Abstract
Pertussis toxin (PTx) is a major virulence factor of Bordetella pertussis and an AB5-type exotoxin that disrupts host signaling. Its enzymatic A subunit ADP-ribosylates the α-subunit of inhibitory G proteins (Gαi), preventing them from mediating receptor-induced inhibition of adenylyl cyclase (AC). This leads to unrestrained cAMP accumulation in host cells, a canonical mechanism underlying many pertussis disease manifestations. PTx works in concert with the bacterium’s adenylate cyclase toxin (ACT) to subvert immune defenses and establish infection. Interestingly, PTx exerts both cAMP-dependent and cAMP-independent effects. In addition to the well-known cAMP-mediated pathway, PTx’s B oligomer can engage host cell surface receptors to trigger signaling cascades independent of the A subunit’s catalytic activity. Such B oligomer-mediated pathways modulate cellular responses in the absence of ADP-ribosylation. This review provides a comprehensive analysis of PTx’s dual functionality, distinguishing its Gi protein-dependent elevation of cAMP from the noncanonical activities of the B oligomer. It also highlights how disruption of constitutive Gi signaling and the interplay between PTx and ACT shape host–pathogen interaction in pertussis pathogenesis.
Key Contribution:
This review summarizes the molecular mechanisms of PTx action, focusing on canonical inactivation of Gi proteins by ADP-ribosylation, the pivotal role of constitutive Gi signaling, and the toxin’s synergistic action with ACT.
1. Pertussis and Its Major Virulence Factors
Pertussis, commonly known as whooping cough, is an acute respiratory infection caused by the Gram-negative bacterium Bordetella pertussis (B. pertussis) [1]. Pertussis infection typically progresses through three clinical stages. The catarrhal stage is characterized by mild upper respiratory symptoms resembling those of a common cold. It is followed by the paroxysmal stage, during which patients experience spasmodic coughing fits often accompanied by inspiratory “whoop” (hence the name “whooping cough”), vomiting, or transient cyanosis. Finally, in the convalescent stage, the frequency and severity of coughing gradually diminish, although symptoms may persist for several weeks.
B. pertussis is a strictly human pathogen that has evolved a specialized arsenal of virulence factors to colonize the respiratory tract and cause disease [2]. It preferentially attaches to the ciliated epithelium of the upper airway, where the bacteria multiply on the surface of host cells, evade mucociliary clearance and immune defenses, and ultimately damage the respiratory epithelium.
Pertussis toxin (PTx) is a major virulence factor secreted by B. pertussis and is a key contributor to the pathogenesis of whooping cough. The diverse pathological effects of pertussis are primarily attributed to PTx, a view consolidated from several biological activities that were historically described and investigated as separate phenomena: histamine-sensitizing factor (HSF) [3], lymphocytosis-promoting factor (LPF) [4], and islet-activating protein (IAP) [5]. The definitive link between these activities was established by Munoz et al. [6], who examined a highly pure, crystalline protein that had been previously isolated from the B. pertussis Tohama I strain [7]. Their research demonstrated conclusively that this single protein, which they named pertussigen (now known as PTx), was responsible for all three effects, establishing that nanogram amounts could induce the characteristic physiological changes in mice (e.g., leukocytosis and increased insulin secretion), while microgram doses were lethal [6]. Early physiological studies by Katada and Ui further showed that pretreatment of rats with B. pertussis vaccine abolished α2-adrenergic receptor-mediated inhibition of insulin secretion [8]. This key finding revealed that PTx selectively disrupts Gi-coupled receptor function in vivo, a concept later summarized by Katada [9]. While PTx is central to many systemic symptoms, it acts in concert with other toxins released by B. pertussis, such as adenylate cyclase toxin (ACT), tracheal cytotoxin (TCT), dermonecrotic toxin (DNT), and lipooligosaccharide (LOS), that collectively orchestrate the disease’s complex pathology [10,11,12,13,14,15] (Figure 1).
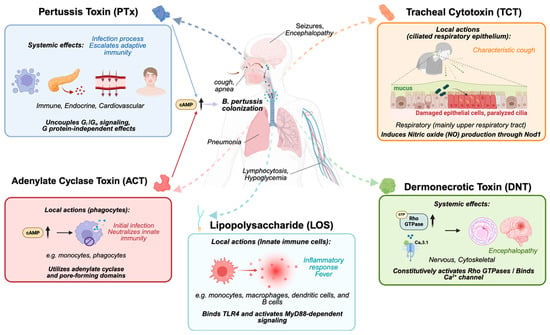
Figure 1.
An overview of the toxins produced by B. pertussis and their target organs/systems. PTx acts primarily as a systemic effector, uncoupling Gi/o protein signaling and modulating immune, endocrine, and cardiovascular responses, thereby modifying adaptive immunity and contributing to lymphocytosis and hypoglycemia. ACT functions locally on phagocytes, where calmodulin-activated cAMP bursts disrupt innate immune functions such as phagocytosis and pore formation. TCT targets the ciliated respiratory epithelium, causing epithelial damage, impaired mucociliary clearance, and the characteristic paroxysmal cough. DNT constitutively activates Rho GTPases and modulates Ca2+ channels, inducing cytoskeletal alterations and contributing to neurological symptoms. LOS signals through TLR4–MyD88 pathways and triggers strong local inflammatory responses in innate immune cells. Together, these toxins shape B. pertussis colonization, immune evasion, tissue damage, and systemic disease manifestations.
ACT, also known as adenylate cyclase toxin-hemolysin (CyaA), increases intracellular cAMP levels in host cells, particularly targeting phagocytes through CD11b/CD18 heterodimer [11,12]. It has two main functions: an adenylate cyclase domain that binds to host calmodulin to generate excessive cAMP and a domain that creates cation-selective pores in host cell membranes [16]. This dual action impairs the ability of immune cells to destroy bacteria by inhibiting monocyte-to-macrophage transition, and high amount of ACT can induce cell death [16,17]. While ACT exhibits high-affinity binding to the CD11b/CD18 integrin on phagocytes, its membrane permeability also allows for receptor-independent entry into a wider variety of host cells [18]. This dual-targeting capability complements the systemic effects of PTx, creating a potent “one-two punch” against the host immune response [11]. Both toxins function by dramatically elevating intracellular cAMP to suppress key innate immune functions like phagocytosis and inflammatory cytokine production [19,20]. This synergy is reflected in their evolution; B. pertussis produces less ACT than its relative B. bronchiseptica, likely because the potent, systemic immunosuppression by PTx lessens the need for high quantities of ACT to disable phagocytes [21].
Unlike the other protein toxins, TCT is a fragment of the bacterium’s peptidoglycan cell wall that is released during cell growth [13,22]. It is specifically toxic to the ciliated epithelial cells that line the respiratory tract, causing these cells to stop beating and can lead to their extrusion from the lining, which impairs the clearance of mucus and debris from the lungs and contributes to the characteristic cough of pertussis [23]. DNT is a potent intracellular toxin that permanently activates the host cell’s Rho GTPases, leading to severe disruption of the cellular cytoskeleton [14,24]. More importantly, DNT has been identified as a neurotropic virulence factor that specifically targets nerve cells by binding to the CaV3.1 calcium channel receptor, which might be the cause of pertussis encephalopathy, a devastating neurological complication of whooping cough [25]. As a Gram-negative bacterium, B. pertussis has LOS in its outer membrane, which can be released to act as an endotoxin [15,26]. The unique structure of B. pertussis LOS contributes to the inflammatory response and fever associated with the infection. In addition to these toxins, B. pertussis also produces several other virulence factors, such as filamentous hemagglutinin, fimbriae, and pertactin, which act as adhesins to help the bacteria attach to the cells of the respiratory tract (Table 1).

Table 1.
Major toxins of B. pertussis.
2. The Canonical Actions of PTx: Gαi- and cAMP-Dependent Mechanisms
2.1. Structural Organization of PTx
Structurally, PTx is a heterohexameric complex with a molecular weight of approximately 105 kDa, composed of five distinct subunits: S1 (26 kDa), S2 (22 kDa), S3 (22 kDa), two copies of S4 (12 kDa), and S5 (10 kDa) [27] (Figure 2A). The holotoxin conforms to the classical AB-type toxin model, in which the catalytically active A protomer (S1) possesses ADP-ribosyltransferase activity [46], while the B oligomer, formed by the S2, S3, S4, and S5 subunits, mediates high-affinity binding to glycoprotein receptors on the host cell surface [47].
Upon binding to glycoprotein receptors, the holotoxin is internalized via endocytosis and retrogradely transported through the endosomal–Golgi network, ultimately reaching the endoplasmic reticulum (ER) [48]. Within the ER, ATP binding to the B oligomer triggers the dissociation of the S1 subunit from the complex [49]. The liberated S1 subunit is thermolabile at physiological temperature, rapidly adopting a partially disordered conformation that prevents its reassociation with the B oligomer. This unfolded state enables S1 to hijack the ER-associated degradation pathway, facilitating its retro-translocation across the ER membrane into the cytosol [50]. In the cytoplasm, glutathione reduces the conserved C41–C201 disulfide bond within S1, exposing the catalytic core and thereby activating the enzyme [28].
This modular architecture reflects a clear division of labor between the binding (B) and catalytic (A) components: the B oligomer ensures precise cellular targeting and delivery, whereas the S1 subunit, once in the cytosol, enzymatically modifies host G proteins. Such structural organization underlies the efficient intoxication of host cells and forms the molecular basis of the canonical ADP-ribosylation mechanism of PTx.
2.2. Enzymatic Mechanism of Gαi ADP-Ribosylation
In host cells, heterotrimeric G proteins (Gαβγ) serve as central mediators of GPCR signaling [51]. The Gα subunit binds and hydrolyzes guanine nucleotides, while the Gβγ complex acts as a membrane anchor that regulates multiple downstream effectors. In the resting state, Gα remains bound to GDP and associates with Gβγ to form an inactive trimeric complex. Upon receptor activation by an agonist, the GPCR catalyzes the exchange of GDP for GTP on Gα, promoting its dissociation from Gβγ [52]. The released Gα–GTP and free Gβγ subunits then modulate a variety of effectors [51]. Among G protein subfamilies, the Gi/o family plays an inhibitory regulatory role: activated Gαi suppresses AC catalytic activity to reduce intracellular cAMP levels.
The primary molecular targets of S1 are the α-subunits of inhibitory heterotrimeric G proteins (Gi, Go) and the visual transduction protein transducin (Gt) [5,30,53]. PTx catalyzes the transfer of ADP-ribose from NAD+ to a conserved cysteine residue, typically four residues from the C-terminus, of these α subunits [30,53]. Notably, PTx prefers to catalyze trimeric Gi proteins that consist of Gαβγ subunits, rather than the Gα monomer alone [54,55,56]. This covalent modification on the cysteine residue of Gαi/o/t subunits prevents their coupling to receptors, thereby disabling receptor-mediated GDP/GTP exchange. In cell-free systems, PTx activity requires NAD+ and ATP, with nicotinamide as a reaction byproduct [31]. Radiolabeled NAD+ tracing confirmed that the ADP-ribose moiety, but not nicotinamide, is incorporated into a 41 kDa membrane protein, later identified as Gαi, with the modification correlating with enhanced AC activity [32,57].

Figure 2.
Structural features of PTx and its interaction with NAD+, Gαi1, and Gβ1. (A) The full PTx structure (PDB code: 1PRT) is shown, including the S1 catalytic subunit (blue) and the B-oligomer, composed of S2 (gray), S3 (pale orange), S4 (pink), and S5 (black) in a 1:1:2:1 ratio. In the cytosol, ATP facilitates dissociation of the S1 subunit (PDB code: 7SKY) from the B-oligomer, coupled with the cleavage of the disulfide bond between C41 and C201, enabling NAD+ and Gα to access the catalytic domain. (B) Key residues involved in ADP-ribosyltransferase activity (R9, Y10, W26, H35, and E129) are shown in green. Residues critical for Gα binding (Y59 and Y63; [58]) are highlighted in pink. (C) Docking simulations using AlphaFold3 incorporated S1, Gαi1, Gβ1, Gγ2, NAD+, and GDP. Consistent structural models were observed only when Gβ1 and Gγ2 were included, consistent with prior studies [29,47,59]. (D) Key residues forming hydrogen bonds between S1 and Gαi1 in the selected docking model (C) are shown. (E) Key residues forming hydrogen bonds between D17 and N21 residues of S1 and R52 of Gβ1 are shown.
Figure 2.
Structural features of PTx and its interaction with NAD+, Gαi1, and Gβ1. (A) The full PTx structure (PDB code: 1PRT) is shown, including the S1 catalytic subunit (blue) and the B-oligomer, composed of S2 (gray), S3 (pale orange), S4 (pink), and S5 (black) in a 1:1:2:1 ratio. In the cytosol, ATP facilitates dissociation of the S1 subunit (PDB code: 7SKY) from the B-oligomer, coupled with the cleavage of the disulfide bond between C41 and C201, enabling NAD+ and Gα to access the catalytic domain. (B) Key residues involved in ADP-ribosyltransferase activity (R9, Y10, W26, H35, and E129) are shown in green. Residues critical for Gα binding (Y59 and Y63; [58]) are highlighted in pink. (C) Docking simulations using AlphaFold3 incorporated S1, Gαi1, Gβ1, Gγ2, NAD+, and GDP. Consistent structural models were observed only when Gβ1 and Gγ2 were included, consistent with prior studies [29,47,59]. (D) Key residues forming hydrogen bonds between S1 and Gαi1 in the selected docking model (C) are shown. (E) Key residues forming hydrogen bonds between D17 and N21 residues of S1 and R52 of Gβ1 are shown.

Mutagenesis and crystallographic studies have revealed how a network of key residues in the S1 subunit exert its enzymatic activity (Table 2). Its catalytic core has a conserved structural feature involving Y8 and E129 [28], which can also be found in cholera toxin (Y6 and E112) [60], exotoxin A (Y439 and E553) [61], and diphtheria toxin (Y20 and E148) [62]. Within this pocket, E129 acts as the key essential glutamate to destabilize NAD+ [63,64,65,66], while H35 primes the target cysteine on Gα for nucleophilic attack [67,68]. The integrity of this active site is further supported by Q127 [58,69] and S52 [67], which help maintain its precise geometry. The NAD+ is bound by interactions with R9, Y10, W26, H35, L36, Q42, S52, T53, Y63, and Y130 [63,65,70,71,72,73,74] (Figure 2B). For recognizing the Gαi protein, the subunit relies on hydrophobic contacts from Y59 and Y63 [58], along with the broader 195–204 region [75]. On the substrate side, a conserved cysteine four amino acids away from the carboxyl terminus of the Gαi subunit is essential for modification, substitutions or positional shifts weaken or even abolish ADP-ribosylation [58]. Structural modeling using AlphaFold3 [76] (Figure 2C) (using each of PTx S1, Gαi1, Gβ1, Gγ2, NAD, and GDP for docking) further suggests that residues D350, C351, and G352 in the C-terminal helix of Gαi1 could form potential hydrogen bonds with Y63, R67, Q127, and Q205 of S1, orienting the acceptor cysteine into the catalytic pocket (Figure 2D). Through ADP-ribosylation, PTx (S1) irreversibly prevents Gi/o-mediated signaling, potentially increasing the intracellular cAMP level, which is a key biochemical event underlying many downstream effects in pertussis pathogenesis. Collectively, these structural and biochemical insights delineate how PTx catalyzes a precise and irreversible ADP-ribosylation reaction that selectively targets Gi proteins, abolishes inhibitory signaling, and drives persistent cAMP elevation—defining the canonical mechanism of PTx action.
Table 2.
Review of key functional residues/regions in S1 of PTx.
2.3. cAMP-Mediated Effects and Pathophysiological Outcomes
By inactivating Gi and thereby releasing the inhibitory brake on AC, PTx drives a persistent rise in intracellular cAMP (Figure 3). This chronic cAMP elevation engages two major cAMP-responsive signaling branches, protein kinase A (PKA) [79] and the exchange proteins directly activated by cAMP (EPAC) [80], which together reprogram multiple immune and metabolic pathways.
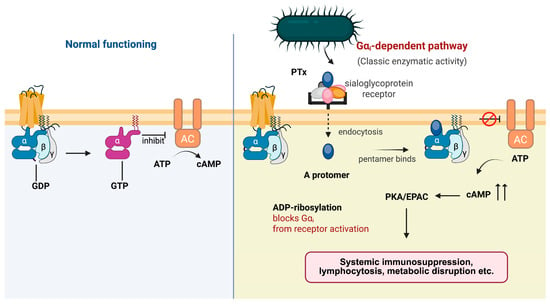
Figure 3.
Canonical cAMP signaling cascade triggered by PTx. Under normal conditions, Gαi couples to its receptor and inhibits AC, thereby restraining intracellular cAMP production. Upon exposure to PTx, the A protomer enters host cells and catalyzes ADP-ribosylation of Gαi, preventing receptor-mediated activation of the G protein. Loss of functional Gαi releases tonic inhibition of AC and results in excessive cAMP accumulation. Elevated cAMP activates downstream PKA and EPAC pathways, leading to systemic immunosuppression, lymphocytosis, and metabolic disruption.
In immune cells, elevated cAMP functions as a potent immunosuppressive signal. Increased cAMP suppresses leukocyte integrin activation as well as adhesion and migratory capacity through PKA, whereas EPAC activation inhibits the production of pro-inflammatory cytokines and chemokines [20]. In the endocrine system, elevated cAMP stimulates insulin secretion from pancreatic β-cells, a mechanism that explains the hypoglycemia frequently observed during severe pertussis infection [81]. Similarly, in vascular and neuronal tissues, the loss of Gi-mediated inhibitory tone heightens responsiveness to vasoactive amines such as histamine, leading to exaggerated vasodilation and histamine hypersensitivity—classical bioassay readouts of PTx activity. Gαi1 and Gαi3 constitute the key molecular targets mediating this response, as genetic ablation of either subunit phenocopies PTx exposure and renders mice spontaneously hypersensitive to vasoactive amine sensitization (VAAS) [82].
3. Synergistic Interplay and Dominant Dynamics Between PTx and ACT
The canonical enzymatic mechanism of PTx establishes the fundamental basis for Gi inactivation and sustained elevation of intracellular cAMP. However, in infected hosts, this process rarely acts in isolation. B. pertussis secretes multiple toxins that converge upon the same second-messenger network, among which ACT plays a particularly prominent role. Together, PTx and ACT constitute a synergistic dual-toxin strategy that amplifies and maintains intracellular cAMP accumulation across distinct immune cell populations. This section describes how these two toxins cooperatively disrupt cAMP homeostasis, the resulting immunopathological consequences, and the key determinants that modulate their interplay.
3.1. Toxin Dominance and Disease Subtypes: From PTx-Dominant to ACT-Dominant Infections
Currently, no standardized method exists to directly quantify the relative secretion levels of PTx and ACT. Because the expression of both toxins is shaped by the BvgAS regulatory state, strain genotype, host environment, and stage of infection, their relative dominance can only be inferred indirectly through multiple complementary readouts: (1) transcriptional and protein-level measurements, such as comparative expression of ptxA/B/E and cyaA [83,84]; (2) functional assays, including PTx-mediated Gαi ADP-ribosylation activity and the measurement of intracellular cAMP peaks induced by PTx or ACT [85,86]; (3) host response signatures, such as leukocyte apoptosis, phagocytic suppression, and characteristic immunopathological features observed in experimental or clinical models.
In the context of cell death, ACT exhibits clear predominance. ACT induces rapid apoptosis in macrophages, neutrophils, and dendritic cells [87]. In the mouse lung, strains producing ACT trigger pronounced macrophage and neutrophil apoptosis, whereas ACT-deficient mutants fail to do so [88]. In contrast, PTx does not directly induce apoptosis in phagocytes; its major effects in lymphoid and myeloid cells manifest as disruptions in migration and homing rather than acute loss of survival signals. The extremely high intracellular cAMP levels generated by ACT profoundly impair macrophage and neutrophil phagocytic capacity [89] and suppress the oxidative burst response [90]. Correspondingly, ACT-deficient strains restore normal phagocytic function in the same experimental models [91]. In tissues affected by ACT, one observes a characteristic pattern of acute pulmonary inflammation, epithelial barrier damage, exudative injury, and paradoxically poor inflammatory cell infiltration, a phenotype marked by severe local tissue injury but ineffective immune clearance.
At the level of immunopathology, PTx disrupts basal inhibitory signaling across multiple tissues through irreversible ADP-ribosylation of Gαi/o, thereby provoking a broad spectrum of immune and physiological dysregulation. The most characteristic in vivo readout is the marked peripheral lymphocytosis, whose magnitude strongly correlates with disease severity and strictly depends on the enzymatic activity of PTx [92]. Comparative studies with B. parapertussis, a closely related species lacking PTx, similarly demonstrate a greatly diminished or absent lymphocytosis response [93]. Another PTx-specific immunopathological hallmark is VAAS, a phenomenon that underlies the classical bioassay used to assess the potency of pertussis vaccines and purified toxin preparations [94].
Taken together, leukocyte apoptosis, phagocytic suppression, and distinct immunopathological signatures provide reliable markers to discriminate PTx-dominant from ACT-dominant effects. PTx primarily drives systemic immune dysregulation and defects in cell trafficking, whereas ACT triggers acute cellular injury and profound suppression of phagocytic function.
3.2. Determinants of Dominance: Temporal Window and Dose Ratio
The relative dominance of PTx and ACT is not fixed but shifts dynamically over the course of infection, shaped by temporal windows and dosage. ACT exerts its effects on a remarkably rapid timescale, typically within minutes to hours after contacting host cells. As a single-component RTX toxin secreted directly through the type I secretion system, ACT rapidly binds to target cell membranes and, upon calmodulin activation, immediately triggers a sharp, localized burst of cAMP. This rapid signaling spike directly suppresses macrophage and neutrophil phagocytosis, oxidative burst activity, and migratory capacity, producing a characteristic frontline immune paralysis. In B. pertussis cultures, ACT secretion is detectable at 60 min. and plateaus by 6 h [95]. Experimentally, elevated cAMP can be detected within seconds post infection [96], oxidative burst is inhibited within 5 min [97], phagocytic suppression becomes evident within 1 h [98], and apoptosis appears by 4 h [99].
In contrast, the bioactivation of PTx is markedly delayed. Studies in cells and tissues show that PTx requires several hours of uptake and intracellular trafficking before it can ADP-ribosylate Gi proteins and induce stable signaling blockade. For example, in renal tubule and thyroid tissue slices, and specifically in isolated medullary collecting tubules and medullary thick ascending limbs, PTx requires at least 3–6 h of preincubation to ADP-ribosylate Gαi; brief 1 h exposure neither induces detectable Gi inactivation nor alters arginine vasopressin-dependent cAMP production [100]. In B. pertussis culture, PTx secretion increases most sharply between 12 and 24 h and then stabilizes [101] (Figure 4). This temporal division of labor ensures that ACT acts first to disable local immune defenses, while PTx subsequently reinforces systemic immune dysregulation through long-range signaling mechanisms, thereby sustaining persistent infection.
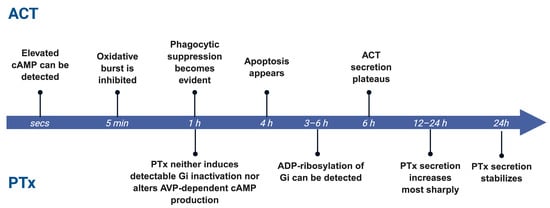
Figure 4.
Temporal separation of ACT and PTx activity. ACT acts rapidly within minutes to hours, whereas PTx shows delayed activity requiring prolonged exposure.
The effective environmental concentration of ACT typically falls within the tens to hundreds of ng/mL range; 60–80 ng/mL in in vitro cultures, and ~12–20 ng/mL in nasopharyngeal aspirates from infected infants [95]. In contrast, PTx exhibits strong biological activity at substantially lower concentrations, often in the ~1–10 ng/mL range, as demonstrated by its disruption of Gαi signaling [102]. PTx also displays much greater diffusibility and systemic “spillover” capacity: even a small subpopulation of PTx-producing bacteria can secrete the toxin at levels sufficient to act on distant host cells and tissues, amplifying its overall impact. Experimental observations show that, in mixed infections, a minority of PTx-secreting strains can enhance the colonization capacity of neighboring non-producing strains; wild-type PTx+ B. pertussis can secrete diffusible toxin in vivo that compensates for the colonization defect of PTx-deficient mutants [103]. In contrast, the effects of the cytolytic ACT are highly concentration dependent and remain confined to the microenvironment where the toxin directly contacts host cells. In mixed infections, PTx+ bacteria can “cover” or complement PTx− strains, whereas ACT activity remains restricted to dense, localized lesions [11].
3.3. Pathological and Therapeutic Implications
In pathophysiological states dominated by PTx, systemic manifestations are the most pronounced. In infants, severe lymphocytosis and pulmonary hypertension are strongly associated with PTx activity [104,105]. Moreover, passive immunization targeting PTx reduces disease severity and improves systemic outcomes [106].
In addition to supportive care, clinical management may include (1) early passive immunization or administration of anti-PTx antibodies to block toxin entry into host cells [107]; (2) exchange transfusion or leukapheresis for life-threatening hyperleukocytosis, enabling rapid reduction in pulmonary arterial pressure [108]; and (3) antihistamines mitigate PTx-mediated vascular hypersensitivity and hypotension [109]. Together, these interventions target the PTx-induced inactivation of Gi proteins and the resulting systemic imbalances in signal transduction.
In ACT-dominant infections, localized immune dysfunction is a defining feature. For example, patients infected with B. parapertussis typically exhibit mild symptoms and rarely develop marked leukocytosis [93]. Studies have shown that anti-ACT antibodies can restore neutrophil phagocytosis and bactericidal activity [110]. Moreover, several TLR agonists and mucosal adjuvants, such as TLR2/TLR4 agonists, can elicit strong Th1/Th17 responses and mucosal IgA production in experimental models, thereby reducing B. pertussis colonization [111,112]; conceptually, these responses could counteract ACT-mediated local immunosuppression, although most evidence remains indirect.
4. Suppression of Basal Gi Activity by PTx
In the classical model, GPCRs are thought to engage Gi proteins only upon ligand binding, thereby inhibiting AC and reducing intracellular cAMP levels. However, accumulating evidence indicates that many cell types maintain a measurable level of Gi-mediated inhibitory tone even in the absence of exogenous agonists. This so-called constitutive Gi signaling, or basal Gi activity, is now recognized as an intrinsic “tension” essential for maintaining signaling homeostasis and metabolic equilibrium [113].
4.1. Experimental Evidence for Basal GPCR Signaling and PTx Sensitivity
A key prediction of constitutive Gi signaling is that, in the presence of a pre-existing inhibitory tone, PTx should elevate basal cAMP levels, resulting in a detectable increase in cAMP even in the absence of GPCR agonists. We validated this concept using a cAMP biosensor assay in HEK293 cells transiently expressing the cAMP-Glosensor-22F reporter (a cAMP-sensitive bioluminescent sensor). Cells were transfected with either an empty vector or plasmids encoding representative GPCRs with distinct coupling preferences, including the Gs-coupled β2-adrenergic (β2-AR) and D1 receptors (D1R), Gi-coupled dopamine D2 receptor (D2R), and melatonin MT1 and MT2 receptors (MT1R and MT2R; known to exhibit constitutive Gi activity) [114].
In cells transfected with MT1R or MT2R, we observed that PTx treatment led to a marked increase in basal cAMP levels (Figure 5A; bottom left). This effect is likely due to the constitutive activity of MT1R and MT2R on Gi activity; disruption of constitutive Gi signaling by PTx relieved the inhibitory constraint on AC, resulting in elevated cAMP. Furthermore, in cells with increased availability of Gαi1 by co-transfection, the PTx-induced increase in cAMP became more pronounced (Figure 5A; bottom right). This suggests that the magnitude of constitutive inhibition of AC is dependent on the level of Gi available to MT1R and MT2R. Interestingly, another Gi-coupled receptor, D2R, did not exhibit any increase in cAMP upon PTx treatment, suggesting that not all GPCRs possess constitutive activity (Figure 5A, bottom).
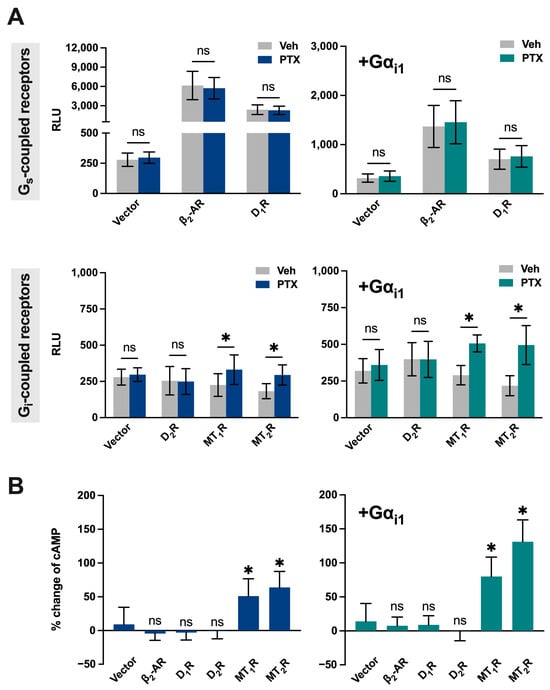
Figure 5.
PTx-induced cAMP elevation depends on receptor constitutive activity and Gi abundance. (A) Left: Baseline cAMP levels (luminescence in RLU) in HEK293 cells transiently transfected with cAMP-Glosensor-22F (22F), one of the indicated GPCRs, and vector (pcDNA3.1) with or without PTx pretreatment. Each construct was used at 0.4 µg per well (total 1.2 µg DNA/well in a 6-well plate). Top: β2-AR or D1R. Bottom: D2R, MT1R, or MT2R. Right: Baseline cAMP levels measured same as the left, except that cells were co-transfected with Gαi1. (B) Percent increase in cAMP upon PTX treatment. Left: Cells with endogenous Gi background. Right: Cells overexpressing Gαi1. Data are extracted from panel A; n = 3; data are shown as mean ± SD. An asterisk (*) indicates a significant difference (p ≤ 0.05), and ‘ns’ indicates no significance (p > 0.05).
Constitutive activity was also observed with the Gs-coupled β2-AR and D1R since their expression significantly increased basal cAMP levels, which were unaffected by PTx treatment (Figure 5A, top left). The lack of effect by PTx reflected that these two receptors are predominantly coupled to Gs. Nevertheless, the constitutive activities of β2-AR and D1R were attenuated upon increased expression of Gαi1 (Figure 5A, top right). Overall, these results indicate that PTx can directly cause significant changes in cAMP levels, particularly in cells expressing constitutively active Gi-coupled receptors and/or with a high Gi availability back-ground (Figure 5B). Our findings align with a growing body of research that suggests that certain GPCRs exhibit constitutive activity, especially among orphan, melatonin, and chemokine receptors [114,115,116].
4.2. Mechanistic Models of PTx-Induced Disruption of Basal Gi Control
To explain how PTx disrupts this basal inhibitory tone, we propose two complementary mechanistic models (Figure 6). Firstly, some Gi-coupled GPCRs can exert a persistent inhibitory input to AC even in the absence of exogenous agonists, as demonstrated for the melatonin receptor. This Gi-dependent basal constraint on AC activity is thus sensitive to PTx treatment [117]. For example, the melatonin MT1R displays ligand-independent (constitutive) activity toward Gi. Orphan GPCRs, such as GPR20 also exhibit robust Gi/cAMP-mediated constitutive activity [118]. Importantly, disruption of basal Gi signaling by PTx may not only relieve tonic inhibition of AC but also unmask latent stimulatory signaling pathways. Recent work has demonstrated that certain Gi-coupled GPCRs can exhibit context-dependent coupling to Gs, wherein ADP-ribosylation of Gi reveals a PTx-insensitive stimulatory component. Notably, the melatonin MT1R —while maintaining constitutive Gi-mediated inhibition of AC—was shown to potentiate forskolin-stimulated cAMP accumulation and, under conditions of elevated Gs availability, to directly stimulate cAMP production in a PTx-insensitive manner, highlighting a dual coupling behavior that amplifies cAMP responses following Gi inactivation [119].
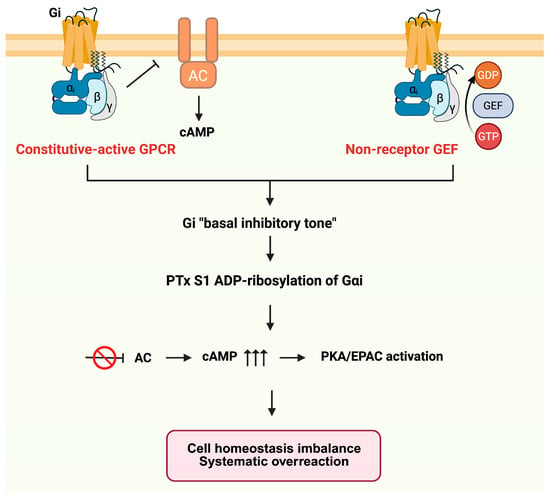
Figure 6.
Mechanistic models of PTx disruption of basal Gi inhibitory tone. Basal Gi activity can arise from constitutively active or autocrine-stimulated Gi-coupled GPCRs (left), or from receptor-independent mechanisms such as non-receptor GEFs or lipid-driven Gi activation (right). PTx-mediated ADP-ribosylation of Gαi eliminates this inhibitory tone, leading to AC disinhibition, elevated cAMP, and downstream PKA/EPAC overactivation. Arrows indicate signaling flow, and the inhibition symbol indicates suppression of AC activity.
In addition, receptors activated by autocrine ligands can maintain continuous Gi signaling. Many cell types employ autocrine loops in which Gi-coupled receptor agonists, such as adenosine [120], prostaglandins [121], or sphingosine-1-phosphate (S1P) [122], are produced locally and can tonically activate Gi pathways. Notably, pro-inflammatory cytokines such as TNF-α can further enhance this basal Gi activity by upregulating S1P production [123], thereby sustaining the activation state of Gi-coupled receptors. Thus, in cells lacking constitutive or autocrine Gi input, PTx has only minimal effects on cAMP levels. If no GPCR engages Gi under resting conditions, there is no persistent inhibitory tone to remove, underscoring that the impact of PTx depends on the presence of such receptor-driven basal signaling.
Secondly, basal Gi activity can also arise independently of ligand-bound receptors through intrinsic regulatory mechanisms of the G protein itself. Gi proteins can be activated by non-receptor guanine nucleotide exchange factors (GEFs), such as the non-receptor GEF Ric-8A [124]. The membrane lipid environment can markedly enhance pre-coupling and binding stability between GPCRs and Gi [125]. Moreover, recent work has demonstrated that not all forms of constitutive Gi activity are functionally equivalent. Using systematic chimeric and mutational analyses, Chung et al. showed that receptor-activated Gαi adopts a distinct and more stable inhibitory conformation toward AC than classical GTPase-deficient mutants [126]. Several recent orphan GPCR structures (e.g., GPR3 and GPR55) further demonstrate that GPCRs can be directly activated by endogenous lipids [127,128].
Other studies have shown that the cellular abundance of Gi has functional consequences. For example, in end-stage human heart failure, upregulation of Gαi2 is correlated with markedly reduced AC activity [129], a phenomenon consistent with the enhanced PTx effect observed when Gαi is overexpressed.
5. Gαi-Dependent but cAMP-Independent Signaling Pathways
PTx not only disrupts the Gi-mediated inhibition of AC but also interferes with a broad array of Gi-dependent yet cAMP-independent signaling cascades. The central event is that PTx, through ADP-ribosylation of Gαi, prevents dissociation of the heterotrimeric G protein complex, thereby blocking the release of Gβγ subunits. Consequently, many signaling pathways initiated by free Gβγ are globally silenced.
These noncanonical Gi pathways are essential for cell growth, migration, and survival, and their loss explains multiple pathological manifestations observed after PTx exposure. The following sections describe how this inhibition impacts key pathways—particularly the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway, the phosphoinositide 3-kinase–protein kinase B–mechanistic target of rapamycin (PI3K–Akt–mTOR) axis, and the phospholipase Cβ–Ca2+–Ras-related protein 1–talin/kindlin (PLCβ–Ca2+–Rap1–talin/kindlin) signaling axis (Figure 7), and how these disruptions are manifested at the physiological level as impaired chemotaxis, leukocytosis, and histamine sensitization.
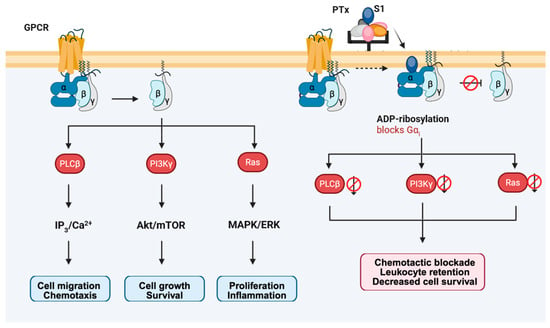
Figure 7.
PTx-mediated blockade of Gi-dependent, cAMP-independent signaling pathways. Under normal conditions, activation of Gi/o-coupled GPCRs leads to dissociation of the heterotrimer and release of Gβγ subunits, which initiate three major signaling modules: PLCβ–IP3/Ca2+–Rap1 for chemotaxis, PI3Kγ–Akt/mTOR for growth and survival, and Ras–MAPK/ERK for proliferation and inflammation. PTx ADP-ribosylates Gαi and prevents heterotrimer dissociation, thereby blocking Gβγ release and globally silencing these noncanonical Gi pathways. As a result, cells exhibit impaired chemotaxis, leukocyte retention, diminished survival signaling, and defective proliferative responses.
5.1. Disruption of MAPK/ERK Signaling
PTx provides a classic example of a Gi-dependent yet cAMP-independent modulation of MAPK/ERK signaling. In normal GPCR signaling, the Gβγ subunits released from activated Gi/o proteins act as the proximal triggers for Ras or PI3K activation, thereby initiating ERK1/2 phosphorylation and downstream proliferative responses [130,131]. This Gi–Gβγ–MAPK coupling has been directly demonstrated for Gi-coupled receptors such as the melatonin MT1R and MT2R, where ERK and c-Jun N-terminal kinase (JNK) activation proceeds via a PTx–sensitive, Gβγ-, Ras/Rac-, and Src-dependent mechanism [132]. This inhibition is particularly evident in GPCR-mediated responses. Agonists targeting α2-adrenergic receptors (α2AR) [133], opioid receptors [134], or chemokine receptors [135] normally activate ERK through Gβγ–Ras or Gβγ–PI3K signaling modules. Pretreatment with PTx abolishes this ERK activation. In contrast, receptors that signal through Gs remain capable of activating ERK even in the presence of PTx [136].
In cardiac fibroblasts, angiotensin II couples the AT1 receptor to Gi proteins, and the released Gβγ subunits sequentially activate Src family tyrosine kinases, the Shc–Grb2–Sos complex, and the Ras/Raf-1 module, thereby driving rapid ERK phosphorylation and promoting DNA synthesis [137]. The insulin-like growth factor-I receptor (IGF-IR) can directly recruit and activate Gi-family heterotrimeric G proteins, leading to the dissociation of Gα from Gβγ; the liberated Gβγ, together with subsequently recruited β-arrestin, functions as a scaffold to facilitate ERK/MAPK activation [138].
5.2. Impact on the PI3K–Akt–mTOR Pathway
The PI3K–Akt–mTOR axis, which is tightly integrated with MAPK signaling, is likewise regulated by Gi–Gβγ inputs. Under normal conditions, activation of GPCRs leads to the release of Gβγ subunits that recruit and activate PI3Kγ [139,140], driving Akt phosphorylation and downstream mTOR signaling to support cellular metabolism, survival, and growth [139,141]. PTx blocks this process by ADP-ribosylating Gαi and preventing Gβγ release, thereby uncoupling these receptors from the PI3K–Akt–mTOR pathway and markedly attenuating intracellular growth and survival signals [142].
This dependence is well documented in neuronal models (e.g., PC12 cells and cortical neurons) in the nervous system. Nerve growth factor (NGF)-driven relief of Tuberous Sclerosis Complex 2 (TSC2) inhibition (via PI3K/Akt) includes a PTx-sensitive Gi/o/Gβγ component, thus positioning Gi/o upstream of the TSC2 gate that controls mTOR signaling output [143]. In PC12 cells, stimulation of the muscarinic M4 receptor (M4R) induces mTOR/p70S6K phosphorylation through a Gi/o-dependent pathway, an effect that is almost completely abolished by PTx treatment [144]. Similarly, PTx attenuates NGF-induced tuberin (TSC2) phosphorylation (most evident at early time points), thereby suppressing mTOR activation; Gβγ scavenging/interference (e.g., transducin) or RGS overexpression produces comparable outcomes. In contrast, epidermal growth factor (EGF) can activate PI3K/Akt independently of Gi, maintaining TSC2 phosphorylation even in the presence of PTx, further underscoring the selective reliance of the NGF–mTOR link on functional Gi signaling. Beyond neuronal NGF signaling, Gi-coupled receptors such as α2-adrenergic and muscarinic receptors can activate the PI3K–Akt pathway via Gβγ release [143].
5.3. Disruption of the PLCβ–Ca2+–Rap1–Talin/Kindlin Pathway
Beyond regulating the MAPK/ERK and PI3K–Akt–mTOR axes, Gi–Gβγ signaling also controls cell polarization, adhesion, and migration through activation of the PLCβ–Ca2+–Rap1–talin/kindlin module [145].
Under physiological conditions, activation of Gi-coupled chemokine receptors such as CXCR1/2 and FPR leads to the release of Gβγ subunits, which stimulate PLCβ2/3 to generate IP3, thereby inducing intracellular Ca2+ mobilization and activating PKC [145,146]. Ca2+ and diacylglycerol (DAG) generated downstream of chemokine receptor–PLC signaling primarily activate calcium and diacylglycerol-regulated guanine nucleotide exchange factor I (CalDAG-GEFI), a Rap1 guanine nucleotide exchange factor, thereby inducing Rap1 activation. Active Rap1 then interacts with Rap1–GTP–interacting adaptor molecule (RIAM), which promotes talin recruitment to CD18. In parallel, kindlin-3 also binds to CD18; together, talin and kindlin-3 induce the high-affinity conformation of β2 integrins, enabling strong ligand binding and supporting leukocyte adhesion and migration [147].
6. Gαi-Independent Activities of PTx: B Oligomer-Mediated Signaling and Immune Modulation
The pathogenic activity of PTx extends far beyond the ADP-ribosyl transferase activity of its S1 subunit. In addition to disrupting both cAMP-dependent and cAMP-independent Gi pathways, the PTx B-oligomer possesses substantial receptor-binding and signal-modulating functions on its own. As a multivalent ligand, the B-oligomer can crosslink multiple receptors on the host cell surface and initiate complex immune signaling even in the absence of catalytic activity [47].
Unlike the enzymatic inactivation mediated by the S1 subunit, the PTx B-oligomer can directly bind to host cell surface receptors and initiate intracellular signaling. As a multivalent ligand, the B-oligomer is capable of crosslinking immune receptors and triggering ligand-independent activation [148]. One of the earliest activities identified was its direct action on the T-cell receptor (TCR) complex. By crosslinking TCRs, the B-oligomer activates the downstream tyrosine kinases Lck and ZAP-70, induces Ca2+ influx, and stimulates transcription factors such as NF-κB and NF-AT, ultimately driving robust secretion of cytokines including IL-2. Consequently, PTx has long been recognized as a potent T-cell mitogen [148,149] (Figure 8).
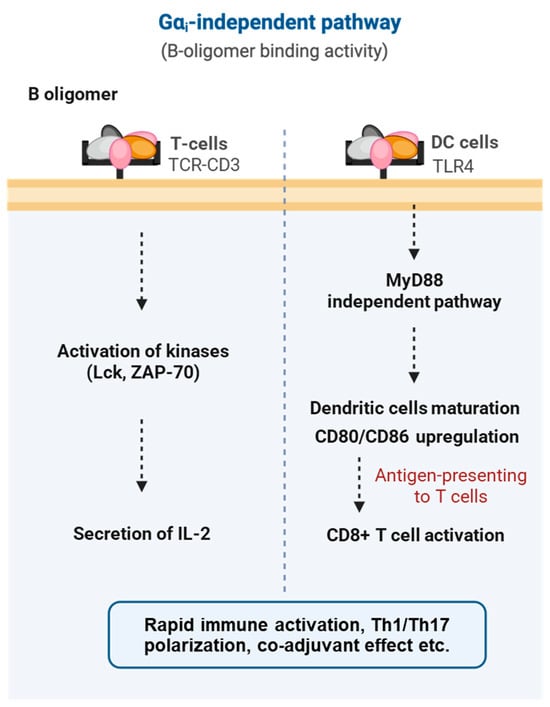
Figure 8.
Gαi-independent, B oligomer-mediated signaling pathways of pertussis toxin. The B-oligomer of pertussis toxin engages multiple immune and structural cell types independently of Gαi ADP-ribosylation. In T cells, B-oligomer binding to the TCR–CD3 complex triggers rapid kinase activation (Lck, ZAP-70) and promotes IL-2 secretion. In dendritic cells, interaction with TLR4 activates a MyD88-independent maturation pathway, leading to CD80/CD86 upregulation, enhanced antigen presentation, and subsequent CD8+ T-cell activation.
Beyond T cells, the B-oligomer also interacts with Toll-like receptor 4 (TLR4) on dendritic cells and macrophages, inducing their maturation through a MyD88-independent pathway [150,151]. This process upregulates costimulatory molecules (CD80/CD86) and promotes the production of IL-6 and IL-12, thereby driving a Th1-skewed immune response [151,152]. Overall, these findings demonstrate that the B-oligomer can directly activate both innate and adaptive immunity without requiring ADP-ribosylation, enhancing antigen presentation and promoting polyclonal T-cell expansion [151,153].
7. Conclusions and Future Perspectives
The pathogenic effects of PTx stem from its canonical and noncanonical mechanisms. The S1 subunit of PTx catalyzes the ADP-ribosylation of Gαi/o, irreversibly uncoupling them from GPCRs. This modification locks Gαi in an inactive, GDP-bound state, preventing the Gi/o family from inhibiting AC activity and leading to elevated intracellular cAMP levels. Excessive cAMP activates PKA and EPAC, disrupting cellular functions and suppressing immune activity, contributing to symptoms like leukocytosis and metabolic dysregulation in pertussis.
PTx also affects cAMP-independent signaling by locking Gαi in the “off” state, which restricts the release of Gβγ subunits that normally mediate various signaling events, including PI3K activation and MAPK cascades. This silencing of Gβγ-dependent processes explains PTx’s inhibition of chemokine-driven leukocyte responses and impairs neutrophil functions, hindering bacterial clearance. Additionally, the B oligomer of PTx serves as an immunomodulatory agent, acting as a T-cell mitogen and engaging TLR4 on dendritic cells and macrophages, contributing to the immune regulatory landscape during infection. PTx’s dual effects—pro-inflammatory via TLR4 activation and immunosuppressive through cAMP elevation—illustrate its complex role in immune modulation.
Constitutive Gi signaling is another key target of PTx. By inactivating Gi/o proteins, PTx removes the tonic inhibitory signaling from Gi-coupled receptors, altering functional states in immune and endocrine cells. This contributes to dysregulation in pertussis, highlighting the active role of Gi proteins in maintaining homeostasis rather than being mere switches.
PTx’s virulence is enhanced in conjunction with ACT, as both toxins elevate cAMP but act through different mechanisms, leading to a coordinated suppression of bacterial clearance and dysregulated inflammation. This synergy highlights the complex interplay between PTx and ACT in modulating host immune responses and bacterial clearance.
Overall, PTx serves as a model for understanding AB-type bacterial toxins and their effects on host signaling pathways. Continued research on PTx will advance efforts against pertussis and improve our understanding of G protein signaling and immune regulation in health and disease.
8. Materials and Methods
8.1. Structural Visualization and Molecular Docking
The structural visualizations in Figure 2 were generated using PyMOL 3.1.4 (Schrödinger, LLC, New York, NY, USA) software. The structures of the full PTx and the S1 subunit in its ATP-activated state (Figure 2A) were visualized using the PDB entry 1PRT and 7SKY, respectively. For the visualization of the NAD+ binding site (Figure 2B), key residues were highlighted using the 7SKY PDB file entry. The structural complex model shown in Figure 2C–E was generated using the AlphaFold web server [76] (accessible at https://alphafoldserver.com; accessed on 1 August 2025). The amino acid sequences for the PTx S1 subunit, Gαi1, Gβ1, and Gγ2, and NAD and GDP molecules were provided as inputs for the complex simulation. From the five top-ranked models generated by the server, the one that showed the highest consistency with reported S1-Gαi interactions [58,75] was manually selected for subsequent visualization and analysis.
8.2. Cell Culture and Transfection
Human embryonic kidney 293 cells (HEK293; ATCC CRL-1573, American Type Culture Collection, Rockville, MD, USA) were maintained under standard conditions. For cAMP assays, cells were seeded into 6-well plates at a density of 3.5 × 105 cells/mL in 2 mL medium per well, yielding 7 × 105 cells/well. On the same day, transient transfection was performed using polyethyleneimine Star™ (PEI; Tocris Bioscience, Bristol, UK). Each well received 400 ng of the cAMP-Glosensor-22F plasmid (Promega, Madison, WI, USA), 400 ng of a single GPCR-encoding plasmid (either β2-AR, D1R, D2R, MT1R, or MT2R; UMR cDNA Resource Center, Rolla, MO, USA), and 400 ng of either Gαi1 or empty vector control (pcDNA3.1).
8.3. cAMP Bioluminescence Assay
Twenty-four hours post transfection, cells were trypsinized and seeded into white-bottom 96-well plates at a density of 4 × 104 cells/well. Pertussis toxin (PTx; List Biological Laboratories, Campbell, CA, USA) was added at a final concentration of 100 ng/mL, and cells were incubated for an additional 24 h. Prior to assay initiation, growth medium was replaced with a labeling medium composed of HBSS buffer supplemented with 20 mM HEPES (pH 7.5) and 2% (v/v) GloSensor™ cAMP Reagent stock solution. Plates were incubated for 30 min at 37 °C, followed by 15–20 min of equilibration at room temperature. Bioluminescence was measured using a SpectraMax microplate reader (Molecular Devices, San Jose, CA, USA) with an emission wavelength of 570 nm.
8.4. Statistical Analysis
Data shown in the figures are the mean ± SD of three independent experiments performed in triplicate. The data sets were analyzed by using multiple unpaired t-tests, two-tailed, with 95% confidence (ns indicates not significant; *, p ≤ 0.05).
8.5. Materials
All experiments were performed using Human embryonic kidney 293 cells (HEK293, ATCC CRL-1573) obtained from the American Type Culture Collection (Rockville, MD, USA). Polyethyleneimine StarTM (PEI) was purchased from Tocris, while plasmids were purchased from UMR cDNA Resource Center (Rolla, MO, USA). Pertussis toxin (PTx) was purchased from List Biological Laboratories (Campbell, CA, USA), and reagents for cell culture were purchased from Thermofisher Scientific (Waltham, MA, USA).
8.6. Use of Generative AI
Generative AI was used to assist in the editing and formatting of this manuscript. No AI tools were used in data analysis or interpretation.
Author Contributions
Conceptualization, Q.T. and Y.H.W.; data curation, Q.T. and Y.H.; experimentation, H.Y.C.; writing Q.T., H.Y.C. and Y.H.W.; supervision, Y.H.W.; funding acquisition, Y.H.W. All authors have read and agreed to the published version of the manuscript.
Funding
This study was partly supported by Research Grants Council of Hong Kong (T13-605/18W), Areas of Excellence Scheme of the University Grants Committee (AoE/M-604/16), and the Innovation and Technology Commission of the HKSAR Government ([INNOHK18SC01 and ITCPD/17-9). Peking University and the National Natural Science Foundation of China.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
We thank Anthony Chan for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AB | AB-type bacterial toxin |
| AC | Adenylyl cyclase |
| ACT | Adenylate cyclase toxin |
| ADP | Adenosine diphosphate |
| AoE | Areas of Excellence Scheme |
| ATP | Adenosine triphosphate |
| AT1R | Angiotensin II type 1 receptor |
| β2-AR | Beta-2 adrenergic receptor |
| β-NAD+ | Beta-nicotinamide adenine dinucleotide |
| BvgAS | Bordetella virulence gene regulatory system |
| Ca2+ | Calcium ion |
| CalDAG-GEFI | Calcium and diacylglycerol-regulated guanine nucleotide exchange factor I |
| cAMP | Cyclic adenosine monophosphate |
| CCR1 | C-C chemokine receptor 1 |
| CR3 | Complement receptor 3 |
| CTX | Cholera toxin |
| DAG | Diacylglycerol |
| D1R | Dopamine D1 receptor |
| D2R | Dopamine D2 receptor |
| DAP | Diaminopimelic acid |
| DNT | Dermonecrotic toxin |
| DT | Diphtheria toxin |
| EGF | Epidermal growth factor |
| EPAC | Exchange protein directly activated by cAMP |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| ETA | Exotoxin A |
| FPR | Formyl peptide receptor |
| GlcNAc | N-acetylglucosamine |
| Gα | G protein alpha subunit |
| Gαi | Alpha subunit of inhibitory G protein |
| Gβγ | G protein beta-gamma complex |
| GEF | Guanine nucleotide exchange factor |
| Gi | Inhibitory G protein |
| Gi/o | Inhibitory G protein family (Gi and Go) |
| GPCR | G protein-coupled receptor |
| GPR | G protein-coupled receptor |
| Gs | Stimulatory G protein |
| Gt | G protein transducin |
| HBSS | Hank’s balanced salt solution |
| HEK293 | Human embryonic kidney 293 cells |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| HSF | Histamine-sensitizing factor |
| IAP | Islet-activating protein |
| IGF-IR | Insulin-like growth factor I receptor |
| IL | Interleukin |
| IP3 | Inositol 1,4,5-trisphosphate |
| JNK | c-Jun N-terminal kinase |
| kDa | Kilodalton |
| LOS | Lipooligosaccharide |
| LPF | Lymphocytosis-promoting factor |
| MAPK | Mitogen-activated protein kinase |
| MD-2 | Myeloid differentiation factor 2 |
| mTOR | Mechanistic target of rapamycin |
| MT1R | Melatonin receptor 1 |
| MT2R | Melatonin receptor 2 |
| MurNAc | N-acetylmuramic acid |
| MyD88 | Myeloid differentiation primary response protein 88 |
| NAD+ | Nicotinamide adenine dinucleotide |
| NF-AT | Nuclear factor of activated T cells |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGF | Nerve growth factor |
| NOD1 | Nucleotide-binding oligomerization domain 1 |
| PBS | Phosphate-buffered saline |
| PC12 | Pheochromocytoma 12 cells |
| PEI | Polyethyleneimine |
| PI3K | Phosphoinositide 3-kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PLCβ | Phospholipase C beta |
| PTx | Pertussis toxin |
| Q/EXE motif | Glutamine-X-glutamic acid conserved sequence pattern where X represents any amino acid |
| Rap1 | Ras-related protein 1 |
| RIAM | Rap1–GTP–interacting adaptor molecule |
| RLU | Relative luminescence unit |
| RTX | Repeats-in-toxin |
| SD | Standard deviation |
| STS motif | Serine-threonine-serine sequence motif |
| S1 | Catalytic subunit of pertussis toxin |
| S1P | Sphingosine-1-phosphate |
| Src | Proto-oncogene tyrosine-protein kinase Src |
| TCR | T-cell receptor |
| TCT | Tracheal cytotoxin |
| Th1 | T helper type 1 |
| Th17 | T helper type 17 |
| TLR | Toll-like receptor |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor alpha |
| TRIF | TIR domain-containing adaptor inducing interferon-β |
| TSC2 | Tuberous sclerosis complex 2 |
| VAAS | Vasoactive amine sensitization |
| WT | Wild type |
References
- Mattoo, S.; Cherry, J.D. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin. Microbiol. Rev. 2005, 18, 326–382. [Google Scholar] [CrossRef]
- Diavatopoulos, D.A.; Cummings, C.A.; Schouls, L.M.; Brinig, M.M.; Relman, D.A.; Mooi, F.R. Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. PLoS Pathog. 2005, 1, e45. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.B.; Vaughan, J.H.; Tan, E.M. Immunologic and biochemical properties of the histamine-sensitizing factor from Bordetella pertussis. J. Immunol. 1975, 114, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Morse, S.I. Lymphocytosis-promoting factor of Bordetella pertussis: Isolation, characterization, and biological activity. J. Infect. Dis. 1977, 136, S234–S238. [Google Scholar] [CrossRef]
- Bokoch, G.M.; Katada, T.; Northup, J.K.; Hewlett, E.L.; Gilman, A.G. Identification of the predominant substrate for ADP-ribosylation by islet activating protein. J. Biol. Chem. 1983, 258, 2072–2075. [Google Scholar] [CrossRef]
- Munoz, J.J.; Arai, H.; Bergman, R.K.; Sadowski, P.L. Biological activities of crystalline pertussigen from Bordetella pertussis. Infect. Immun. 1981, 33, 820–826. [Google Scholar] [CrossRef]
- Arai, H.; Munoz, J.J. Crystallization of pertussigen from Bordetella pertussis. Infect. Immun. 1981, 31, 495–499. [Google Scholar] [CrossRef]
- Katada, T.; Ui, M. Islet-activating protein. Enhanced insulin secretion and cyclic AMP accumulation in pancreatic islets due to activation of native calcium ionophores. J. Biol. Chem. 1979, 254, 469–479. [Google Scholar] [CrossRef]
- Katada, T. The Inhibitory G protein Gi identified as pertussis toxin-catalyzed ADP-ribosylation. Biol. Pharm. Bull. 2012, 35, 2103–2111. [Google Scholar] [CrossRef]
- Scanlon, K.; Skerry, C.; Carbonetti, N. Role of major toxin virulence factors in pertussis infection and disease pathogenesis. Adv. Exp. Med. Biol. 2019, 1183, 35. [Google Scholar] [CrossRef] [PubMed]
- Carbonetti, N.H.; Artamonova, G.V.; Andreasen, C.; Bushar, N. Pertussis toxin and adenylate cyclase toxin provide a one-two punch for establishment of Bordetella pertussis infection of the respiratory tract. Infect. Immun. 2005, 73, 2698–2703. [Google Scholar] [CrossRef]
- Carbonetti, N.H. Pertussis toxin and adenylate cyclase toxin: Key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 2010, 5, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Tyler, A.N.; Marshall, G.R.; Goldman, W.E. Tracheal cytotoxin structural requirements for respiratory epithelial damage in pertussis. Mol. Microbiol. 1995, 16, 733–743. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Fukui, A.; Kashimoto, T.; Nagao, K.; Oka, K.; Miyake, M.; Horiguchi, Y. Bordetella dermonecrotic toxin undergoes proteolytic processing to be translocated from a dynamin-related endosome into the cytoplasm in an acidification-independent manner. J. Biol. Chem. 2004, 279, 2866–2872. [Google Scholar] [CrossRef] [PubMed]
- Fedele, G.; Nasso, M.; Spensieri, F.; Palazzo, R.; Frasca, L.; Watanabe, M.; Ausiello, C.M. Lipopolysaccharides from Bordetella pertussis and Bordetella parapertussis differently modulate human dendritic cell functions resulting in divergent prevalence of Th17-polarized responses. J. Immunol. 2008, 181, 208–216. [Google Scholar] [CrossRef]
- Novak, J.; Cerny, O.; Osickova, A.; Linhartova, I.; Masin, J.; Bumba, L.; Sebo, P.; Osicka, R. Structure-function relationships underlying the capacity of Bordetella adenylate cyclase toxin to disarm host phagocytes. Toxins 2017, 9, 300. [Google Scholar] [CrossRef]
- Ahmad, J.N.; Holubova, J.; Benada, O.; Kofronova, O.; Stehlik, L.; Vasakova, M.; Sebo, P. Bordetella adenylate cyclase toxin inhibits monocyte-to-macrophage transition and dedifferentiates human alveolar macrophages into monocyte-like cells. mBio 2019, 10, e01743-19. [Google Scholar] [CrossRef]
- Veneziano, R.; Rossi, C.; Chenal, A.; Devoisselle, J.-M.; Ladant, D.; Chopineau, J. Bordetella pertussis adenylate cyclase toxin translocation across a tethered lipid bilayer. Proc. Natl. Acad. Sci. USA 2013, 110, 20473–20478. [Google Scholar] [CrossRef]
- Bagley, K.C.; Abdelwahab, S.F.; Tuskan, R.G.; Fouts, T.R.; Lewis, G.K. Pertussis toxin and the adenylate cyclase toxin from Bordetella pertussis activate human monocyte-derived dendritic cells and dominantly Inhibit cytokine production through a cAMP-dependent pathway. J. Leukoc. Biol. 2002, 72, 962–969. [Google Scholar] [CrossRef]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic AMP: Master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Wolber, A.R.; McKay, L.S.; Mote, K.B.; Johnson, R.M.; Inatsuka, C.S.; Cotter, P.A. Nuanced differences in adenylate cyclase toxin production, acylation, and secretion may contribute to the evolution of virulence in Bordetella species. mBio 2025, 16, e0108225. [Google Scholar] [CrossRef] [PubMed]
- Goldman, W.E.; Cookson, B.T. Structure and functions of the Bordetella tracheal cytotoxin. Tokai J. Exp. Clin. Med. 1988, 13, 187–191. [Google Scholar]
- Kessie, D.K.; Lodes, N.; Oberwinkler, H.; Goldman, W.E.; Walles, T.; Steinke, M.; Gross, R. Activity of tracheal cytotoxin of Bordetella pertussis in a human tracheobronchial 3D tissue model. Front. Cell. Infect. Microbiol. 2021, 10, 614994. [Google Scholar] [CrossRef]
- Masuda, M.; Minami, M.; Shime, H.; Matsuzawa, T.; Horiguchi, Y. In vivo modifications of small GTPase Rac and Cdc42 by Bordetella dermonecrotic toxin. Infect. Immun. 2002, 70, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Teruya, S.; Hiramatsu, Y.; Nakamura, K.; Fukui-Miyazaki, A.; Tsukamoto, K.; Shinoda, N.; Motooka, D.; Nakamura, S.; Ishigaki, K.; Shinzawa, N.; et al. Bordetella dermonecrotic toxin is a neurotropic virulence factor that uses CaV3.1 as the cell surface receptor. mBio 2020, 11, e03146-19. [Google Scholar] [CrossRef]
- Gorman, A.; Golovanov, A.P. Lipopolysaccharide structure and the phenomenon of low endotoxin recovery. Eur. J. Pharm. Biopharm. 2022, 180, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Nogimori, K.; Murai, S.; Yajima, M.; Ito, K.; Katada, T.; Ui, M.; Ishii, S. Subunit structure of islet-activating protein, pertussis toxin, in conformity with the A-B model. Biochemistry 1982, 21, 5516–5522. [Google Scholar] [CrossRef]
- Stein, P.E.; Boodhoo, A.; Armstrong, G.D.; Cockle, S.A.; Klein, M.H.; Read, R.J. The crystal structure of pertussis toxin. Structure 1994, 2, 45–57. [Google Scholar] [CrossRef]
- Brennan, M.J.; David, J.L.; Kenimer, J.G.; Manclark, C.R. Lectin-like binding of pertussis toxin to a 165-kilodalton chinese hamster ovary cell glycoprotein. J. Biol. Chem. 1988, 263, 4895–4899. [Google Scholar] [CrossRef]
- West, R.E.; Moss, J.; Vaughan, M.; Liu, T.; Liu, T.Y. Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site. J. Biol. Chem. 1985, 260, 14428–14430. [Google Scholar] [CrossRef]
- Katada, T.; Ui, M. Direct modification of the membrane adenylate cyclase system by islet-activating protein due to ADP-ribosylation of a membrane protein. Proc. Natl. Acad. Sci. USA 1982, 79, 3129–3133. [Google Scholar] [CrossRef]
- Katada, T.; Ui, M. ADP ribosylation of the specific membrane protein of C6 cells by islet-activating protein associated with modification of adenylate cyclase activity. J. Biol. Chem. 1982, 257, 7210–7216. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, S.; Imagawa, Y.; Tokunaga, E.; Ohtomo, N. Increase in intradermal vascular permeability caused by pertussis toxin from Bordetella pertussis. Microbiol. Immunol. 1987, 31, 531–539. [Google Scholar] [CrossRef]
- Glaser, P.; Sakamoto, H.; Bellalou, J.; Ullmann, A.; Danchin, A. Secretion of cyclolysin, the calmodulin-sensitive adenylate cyclase-haemolysin bifunctional protein of Bordetella pertussis. EMBO J. 1988, 7, 3997–4004. [Google Scholar] [CrossRef]
- Gray, M.; Szabo, G.; Otero, A.S.; Gray, L.; Hewlett, E. Distinct mechanisms for K+ efflux, intoxication, and hemolysis by Bordetella pertussis AC toxin. J. Biol. Chem. 1998, 273, 18260–18267. [Google Scholar] [CrossRef]
- Hewlett, E.L.; Donato, G.M.; Gray, M.C. Macrophage cytotoxicity produced by adenylate cyclase toxin from Bordetella pertussis: More than just making cyclic AMP! Mol. Microbiol. 2006, 59, 447–459. [Google Scholar] [CrossRef]
- Vojtova, J.; Kamanova, J.; Sebo, P. Bordetella adenylate cyclase toxin: A swift saboteur of host defense. Curr. Opin. Microbiol. 2006, 9, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Collier, J.L.; Kolodziej, E.W.; Marshall, G.R.; Goldman, W.E. Bordetella pertussis tracheal cytotoxin and other muramyl peptides: Distinct structure-activity relationships for respiratory epithelial cytopathology. Proc. Natl. Acad. Sci. USA 1993, 90, 2365–2369. [Google Scholar] [CrossRef]
- Cowell, J.L.; Hewlett, E.L.; Manclark, C.R. Intracellular localization of the dermonecrotic toxin of Bordetella Pertussis. Infect. Immun. 1979, 25, 896–901. [Google Scholar] [CrossRef]
- Horiguchi, Y.; Inoue, N.; Masuda, M.; Kashimoto, T.; Katahira, J.; Sugimoto, N.; Matsuda, M. Bordetella bronchiseptica dermonecrotizing toxin induces reorganization of actin stress fibers through deamidation of Gln-63 of the GTP-binding protein Rho. Proc. Natl. Acad. Sci. USA 1997, 94, 11623–11626. [Google Scholar] [CrossRef] [PubMed]
- Marr, N.; Hajjar, A.M.; Shah, N.R.; Novikov, A.; Yam, C.S.; Caroff, M.; Fernandez, R.C. Substitution of the Bordetella pertussis lipid a phosphate groups with glucosamine is required for robust NF-κB activation and release of proinflammatory cytokines in cells expressing human but not murine toll-like receptor 4-MD-2-CD14. Infect. Immun. 2010, 78, 2060–2069. [Google Scholar] [CrossRef]
- Maeshima, N.; Fernandez, R.C. Recognition of lipid a variants by the TLR4-MD-2 receptor complex. Front. Cell. Infect. Microbiol. 2013, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Manivannan, K.; Mohamed, Y.F.; Fernandez, R.C. Determining the Bordetella LPS structural features that influence TLR4 downstream signaling. Front. Microbiol. 2025, 16, 1540534. [Google Scholar] [CrossRef]
- Caroff, M.; Brisson, J.; Martin, A.; Karibian, D. Structure of the Bordetella pertussis 1414 endotoxin. FEBS Lett. 2000, 477, 8–14. [Google Scholar] [CrossRef]
- Caroff, M.; Aussel, L.; Zarrouk, H.; Martin, A.; Richards, J.C.; Thérisod, H.; Perry, M.B.; Karibian, D. Structural variability and originality of the Bordetella endotoxins. J. Endotoxin Res. 2001, 7, 63–68. [Google Scholar]
- Katada, T.; Tamura, M.; Ui, M. The a protomer of islet-activating protein, pertussis toxin, as an active peptide catalyzing ADP-ribosylation of a membrane protein. Arch. Biochem. Biophys. 1983, 224, 290–298. [Google Scholar] [CrossRef]
- Tamura, M.; Nogimori, K.; Yajima, M.; Ase, K.; Ui, M. A role of the B-oligomer moiety of islet-activating protein, pertussis toxin, in development of the biological effects on intact cells. J. Biol. Chem. 1983, 258, 6756–6761. [Google Scholar] [CrossRef] [PubMed]
- Plaut, R.D.; Carbonetti, N.H. Retrograde transport of pertussis toxin in the mammalian cell. Cell. Microbiol. 2008, 10, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Hazes, B.; Boodhoo, A.; Cockle, S.A.; Read, R.J. Crystal structure of the pertussis toxin–ATP complex: A molecular sensor. J. Mol. Biol. 1996, 258, 661–671. [Google Scholar] [CrossRef]
- Banerjee, T.; Cilenti, L.; Taylor, M.; Showman, A.; Tatulian, S.A.; Teter, K. Thermal unfolding of the pertussis toxin S1 subunit facilitates toxin translocation to the cytosol by the mechanism of endoplasmic reticulum-associated degradation. Infect. Immun. 2016, 84, 3388. [Google Scholar] [CrossRef]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef]
- Bi, M.; Wang, X.; Wang, J.; Xu, J.; Sun, W.; Adediwura, V.A.; Miao, Y.; Cheng, Y.; Ye, L. Structure and function of a near fully-activated intermediate GPCR-Gαβγ complex. Nat. Commun. 2025, 16, 1100. [Google Scholar] [CrossRef]
- Hoshino, S.; Kikkawa, S.; Takahashi, K.; Itoh, H.; Kaziro, Y.; Kawasaki, H.; Suzuki, K.; Katada, T.; Ui, M. Identification of sites for alkylation by N-ethylmaleimide and pertussis toxin-catalyzed ADP-ribosylation on GTP-binding proteins. FEBS Lett. 1990, 276, 227–231. [Google Scholar] [CrossRef]
- Neer, E.J.; Lok, J.M.; Wolf, L.G. Purification and properties of the inhibitory guanine nucleotide regulatory unit of brain adenylate cyclase. J. Biol. Chem. 1984, 259, 14222–14229. [Google Scholar] [CrossRef]
- Huff, R.M.; Neer, E.J. Subunit interactions of native and ADP-ribosylated α 39 and α 41, two guanine nucleotide-binding proteins from bovine cerebral cortex. J. Biol. Chem. 1986, 261, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Katada, T.; Oinuma, M.; Ui, M. Two guanine nucleotide-binding proteins in rat brain serving as the specific substrate of islet-activating protein, pertussis toxin. Interaction of the α subunits with βγ Subunits in development of their biological activities. J. Biol. Chem. 1986, 261, 8182–8191. [Google Scholar] [CrossRef] [PubMed]
- Katada, T.; Oinuma, M.; Ui, M. Mechanisms for inhibition of the catalytic activity of adenylate cyclase by the guanine nucleotide-binding proteins serving as the substrate of islet-activating protein, pertussis toxin. J. Biol. Chem. 1986, 261, 5215–5221. [Google Scholar] [CrossRef] [PubMed]
- Sakari, M.; Tran, M.T.; Rossjohn, J.; Pulliainen, A.T.; Beddoe, T.; Littler, D.R. Crystal structures of pertussis toxin with NAD+ and analogs provide structural insights into the mechanism of its cytosolic ADP-ribosylation activity. J. Biol. Chem. 2022, 298, 101892. [Google Scholar] [CrossRef]
- Clark, C.G.; Armstrong, G.D. Lymphocyte receptors for pertussis toxin. Infect. Immun. 1990, 58, 3840–3846. [Google Scholar] [CrossRef]
- Zhang, R.G.; Scott, D.L.; Westbrook, M.L.; Nance, S.; Spangler, B.D.; Shipley, G.G.; Westbrook, E.M. The three-dimensional crystal structure of cholera toxin. J. Mol. Biol. 1995, 251, 563–573. [Google Scholar] [CrossRef]
- Allured, V.S.; Collier, R.J.; Carroll, S.F.; McKay, D.B. Structure of exotoxin a of Pseudomonas aeruginosa at 3.0-angstrom resolution. Proc. Natl. Acad. Sci. USA 1986, 83, 1320–1324. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. [Google Scholar] [CrossRef]
- Burnette, W.N.; Cieplak, W.; Mar, V.L.; Kaljot, K.T.; Sato, H.; Keith, J.M. Pertussis toxin S1 mutant with reduced enzyme activity and a conserved protective epitope. Science 1988, 242, 72–74. [Google Scholar] [CrossRef]
- Pizza, M.; Bartoloni, A.; Prugnola, A.; Silvestri, S.; Rappuoli, R. Subunit S1 of pertussis toxin: Mapping of the regions essential for ADP-ribosyltransferase activity. Proc. Natl. Acad. Sci. USA 1988, 85, 7521–7525. [Google Scholar] [CrossRef]
- Barbieri, J.T.; Mende-Mueller, L.M.; Rappuoli, R.; Collier, R.J. Photolabeling of Glu-129 of the S-1 Subunit of pertussis toxin with NAD. Infect. Immun. 1989, 57, 3549–3554. [Google Scholar] [CrossRef]
- Antoine, R.; Tallett, A.; van Heyningen, S.; Locht, C. Evidence for a catalytic role of glutamic acid 129 in the NAD-glycohydrolase activity of the pertussis toxin S1 subunit. J. Biol. Chem. 1993, 268, 24149–24155. [Google Scholar] [CrossRef]
- Antoine, R.; Locht, C. The NAD-glycohydrolase activity of the pertussis toxin S1 subunit. Involvement of the catalytic HIS-35 residue. J. Biol. Chem. 1994, 269, 6450–6457. [Google Scholar] [CrossRef]
- Xu, Y.; Barbançon-Finck, V.; Barbieri, J.T. Role of histidine 35 of the S1 subunit of pertussis toxin in the ADP-ribosylation of transducin. J. Biol. Chem. 1994, 269, 9993–9999. [Google Scholar] [CrossRef]
- Domenighini, M.; Rappuoli, R. Three conserved consensus sequences identify the NAD-binding site of ADP-ribosylating enzymes, expressed by eukaryotes, bacteria and T-even bacteriophages. Mol. Microbiol. 1996, 21, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Keith, J.M.; Sato, H.; Sato, Y. Construction and characterization of genetically inactivated pertussis toxin. Dev. Biol. Stand. 1991, 73, 63–73. [Google Scholar] [PubMed]
- Pizza, M.; Covacci, A.; Bartoloni, A.; Perugini, M.; Nencioni, L.; De Magistris, M.T.; Villa, L.; Nucci, D.; Manetti, R.; Bugnoli, M.; et al. Mutants of pertussis toxin suitable for vaccine development. Science 1989, 246, 497–500. [Google Scholar] [CrossRef]
- Cortina, G.; Barbieri, J.T. Role of tryptophan 26 in the NAD glycohydrolase reaction of the S-1 subunit of pertussis toxin. J. Biol. Chem. 1989, 264, 17322–17328. [Google Scholar] [CrossRef] [PubMed]
- Scheuring, J.; Berti, P.J.; Schramm, V.L. Transition-state structure for the ADP-ribosylation of recombinant Giα1 subunits by pertussis toxin. Biochemistry 1998, 37, 2748–2758. [Google Scholar] [CrossRef] [PubMed]
- Sakari, M.; Bhadane, R.; Kumar, S.; Azevedo, R.; Malakoutikhah, M.; Masoumi, A.; Littler, D.R.; Härmä, H.; Kopra, K.; Pulliainen, A.T. ADP-ribosyltransferase-based biocatalysis of nonhydrolyzable NAD+ analogs. J. Biol. Chem. 2025, 301, 108106. [Google Scholar] [CrossRef] [PubMed]
- Krueger, K.M.; Barbieri, J.T. Assignment of functional domains involved in ADP-ribosylation and B-oligomer binding within the carboxyl terminus of the S1 subunit of pertussis toxin. Infect. Immun. 1994, 62, 2071–2078. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Locht, C.; Lobet, Y.; Feron, C.; Cieplak, W.; Keith, J.M. The role of cysteine 41 in the enzymatic activities of the pertussis toxin S1 subunit as investigated by site-directed mutagenesis. J. Biol. Chem. 1990, 265, 4552–4559. [Google Scholar] [CrossRef]
- Moss, J.; Stanley, S.J.; Burns, D.L.; Hsia, J.A.; Yost, D.A.; Myers, G.A.; Hewlett, E.L. Activation by thiol of the latent NAD glycohydrolase and ADP-ribosyltransferase activities of Bordetella Pertussis toxin (islet-activating protein). J. Biol. Chem. 1983, 258, 11879–11882. [Google Scholar] [CrossRef]
- Montminy, M. Transcriptional regulation by cyclic AMP. Annu. Rev. Biochem. 1997, 66, 807–822. [Google Scholar] [CrossRef]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Furman, B.L.; Sidey, F.M.; Wardlaw, A.C. Role of insulin in the hypoglycaemic effect of sublethal Bordetella pertussis infection in mice. Br. J. Exp. Pathol. 1986, 67, 305–312. [Google Scholar] [PubMed]
- Diehl, S.A.; McElvany, B.; Noubade, R.; Seeberger, N.; Harding, B.; Spach, K.; Teuscher, C. G proteins Gαi1/3 are critical targets for Bordetella pertussis toxin-induced vasoactive amine sensitization. Infect. Immun. 2014, 82, 773. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, V.; Hegerle, N.; Strati, F.; Njamkepo, E.; Guiso, N. New data on vaccine antigen deficient Bordetella pertussis isolates. Vaccines 2015, 3, 751–770. [Google Scholar] [CrossRef]
- Luu, L.D.W.; Octavia, S.; Zhong, L.; Raftery, M.J.; Sintchenko, V.; Lan, R. Comparison of the whole cell proteome and secretome of epidemic Bordetella pertussis strains from the 2008-2012 australian epidemic under sulfate-modulating conditions. Front. Microbiol. 2018, 9, 2851. [Google Scholar] [CrossRef]
- Cyr, T.; Menzies, A.J.; Calver, J.; Whitehouse, L.W. A quantitative analysis for the ADP-ribosylation activity of pertussis toxin: An enzymatic-HPLC coupled assay applicable to formulated whole cell and acellular pertussis vaccine products. Biol. J. Int. Assoc. Biol. Stand. 2001, 29, 81–95. [Google Scholar] [CrossRef]
- Hoonakker, M.E.; Ruiterkamp, N.; Hendriksen, C.F.M. The cAMP Assay: A functional in vitro alternative to the in vivo histamine sensitization test. Vaccine 2010, 28, 1347–1352. [Google Scholar] [CrossRef]
- Higgs, R.; Higgins, S.C.; Ross, P.J.; Mills, K.H.G. Immunity to the respiratory pathogen Bordetella pertussis. Mucosal Immunol. 2012, 5, 485–500. [Google Scholar] [CrossRef]
- Gueirard, P.; Druilhe, A.; Pretolani, M.; Guiso, N. Role of adenylate cyclase-hemolysin in alveolar macrophage apoptosis during Bordetella pertussis infection in vivo. Infect. Immun. 1998, 66, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Confer, D.L.; Eaton, J.W. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 1982, 217, 948–950. [Google Scholar] [CrossRef]
- Pearson, R.D.; Symes, P.; Conboy, M.; Weiss, A.A.; Hewlett, E.L. Inhibition of monocyte oxidative responses by Bordetella pertussis adenylate cyclase toxin. J. Immunol. 1987, 139, 2749–2754. [Google Scholar] [CrossRef]
- Weingart, C.L.; Weiss, A.A. Bordetella pertussis virulence factors affect phagocytosis by human neutrophils. Infect. Immun. 2000, 68, 1735–1739. [Google Scholar] [CrossRef]
- Pauza, C.D.; Hinds, P.W.; Yin, C.; McKechnie, T.S.; Hinds, S.B.; Salvato, M.S. The lymphocytosis-promoting agent pertussis toxin affects virus burden and lymphocyte distribution in the SIV-infected rhesus macaque. AIDS Res. Hum. Retroviruses 1997, 13, 87–95. [Google Scholar] [CrossRef]
- Heininger, U.; Stehr, K.; Schmitt-Grohé, S.; Lorenz, C.; Rost, R.; Christenson, P.D.; Uberall, M.; Cherry, J.D. Clinical characteristics of illness caused by Bordetella Parapertussis compared with illness caused by Bordetella pertussis. Pediatr. Infect. Dis. J. 1994, 13, 306–309. [Google Scholar] [CrossRef]
- Ishida, S.; Kurokawa, M.; Asakawa, S.; Iwasa, S. A sensitive assay method for the histamine-sensitizing factor using change in rectal temperature of mice after histamine challenge as a response. J. Biol. Stand. 1979, 7, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Eby, J.C.; Gray, M.C.; Warfel, J.M.; Paddock, C.D.; Jones, T.F.; Day, S.R.; Bowden, J.; Poulter, M.D.; Donato, G.M.; Merkel, T.J.; et al. Quantification of the adenylate cyclase toxin of Bordetella pertussis in vitro and during respiratory infection. Infect. Immun. 2013, 81, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Chenal, A.; Ladant, D. Bioengineering of Bordetella pertussis adenylate cyclase toxin for antigen-delivery and immunotherapy. Toxins 2018, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Cerny, O.; Anderson, K.E.; Stephens, L.R.; Hawkins, P.T.; Sebo, P. cAMP signaling of adenylate cyclase toxin blocks the oxidative burst of neutrophils through Epac-mediated inhibition of phospholipase C activity. J. Immunol. 2017, 198, 1285–1296. [Google Scholar] [CrossRef]
- Kamanova, J.; Kofronova, O.; Masin, J.; Genth, H.; Vojtova, J.; Linhartova, I.; Benada, O.; Just, I.; Sebo, P. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J. Immunol. 2008, 181, 5587–5597. [Google Scholar] [CrossRef]
- Khelef, N.; Guiso, N. Induction of macrophage apoptosis by Bordetella pertussis adenylate cyclase-hemolysin. FEMS Microbiol. Lett. 1995, 134, 27–32. [Google Scholar] [CrossRef]
- Takaichi, K.; Kurokawa, K. Inhibitory guanosine triphosphate-binding protein-mediated regulation of vasopressin action in isolated single medullary tubules of mouse kidney. J. Clin. Investig. 1988, 82, 1437–1444. [Google Scholar] [CrossRef]
- Rambow-Larsen, A.A.; Weiss, A.A. Temporal expression of pertussis toxin and Ptl secretion proteins by Bordetella pertussis. J. Bacteriol. 2004, 186, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Paramonov, V.M.; Sahlgren, C.; Rivero-Müller, A.; Pulliainen, A.T. iGIST-a kinetic bioassay for pertussis toxin based on Its effect on inhibitory GPCR signaling. ACS Sens. 2020, 5, 3438–3448. [Google Scholar] [CrossRef]
- Carbonetti, N.H.; Artamonova, G.V.; Mays, R.M.; Worthington, Z.E.V. Pertussis toxin plays an early role in respiratory tract colonization by Bordetella Pertussis. Infect. Immun. 2003, 71, 6358–6366. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.T.; Carcillo, J.A.; Shanley, T.P.; Wessel, D.L.; Clark, A.; Holubkov, R.; Meert, K.L.; Newth, C.J.L.; Berg, R.A.; Heidemann, S.; et al. Critical pertussis illness in children: A multicenter prospective cohort study. Pediatr. Crit. Care Med. 2013, 14, 356–365. [Google Scholar] [CrossRef]
- Namachivayam, P.; Shimizu, K.; Butt, W. Pertussis: Severe clinical presentation in pediatric intensive care and its relation to outcome. Pediatr. Crit. Care Med. 2007, 8, 207–211. [Google Scholar] [CrossRef]
- Nguyen, A.W.; Wagner, E.K.; Laber, J.R.; Goodfield, L.L.; Smallridge, W.E.; Harvill, E.T.; Papin, J.F.; Wolf, R.F.; Padlan, E.A.; Bristol, A.; et al. A cocktail of humanized anti-pertussis toxin antibodies limits disease in murine and baboon models of whooping cough. Sci. Transl. Med. 2015, 7, 316ra195. [Google Scholar] [CrossRef]
- Ernst, K. Novel strategies to inhibit pertussis toxin. Toxins 2022, 14, 187. [Google Scholar] [CrossRef]
- Rowlands, H.E.; Goldman, A.P.; Harrington, K.; Karimova, A.; Brierley, J.; Cross, N.; Skellett, S.; Peters, M.J. Impact of rapid leukodepletion on the outcome of severe clinical pertussis in young infants. Pediatrics 2010, 126, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Vleeming, W.; Hendriksen, C.F.M.; van de Kuil, A.; van den Hout, J.W.E.; de Wildt, D.J. Mepyramine but not cimetidine or clobenpropit blocks pertussis toxin-induced histamine sensitization in rats. Br. J. Pharmacol. 2000, 129, 1801–1807. [Google Scholar] [CrossRef]
- Weingart, C.L.; Mobberley-Schuman, P.S.; Hewlett, E.L.; Gray, M.C.; Weiss, A.A. Neutralizing antibodies to adenylate cyclase toxin promote phagocytosis of Bordetella Pertussis by human neutrophils. Infect. Immun. 2000, 68, 7152–7155. [Google Scholar] [CrossRef]
- Dunne, A.; Mielke, L.A.; Allen, A.C.; Sutton, C.E.; Higgs, R.; Cunningham, C.C.; Higgins, S.C.; Mills, K.H.G. A novel TLR2 agonist from Bordetella pertussis is a potent adjuvant that promotes protective immunity with an acellular pertussis vaccine. Mucosal Immunol. 2015, 8, 607–617. [Google Scholar] [CrossRef]
- Asokanathan, C.; Corbel, M.; Xing, D. A CpG-containing oligodeoxynucleotide adjuvant for acellular pertussis vaccine improves the protective response against Bordetella pertussis. Hum. Vaccines Immunother. 2013, 9, 325–331. [Google Scholar] [CrossRef]
- Seifert, R.; Wenzel-Seifert, K. Constitutive activity of G-protein-coupled receptors: Cause of disease and common property of wild-type receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2002, 366, 381–416. [Google Scholar] [CrossRef]
- Roka, F.; Brydon, L.; Waldhoer, M.; Strosberg, A.D.; Freissmuth, M.; Jockers, R.; Nanoff, C. Tight association of the human Mel1a-melatonin receptor and Gi: Precoupling and constitutive activity. Mol. Pharmacol. 1999, 56, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.L.; Steurer, M.A.; Aronstam, R.S. Constitutive activity among orphan class-A G protein coupled receptors. PLoS ONE 2015, 10, e0138463. [Google Scholar] [CrossRef]
- Gilliland, C.T.; Salanga, C.L.; Kawamura, T.; Trejo, J.; Handel, T.M. The chemokine receptor CCR1 is constitutively active, which leads to G protein-independent, β-arrestin-mediated internalization. J. Biol. Chem. 2013, 288, 32194–32210. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.Y.; Tsim, S.T.; Wong, Y.H. Stimulation of cAMP accumulation by the cloned xenopus melatonin receptor through Gi and Gz proteins. FEBS Lett. 1995, 372, 99–102. [Google Scholar] [CrossRef]
- Hase, M.; Yokomizo, T.; Shimizu, T.; Nakamura, M. Characterization of an orphan G protein-coupled receptor, GPR20, that constitutively activates Gi proteins. J. Biol. Chem. 2008, 283, 12747–12755. [Google Scholar] [CrossRef] [PubMed]
- Tse, L.H.; Cheung, S.T.; Lee, S.; Wong, Y.H. Real-time determination of intracellular cAMP reveals functional coupling of Gs protein to the melatonin MT1 receptor. Int. J. Mol. Sci. 2024, 25, 2919. [Google Scholar] [CrossRef]
- Chen, Y.; Corriden, R.; Inoue, Y.; Yip, L.; Hashiguchi, N.; Zinkernagel, A.; Nizet, V.; Insel, P.A.; Junger, W.G. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 2006, 314, 1792–1795. [Google Scholar] [CrossRef]
- Schaid, M.D.; Wisinski, J.A.; Kimple, M.E. The EP3 receptor/Gz signaling axis as a therapeutic target for diabetes and cardiovascular disease. AAPS J. 2017, 19, 1276–1283. [Google Scholar] [CrossRef]
- Blom, T.; Bergelin, N.; Meinander, A.; Löf, C.; Slotte, J.P.; Eriksson, J.E.; Törnquist, K. An autocrine sphingosine-1-phosphate signaling loop enhances NF-κB-activation and survival. BMC Cell Biol. 2010, 11, 45. [Google Scholar] [CrossRef]
- Zhang, W.; An, J.; Jawadi, H.; Siow, D.L.; Lee, J.-F.; Zhao, J.; Gartung, A.; Maddipati, K.R.; Honn, K.V.; Wattenberg, B.W.; et al. Sphingosine-1-phosphate receptor-2 mediated NF-κB activation contributes to tumor necrosis factor-α induced VCAM-1 and ICAM-1 expression in endothelial cells. Prostaglandins Other Lipid Mediat. 2013, 106, 62–71. [Google Scholar] [CrossRef]
- Tall, G.G.; Krumins, A.M.; Gilman, A.G. Mammalian Ric-8A (Synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 2003, 278, 8356–8362. [Google Scholar] [CrossRef]
- Yen, H.-Y.; Hoi, K.K.; Liko, I.; Hedger, G.; Horrell, M.R.; Song, W.; Wu, D.; Heine, P.; Warne, T.; Lee, Y.; et al. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 2018, 559, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.K.; Chan, H.Y.; Lee, T.Y.; Wong, Y.H. Inhibition of adenylyl cyclase by GTPase-deficient Gαi is mechanistically different from that mediated by receptor-activated Gαi. Cell Commun. Signal. CCS 2024, 22, 218. [Google Scholar] [CrossRef]
- Russell, I.C.; Zhang, X.; Bumbak, F.; McNeill, S.M.; Josephs, T.M.; Leeming, M.G.; Christopoulos, G.; Venugopal, H.; Flocco, M.M.; Sexton, P.M.; et al. Lipid-dependent activation of the orphan G protein-coupled receptor, GPR3. Biochemistry 2024, 63, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Claff, T.; Ebenhoch, R.; Kley, J.T.; Magarkar, A.; Nar, H.; Weichert, D. Structural basis for lipid-mediated activation of G protein-coupled receptor GPR55. Nat. Commun. 2025, 16, 1973. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Mende, U.; Nose, M.; Schmitz, W.; Scholz, H.; Haverich, A.; Hirt, S.; Döring, V.; Kalmár, P.; Höppner, W. Increased messenger RNA level of the inhibitory G protein α subunit Gi α-2 in human end-stage Heart Failure. Circ. Res. 1992, 70, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Matozaki, T.; Takada, T.; Noguchi, T.; Yamao, T.; Tsuda, M.; Ochi, F.; Fukunaga, K.; Inagaki, K.; Kasuga, M. PI 3-kinase γ and protein kinase C-ζ mediate RAS-independent activation of MAP kinase by a Gi protein-coupled receptor. EMBO J. 1999, 18, 386–395. [Google Scholar] [CrossRef]
- Koch, W.J.; Hawes, B.E.; Allen, L.F.; Lefkowitz, R.J. Direct evidence that Gi-coupled receptor stimulation of mitogen-activated protein kinase is mediated by Gβγ activation of P21ras. Proc. Natl. Acad. Sci. USA 1994, 91, 12706–12710. [Google Scholar] [CrossRef]
- Chan, A.S.L.; Lai, F.P.L.; Lo, R.K.H.; Voyno-Yasenetskaya, T.A.; Stanbridge, E.J.; Wong, Y.H. Melatonin MT1 and MT2 receptors stimulate c-Jun N-terminal kinase via pertussis toxin-sensitive and -insensitive G proteins. Cell. Signal. 2002, 14, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Schaak, S.; Cussac, D.; Cayla, C.; Devedjian, J.; Guyot, R.; Paris, H.; Denis, C. α2 adrenoceptors regulate proliferation of human intestinal epithelial cells. Gut 2000, 47, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Kramer, H.K.; Simon, E.J. μ and δ-opioid receptor agonists induce mitogen-activated protein kinase (MAPK) activation in the absence of receptor internalization. Neuropharmacology 2000, 39, 1707–1719. [Google Scholar] [CrossRef]
- Khater, M.; Wei, Z.; Xu, X.; Huang, W.; Lokeshwar, B.L.; Lambert, N.A.; Wu, G. G protein βγ translocation to the Golgi apparatus activates MAPK via P110γ-P101 heterodimers. J. Biol. Chem. 2021, 296, 100325. [Google Scholar] [CrossRef] [PubMed]
- Bono, F.; Tomasoni, Z.; Mutti, V.; Sbrini, G.; Kumar, R.; Longhena, F.; Fiorentini, C.; Missale, C. G protein-dependent activation of the PKA-ERK1/2 pathway by the striatal dopamine D1/D3 receptor heteromer involves β-arrestin and the tyrosine phosphatase Shp-2. Biomolecules 2023, 13, 473. [Google Scholar] [CrossRef]
- Zou, Y.; Komuro, I.; Yamazaki, T.; Kudoh, S.; Aikawa, R.; Zhu, W.; Shiojima, I.; Hiroi, Y.; Tobe, K.; Kadowaki, T.; et al. Cell type-specific angiotensin II-evoked signal transduction pathways: Critical roles of Gβγ subunit, Src family, and Ras in cardiac fibroblasts. Circ. Res. 1998, 82, 337–345. [Google Scholar] [CrossRef]
- Crudden, C.; Song, D.; Cismas, S.; Trocmé, E.; Pasca, S.; Calin, G.A.; Girnita, A.; Girnita, L. Below the surface: IGF-1R therapeutic targeting and its endocytic journey. Cells 2019, 8, 1223. [Google Scholar] [CrossRef]
- Hawkins, P.T.; Stephens, L.R. PI3Kγ is a key regulator of inflammatory responses and cardiovascular homeostasis. Science 2007, 318, 64–66. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Martin, D.; Galisteo, R.; Molinolo, A.A.; Wetzker, R.; Hirsch, E.; Gutkind, J.S. PI3Kγ mediates kaposi’s sarcoma-associated herpesvirus vGPCR-induced sarcomagenesis. Cancer Cell 2011, 19, 805–813. [Google Scholar] [CrossRef]
- Wu, E.H.T.; Wong, Y.H. Involvement of Gi/o proteins in nerve growth factor-stimulated phosphorylation and degradation of tuberin in PC-12 cells and cortical neurons. Mol. Pharmacol. 2005, 67, 1195–1205. [Google Scholar] [CrossRef]
- Chan, G.P.W.; Wu, E.H.T.; Wong, Y.H. Regulation of mTOR and P70 S6 kinase by the muscarinic M4 receptor in PC12 cells. Cell Biol. Int. 2009, 33, 230–238. [Google Scholar] [CrossRef]
- Bouti, P.; Webbers, S.D.S.; Fagerholm, S.C.; Alon, R.; Moser, M.; Matlung, H.L.; Kuijpers, T.W. β2 integrin signaling cascade in neutrophils: More than a single function. Front. Immunol. 2020, 11, 619925. [Google Scholar] [CrossRef]
- Futosi, K.; Fodor, S.; Mócsai, A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 638–650. [Google Scholar] [CrossRef]
- Bednarczyk, M.; Stege, H.; Grabbe, S.; Bros, M. β2 integrins-multi-functional leukocyte receptors in health and disease. Int. J. Mol. Sci. 2020, 21, 1402. [Google Scholar] [CrossRef] [PubMed]
- Schneider, O.D.; Weiss, A.A.; Miller, W.E. Pertussis toxin utilizes proximal components of the T-cell receptor complex to initiate signal transduction events in T cells. Infect. Immun. 2007, 75, 4040–4049. [Google Scholar] [CrossRef]
- Strnad, C.F.; Carchman, R.A. Human T lymphocyte mitogenesis in response to the B oligomer of pertussis toxin is associated with an early elevation in cytosolic calcium concentrations. FEBS Lett. 1987, 225, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Carbonetti, N.H. Contribution of pertussis toxin to the pathogenesis of pertussis disease. Pathog. Dis. 2015, 73, ftv073. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Yang, D.; Chen, Q.; Leifer, C.A.; Segal, D.M.; Su, S.B.; Caspi, R.R.; Howard, Z.O.M.; Oppenheim, J.J. Induction of dendritic cell maturation by pertussis toxin and its B subunit differentially initiate toll-like receptor 4–dependent signal transduction pathways. Exp. Hematol. 2006, 34, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Ausiello, C.M.; Fedele, G.; Urbani, F.; Lande, R.; Di Carlo, B.; Cassone, A. Native and genetically inactivated pertussis toxins induce human dendritic cell maturation and synergize with lipopolysaccharide in promoting T helper type 1 responses. J. Infect. Dis. 2002, 186, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Alfano, M.; Grivel, J.-C.; Ghezzi, S.; Corti, D.; Trimarchi, M.; Poli, G.; Margolis, L. Pertussis toxin B-oligomer dissociates T cell activation and HIV replication in CD4 T cells released from infected lymphoid tissue. AIDS 2005, 19, 1007–1014. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.