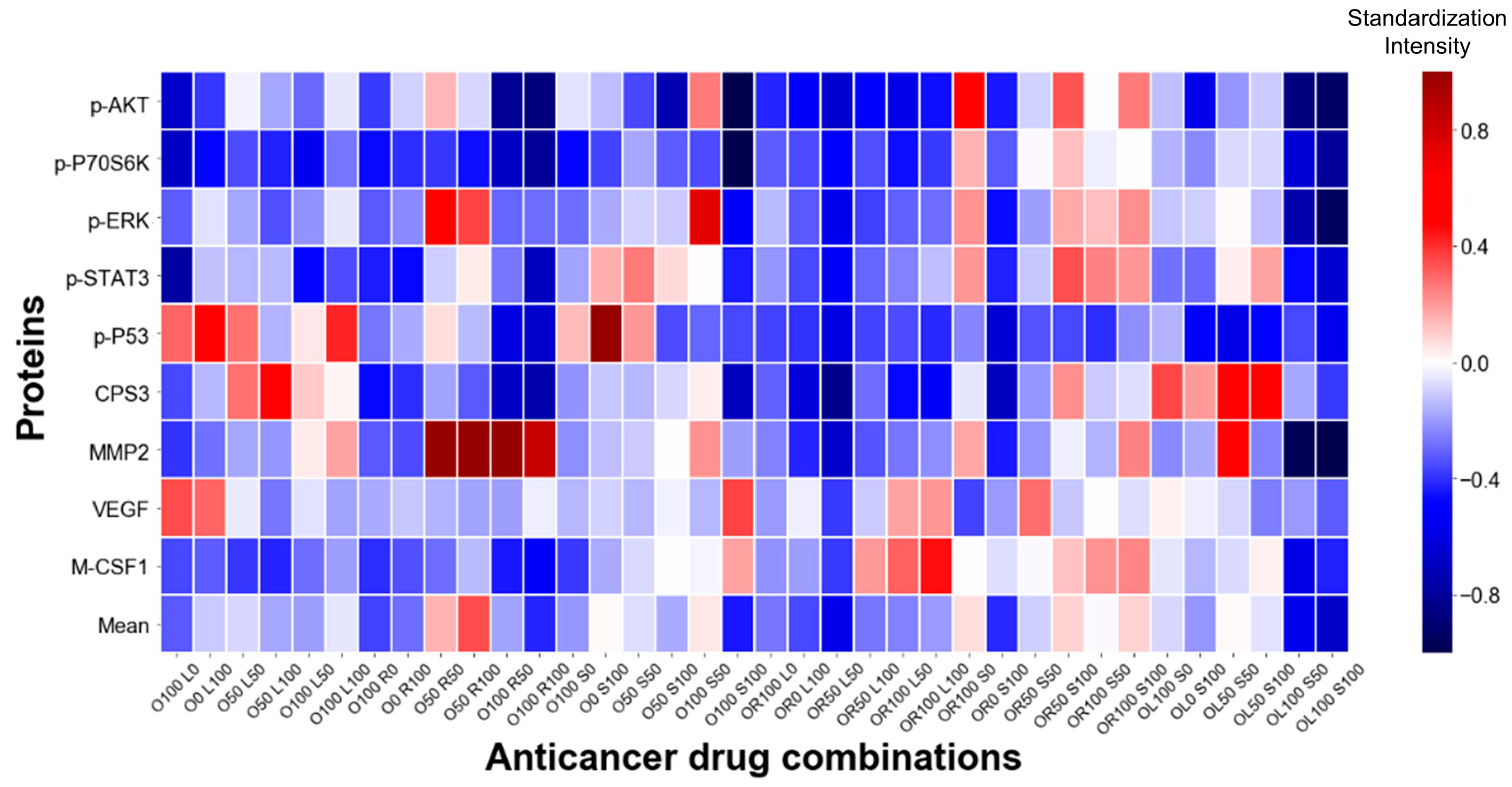{kind=link}
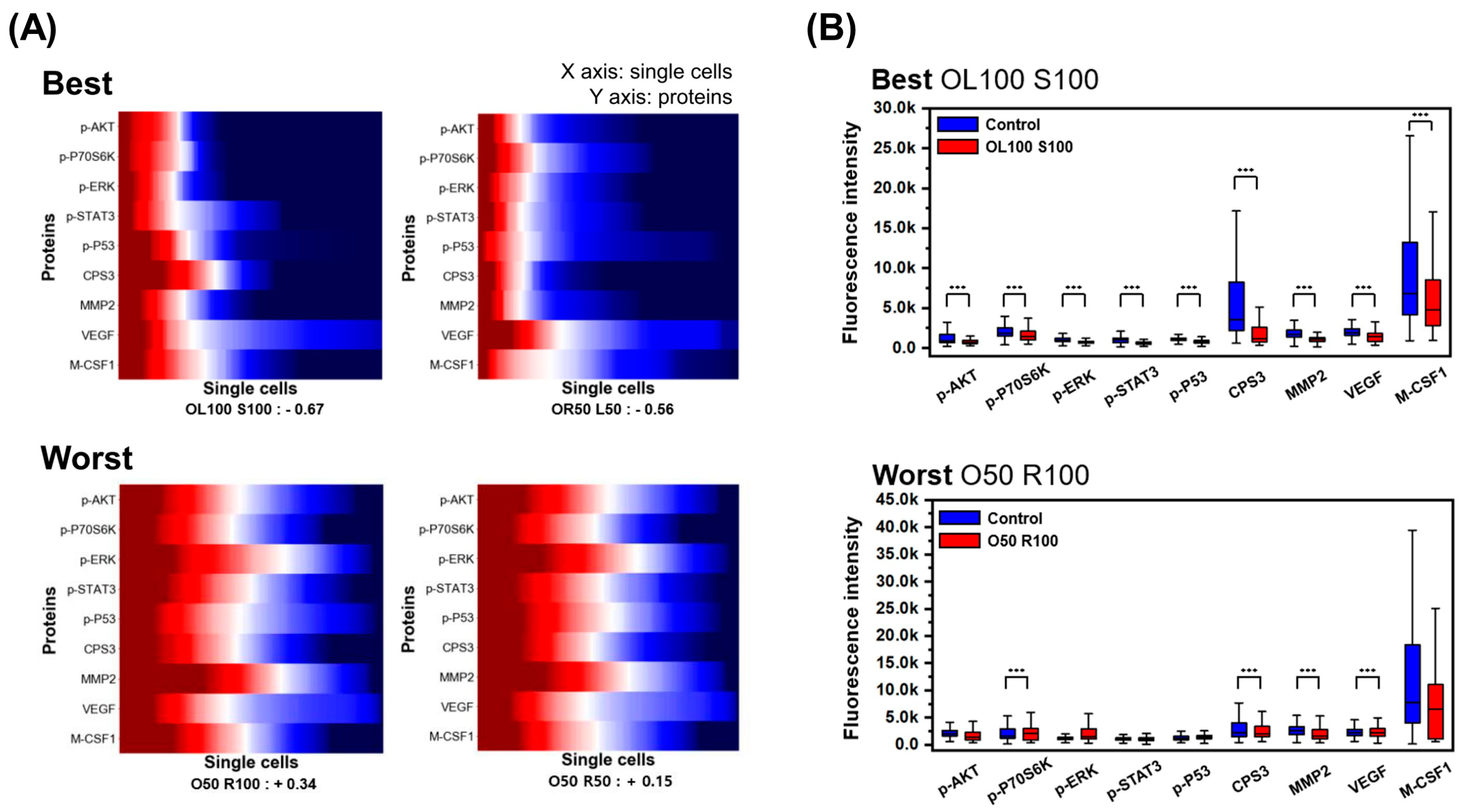{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Device Preparation
2.2. Preparation of the Multiplex Barcode Array
2.3. Cell Line and Reagents
2.4. MTT Assay and Drug IC50 Measurement
2.5. Single-Cell Assay
2.6. Single-Cell Data Analysis
2.7. Heterogeneity Evaluation and Analysis
2.8. ELISA Assay
3. Results
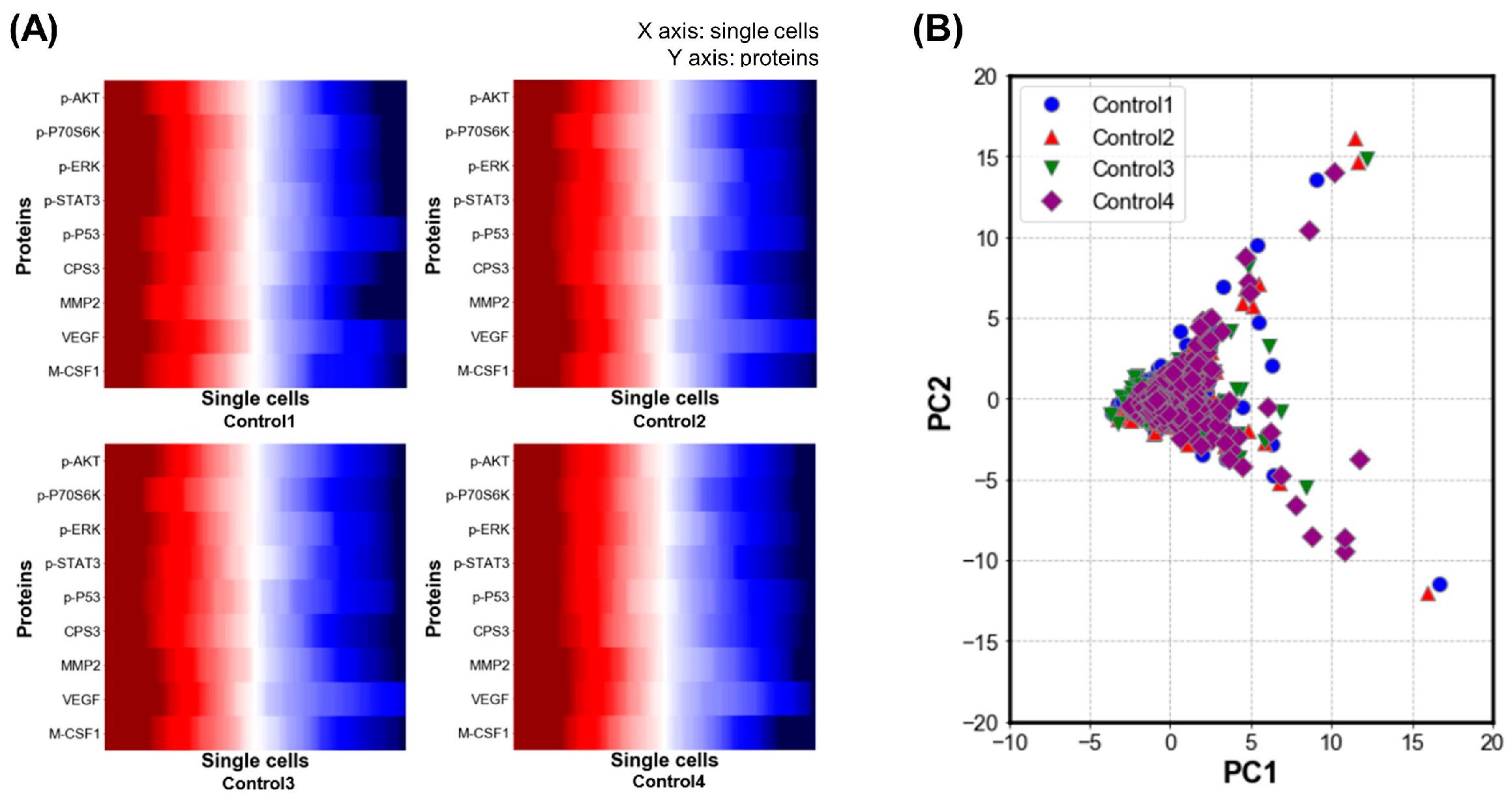
3.1. Validation of Single-Cell Proteomics Assay Chip Using H1650 Lung-Cancer Cell Line
3.2. Single-Cell Analysis Using H1975 Lung-Cancer Cell Line
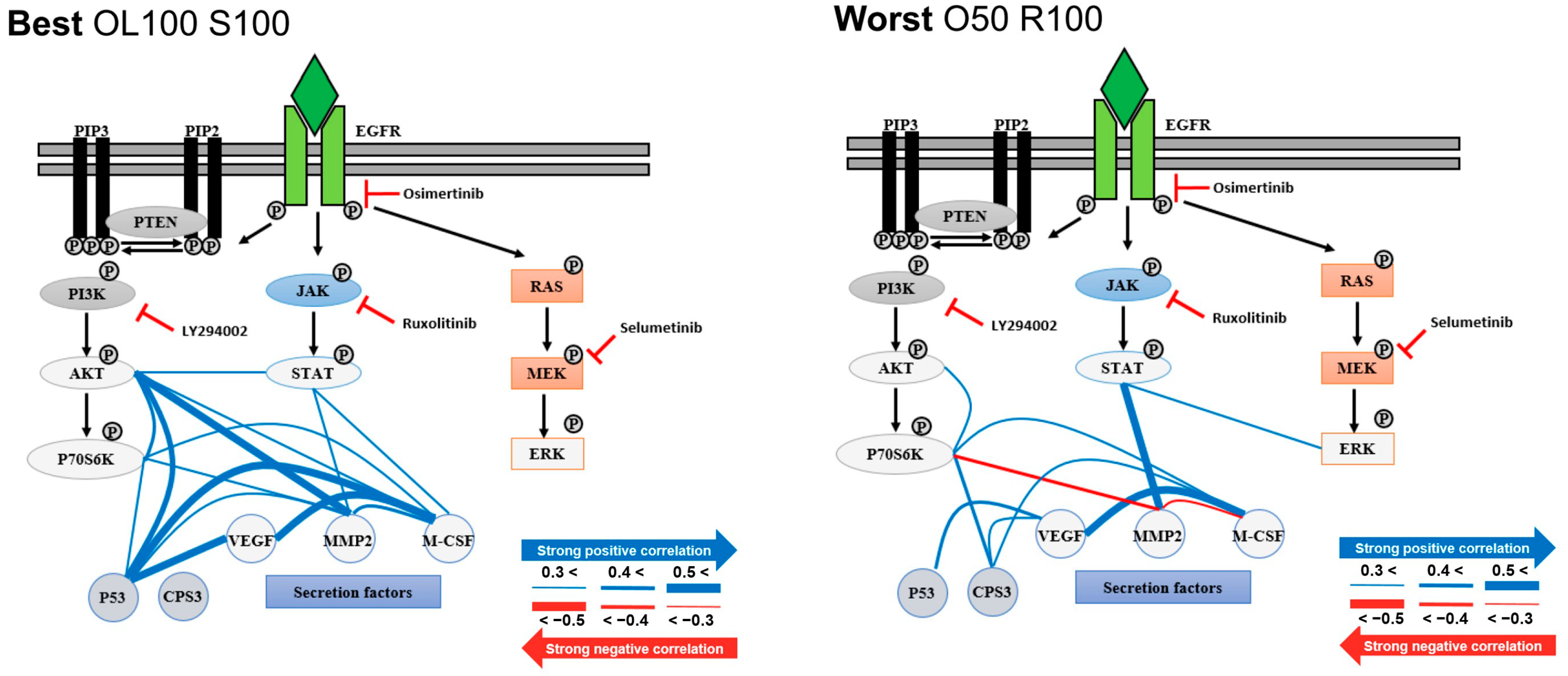
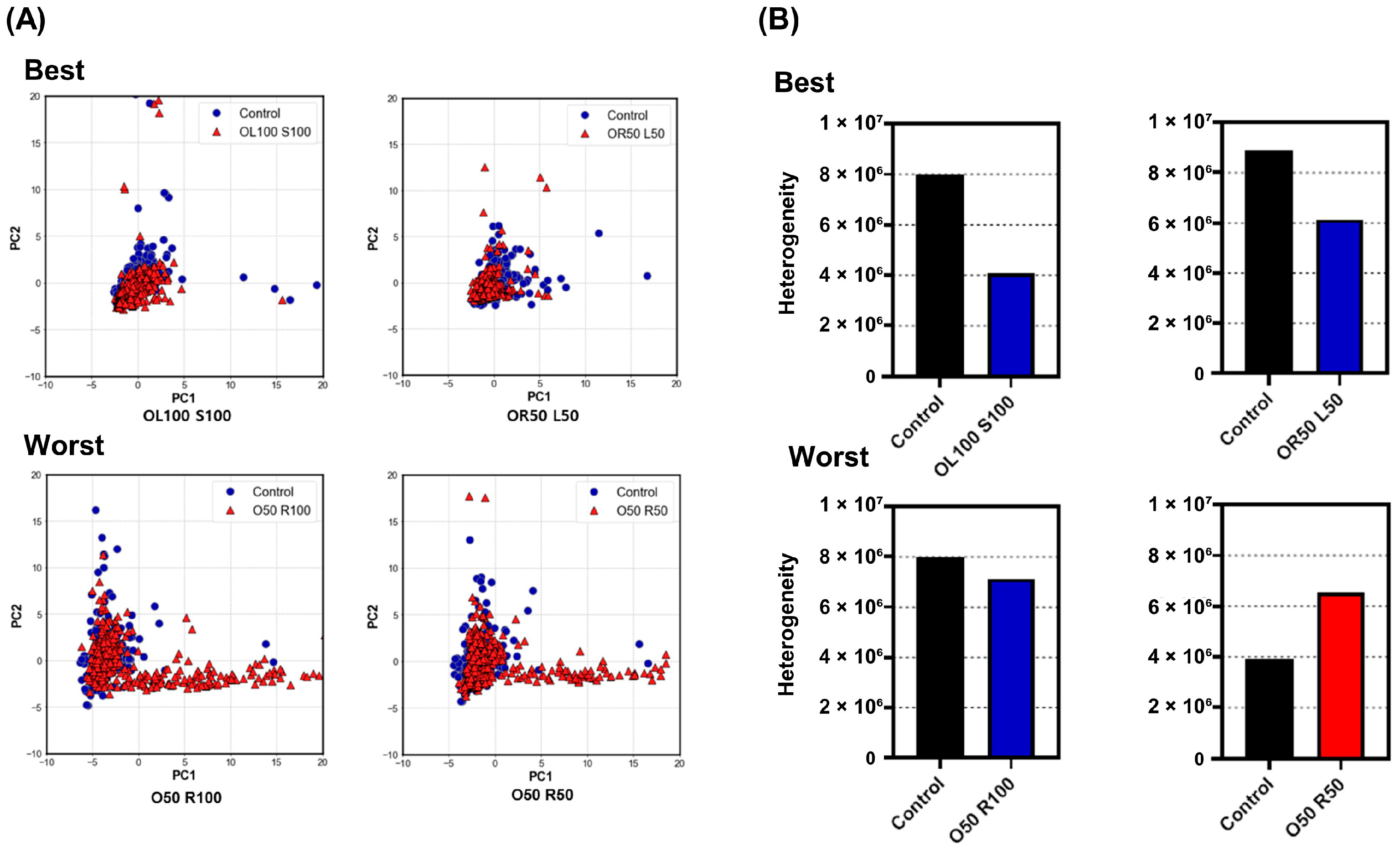
3.3. Evaluation of Heterogeneity
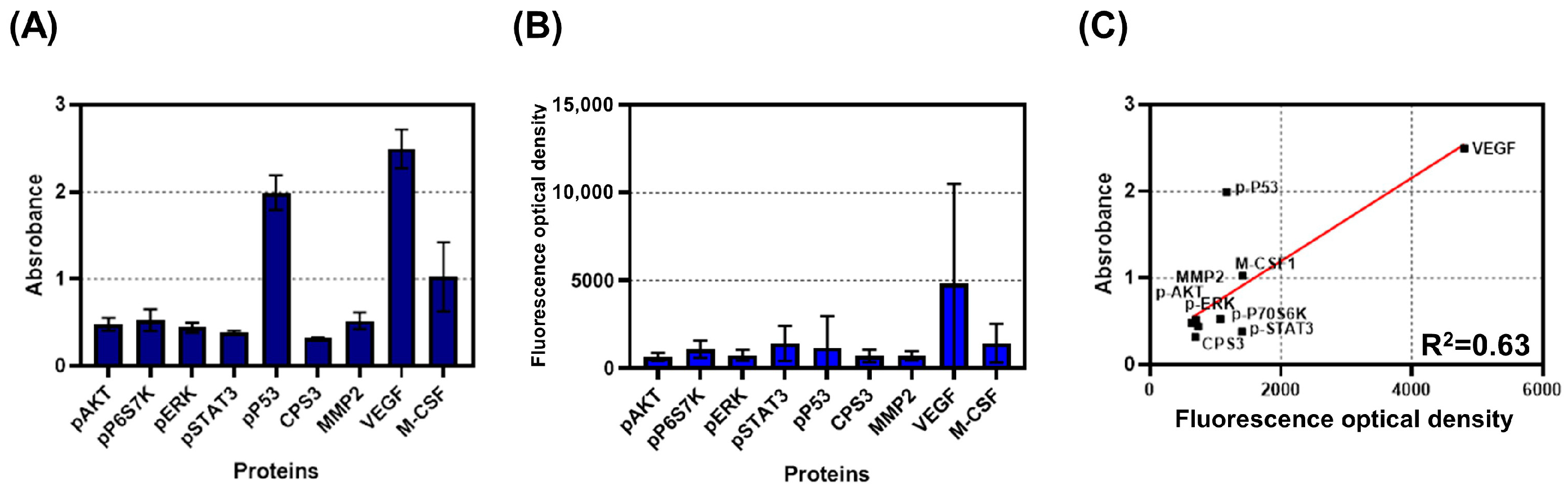
3.4. Comparison between Bulk and Single-Cell Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Saunders, N.A.; Simpson, F.; Thompson, E.W.; Hill, M.M.; Endo-Munoz, L.; Leggatt, G.; Minchin, R.F.; Guminski, A. Role of intratumoural heterogeneity in cancer drug resistance: Molecular and clinical perspectives. EMBO Mol. Med. 2012, 4, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; Swanton, C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol. Oncol. 2014, 8, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Burrell, R.A.; Endesfelder, D.; Novelli, M.R.; Swanton, C. Cancer chromosomal instability: Therapeutic and diagnostic challenges. EMBO Rep. 2012, 13, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Mroz, E.A.; Tward, A.D.; Hammon, R.J.; Ren, Y.; Rocco, J.W. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: Analysis of data from the Cancer Genome Atlas. PLoS Med. 2015, 12, e1001786. [Google Scholar] [CrossRef]
- Lim, L.C.; Lim, Y.M. Proteome Heterogeneity in Colorectal Cancer. Proteomics 2018, 18, 1700169. [Google Scholar] [CrossRef]
- Greenbaum, A.; Martin, D.R.; Bocklage, T.; Lee, J.H.; Ness, S.A.; Rajput, A. Tumor Heterogeneity as a Predictor of Response to Neoadjuvant Chemotherapy in Locally Advanced Rectal Cancer. Clin. Colorectal Cancer 2019, 18, 102–109. [Google Scholar] [CrossRef]
- Kikutake, C.; Yoshihara, M.; Sato, T.; Saito, D.; Suyama, M. Pan-cancer analysis of intratumor heterogeneity associated with patient prognosis using multidimensional measures. Oncotarget 2018, 9, 37689–37699. [Google Scholar] [CrossRef]
- Seoane, J.; De Mattos-Arruda, L. The challenge of intratumour heterogeneity in precision medicine. J. Intern. Med. 2014, 276, 41–51. [Google Scholar] [CrossRef]
- Shalek, A.K.; Benson, M. Single-cell analyses to tailor treatments. Sci. Transl. Med. 2017, 9, 408. [Google Scholar] [CrossRef]
- Zito Marino, F.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular heterogeneity in lung cancer: From mechanisms of origin to clinical implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef]
- Guan, Y.F.; Li, G.R.; Wang, R.J.; Yi, Y.T.; Yang, L.; Jiang, D.; Zhang, X.P.; Peng, Y. Application of next-generation sequencing in clinical oncology to advance personalized treatment of cancer. Chin. J. Cancer 2012, 31, 463–470. [Google Scholar] [CrossRef]
- Waters, K.M.; Liu, T.; Quesenberry, R.D.; Willse, A.R.; Bandyopadhyay, S.; Kathmann, L.E.; Weber, T.J.; Smith, R.D.; Wiley, H.S.; Thrall, B.D. Network analysis of epidermal growth factor signaling using integrated genomic, proteomic and phosphorylation data. PLoS ONE 2012, 7, e34515. [Google Scholar] [CrossRef]
- Boja, E.S.; Rodriguez, H. Proteogenomic convergence for understanding cancer pathways and networks. Clin. Proteom. 2014, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Qin, L.; Wei, W.; Geng, F.; Fan, R.; Shin, Y.S.; Guo, D.; Hood, L.; Mischel, P.S.; Heath, J.R. Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proc. Natl. Acad. Sci. USA 2012, 109, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015, 25, 1499–1507. [Google Scholar] [CrossRef]
- Wei, W.; Shin, Y.S.; Xue, M.; Matsutani, T.; Masui, K.; Yang, H.; Ikegami, S.; Gu, Y.; Herrmann, K.; Johnson, D.; et al. Single-Cell Phosphoproteomics Resolves Adaptive Signaling Dynamics and Informs Targeted Combination Therapy in Glioblastoma. Cancer Cell 2016, 29, 563–573. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, D.M.; Xu, Y.; Liang, Z.; Reddy, M.P.; Wu, D.Y.; Litwin, V. Recommendations for the validation of flow cytometric testing during drug development: II assays. J. Immunol. Methods 2011, 363, 120–134. [Google Scholar] [CrossRef]
- O’Brien, P.J.; Lee, M.; Spilker, M.E.; Zhang, C.C.; Yan, Z.; Nichols, T.C.; Li, W.; Johnson, C.H.; Patti, G.J.; Siuzdak, G. Monitoring metabolic responses to chemotherapy in single cells and tumors using nanostructure-initiator mass spectrometry (NIMS) imaging. Cancer Metab. 2013, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tham, D.; Wei, W.; Shin, Y.S.; Ma, C.; Ahmad, H.; Shi, Q.; Yu, J.; Levine, R.D.; Heath, J.R. Quantitating cell-cell interaction functions with applications to glioblastoma multiforme cancer cells. Nano Lett. 2012, 12, 6101–6106. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Shi, Q.; Remacle, F.; Qin, L.; Shackelford, D.B.; Shin, Y.S.; Mischel, P.S.; Levine, R.D.; Heath, J.R. Hypoxia induces a phase transition within a kinase signaling network in cancer cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1352–E1360. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko-Balasha, N.; Wang, J.; Remacle, F.; Levine, R.D.; Heath, J.R. Glioblastoma cellular architectures are predicted through the characterization of two-cell interactions. Proc. Natl. Acad. Sci. USA 2014, 111, 6521–6526. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Masterman-Smith, M.D.; Graham, N.A.; Jiao, J.; Mottahedeh, J.; Laks, D.R.; Ohashi, M.; DeJesus, J.; Kamei, K.; Lee, K.B.; et al. A microfluidic platform for systems pathology: Multiparameter single-cell signaling measurements of clinical brain tumor specimens. Cancer Res. 2010, 70, 6128–6138. [Google Scholar] [CrossRef] [PubMed]
- Sinkala, E.; Herr, A.E. Single-Cell Western Blotting. Methods Mol. Biol. 2015, 1346, 1–9. [Google Scholar] [CrossRef]
- Xue, M.; Wei, W.; Su, Y.; Kim, J.; Shin, Y.S.; Mai, W.X.; Nathanson, D.A.; Heath, J.R. Chemical methods for the simultaneous quantitation of metabolites and proteins from single cells. J. Am. Chem. Soc. 2015, 137, 4066–4069. [Google Scholar] [CrossRef]
- Jin, D.; Deng, B.; Li, J.X.; Cai, W.; Tu, L.; Chen, J.; Wu, Q.; Wang, W.H. A microfluidic device enabling high-efficiency single cell trapping. Biomicrofluidics 2015, 9, 014101. [Google Scholar] [CrossRef]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis Primers 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Gilligan, D.; Pacey, S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015, 16, e447–e459. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Jiang, T.; Su, C.; Ren, S.; Cappuzzo, F.; Rocco, G.; Palmer, J.D.; van Zandwijk, N.; Blackhall, F.; Le, X.; Pennell, N.A.; et al. A consensus on the role of osimertinib in non-small cell lung cancer from the AME Lung Cancer Collaborative Group. J. Thorac. Dis. 2018, 10, 3909–3921. [Google Scholar] [CrossRef]
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef]
- Casaluce, F.; Sgambato, A.; Maione, P.; Sacco, P.C.; Santabarbara, G.; Gridelli, C. Selumetinib for the treatment of non-small cell lung cancer. Expert Opin. Investig. Drugs 2017, 26, 973–984. [Google Scholar] [CrossRef]
- Giaccone, G.; Sanborn, R.E.; Waqar, S.N.; Martinez-Marti, A.; Ponce, S.; Zhen, H.; Kennealey, G.; Erickson-Viitanen, S.; Schaefer, E. A Placebo-Controlled Phase II Study of Ruxolitinib in Combination with Pemetrexed and Cisplatin for First-Line Treatment of Patients with Advanced Nonsquamous Non-Small-Cell Lung Cancer and Systemic Inflammation. Clin. Lung Cancer 2018, 19, e567–e574. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lei, H.L.; Zhang, Y.F.; Xu, Y.G.; Shen, S.; Ge, Y.R.; Li, H.M.; Xie, J.M. Non-covalent modification of graphene oxide nanocomposites with chitosan/dextran and its application in drug delivery. RSC Adv. 2016, 6, 9328–9337. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Campbell, K.R.; Lun, A.T.; Wills, Q.F. Scater: Pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 2017, 33, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Bro, R.; Smilde, A.K. Principal component analysis. Anal. Methods 2014, 6, 2812–2831. [Google Scholar] [CrossRef]
- Zhang, Z.; Murtagh, F.; Van Poucke, S.; Lin, S.; Lan, P. Hierarchical cluster analysis in clinical research with heterogeneous study population: Highlighting its visualization with R. Ann. Transl. Med. 2017, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Vicent, S.; López-Picazo, J.M.; Toledo, G.; Lozano, M.D.; Torre, W.; Garcia-Corchón, C.; Quero, C.; Soria, J.C.; Martín-Algarra, S.; Manzano, R.G.; et al. ERK1/2 is activated in non-small-cell lung cancer and associated with advanced tumours. Br. J. Cancer 2004, 90, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, T.; Pirinen, R.; Bohm, J.; Johansson, R.; Kosma, V.M. Increased expression of matrix metalloproteinase-2 (MMP-2) predicts tumour recurrence and unfavourable outcome in non-small cell lung cancer. Histol. Histopathol. 2008, 23, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Paskal, W.; Wlodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef]
- Liu, J.; Dang, H.; Wang, X.W. The significance of intertumor and intratumor heterogeneity in liver cancer. Exp. Mol. Med. 2018, 50, e416. [Google Scholar] [CrossRef]
- Obulkasim, A.; Ylstra, B.; van Essen, H.F.; Benner, C.; Stenning, S.; Langley, R.; Allum, W.; Cunningham, D.; Inam, I.; Hewitt, L.C.; et al. Reduced genomic tumor heterogeneity after neoadjuvant chemotherapy is related to favorable outcome in patients with esophageal adenocarcinoma. Oncotarget 2016, 7, 44084–44095. [Google Scholar] [CrossRef]
- Trapnell, C. Defining cell types and states with single-cell genomics. Genome Res. 2015, 25, 1491–1498. [Google Scholar] [CrossRef]
- Yuan, G.C.; Cai, L.; Elowitz, M.; Enver, T.; Fan, G.; Guo, G.; Irizarry, R.; Kharchenko, P.; Kim, J.; Orkin, S.; et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017, 18, 84. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Zhang, Y.; Liu, J. Advancing single-cell proteomics and metabolomics with microfluidic technologies. Analyst 2019, 144, 846–858. [Google Scholar] [CrossRef]
- Pandey, A.; Mann, M. Proteomics to study genes and genomes. Nature 2000, 405, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, K.; Qian, P.Y. Proteomics: Challenges, techniques and possibilities to overcome biological sample complexity. Hum. Genom. Proteom. 2009, 2009. [Google Scholar] [CrossRef] [PubMed]
- Castro-Giner, F.; Gkountela, S.; Donato, C.; Alborelli, I.; Quagliata, L.; Ng, C.K.Y.; Piscuoglio, S.; Aceto, N. Cancer Diagnosis Using a Liquid Biopsy: Challenges and Expectations. Diagnostics 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhao, H. Liquid biopsy in tumors: Opportunities and challenges. Ann. Transl. Med. 2018, 6, S89. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, Y.; Son, M.; Nam, Y.R.; Choi, J.; Heath, J.R.; Yang, S. Microfluidic Single-Cell Proteomics Assay Chip: Lung Cancer Cell Line Case Study. Micromachines 2021, 12, 1147. https://doi.org/10.3390/mi12101147
Jung Y, Son M, Nam YR, Choi J, Heath JR, Yang S. Microfluidic Single-Cell Proteomics Assay Chip: Lung Cancer Cell Line Case Study. Micromachines. 2021; 12(10):1147. https://doi.org/10.3390/mi12101147
Chicago/Turabian StyleJung, Yugyung, Minkook Son, Yu Ri Nam, Jongchan Choi, James R. Heath, and Sung Yang. 2021. "Microfluidic Single-Cell Proteomics Assay Chip: Lung Cancer Cell Line Case Study" Micromachines 12, no. 10: 1147. https://doi.org/10.3390/mi12101147
APA StyleJung, Y., Son, M., Nam, Y. R., Choi, J., Heath, J. R., & Yang, S. (2021). Microfluidic Single-Cell Proteomics Assay Chip: Lung Cancer Cell Line Case Study. Micromachines, 12(10), 1147. https://doi.org/10.3390/mi12101147