Multi-Mycotoxin Method Development Using Ultra-High Liquid Chromatography with Orbitrap High-Resolution Mass Spectrometry Detection in Breakfast Cereals from the Campania Region, Italy

 ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
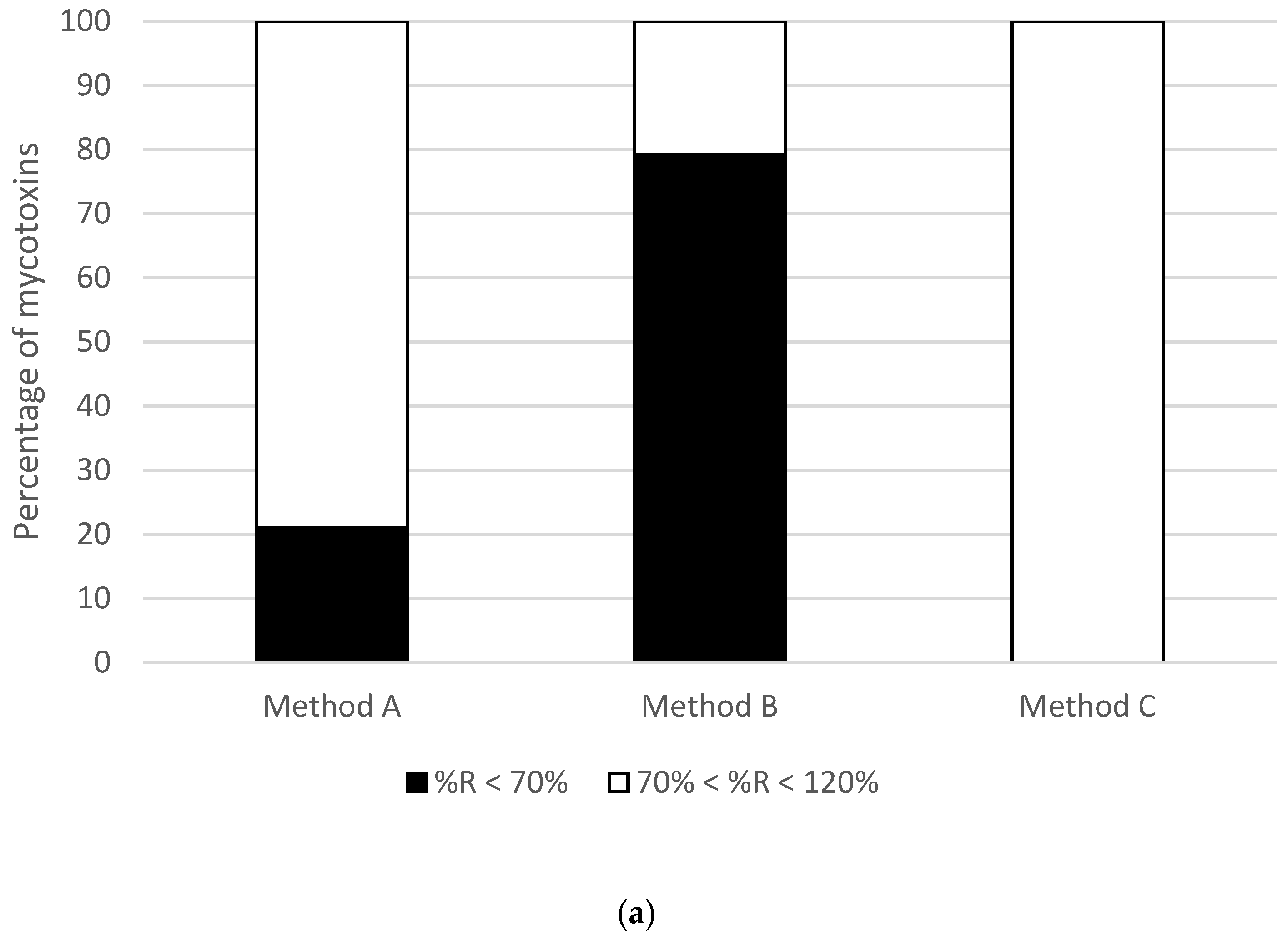
2.1. Optimization of Sample Preparation
2.2. Method Validation
2.3. Analysis of Real Samples
3. Conclusions
4. Material and Methods
4.1. Chemical, Reagents and Materials
4.2. Sampling
4.3. Sample Treatment
4.4. UHPLC-Q-Orbitrap HRMS Analysis
4.5. Method Validation
4.6. Quality Control/Quality Assurance
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wrigley, C.W. An introduction to the cereal grains: Major providers for mankind’s food needs. In Cereal Grains, 1st ed.; Wrigley, C.W., Batey, I.L., Eds.; Woodhead Publishing: Sawston, UK, 2010; pp. 3–23. [Google Scholar]
- FAO; UNICEF; WFP; WHO. The State of Food Security and Nutrition in the World 2022: Repurposing Food and Agricultural Policies to Make Healthy Diets More Affordable; Food & Agriculture Org.: Rome, Italy, 2022. [Google Scholar]
- Pereira, V.L.; Fernandes, J.O.; Cunha, S.C. Mycotoxins in cereals and related foodstuffs: A review on occurrence and recent methods of analysis. Trends Food Sci. Technol. 2014, 36, 96–136. [Google Scholar] [CrossRef]
- Narváez, A.; Rodríguez-Carrasco, Y.; Castaldo, L.; Izzo, L.; Graziani, G.; Ritieni, A. Occurrence and Exposure Assessment of Mycotoxins in Ready-to-Eat Tree Nut Products through Ultra-High Performance Liquid Chromatography Coupled with High Resolution Q-Orbitrap Mass Spectrometry. Metabolites 2020, 10, 344. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Moltó, J.C.; Berrada, H.; Mañes, J. A survey of trichothecenes, zearalenone and patulin in milled grain-based products using GC–MS/MS. Food Chem. 2014, 146, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Izzo, L.; Luz, C.; Ritieni, A.; Mañes, J.; Meca, G. Whey fermented by using Lactobacillus plantarum strains: A promising approach to increase the shelf life of pita bread. J. Dairy Sci. 2020, 103, 5906–5915. [Google Scholar] [CrossRef] [PubMed]
- Karsauliya, K.; Yahavi, C.; Pandey, A.; Bhateria, M.; Sonker, A.K.; Pandey, H.; Sharma, M.; Singh, S.P. Co-occurrence of mycotoxins: A review on bioanalytical methods for simultaneous analysis in human biological samples, mixture toxicity and risk assessment strategies. Toxicon 2022, 218, 25–39. [Google Scholar] [CrossRef]
- European Commission. Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union 2006, 364, 5–24. [Google Scholar]
- Battilani, P.; Palumbo, R.; Giorni, P.; Dall’Asta, C.; Dellafiora, L.; Gkrillas, A.; Toscano, P.; Crisci, A.; Brera, C.; De Santis, B.; et al. Mycotoxin mixtures in food and feed: Holistic, innovative, flexible risk assessment modelling approach. EFSA Support. Publ. 2020, 17, 1757. [Google Scholar] [CrossRef]
- Khaneghah, A.M.; Fakhri, Y.; Raeisi, S.; Armoon, B.; Sant’Ana, A.S. Prevalence and concentration of ochratoxin A, zearalenone, deoxynivalenol and total aflatoxin in cereal-based products: A systematic review and meta-analysis. Food Chem. Toxicol. 2018, 118, 830–848. [Google Scholar] [CrossRef]
- Narváez, A.; Castaldo, L.; Izzo, L.; Pallarés, N.; Rodríguez-Carrasco, Y.; Ritieni, A. Deoxynivalenol contamination in cereal-based foodstuffs from Spain: Systematic review and meta-analysis approach for exposure assessment. Food Control 2022, 132, 108521. [Google Scholar] [CrossRef]
- Izzo, L.; Castaldo, L.; Narváez, A.; Gaspari, A.; Grosso, M.; Rodríguez-Carrasco, Y.; Ritieni, A. Target analysis and retrospective screening of contaminants in ready-to-eat cooked ham samples through UHPLC-Q-Orbitrap HRMS. Food Chem. 2023, 408, 135244. [Google Scholar] [CrossRef]
- Izzo, L.; Rodríguez-Carrasco, Y.; Tolosa, J.; Graziani, G.; Gaspari, A.; Ritieni, A. Target analysis and retrospective screening of mycotoxins and pharmacologically active substances in milk using an ultra-high-performance liquid chromatography/high-resolution mass spectrometry approach. J. Dairy Sci. 2020, 103, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- EFSA CONTAM Panel. Scientific opinion on the risks to human and animal health related to the presence of beauvericin and enniatins in food and feed. EFSA J. 2014, 13, 3802. [Google Scholar]
- Smaoui, S.; Ben Braïek, O.; Ben Hlima, H. Mycotoxins Analysis in Cereals and Related Foodstuffs by Liquid Chromatography-Tandem Mass Spectrometry Techniques. J. Food Qual. 2020, 2020, 8888117. [Google Scholar] [CrossRef]
- Shanakhat, H.; Sorrentino, A.; Raiola, A.; Romano, A.; Masi, P.; Cavella, S. Current methods for mycotoxins analysis and innovative strategies for their reduction in cereals: An overview. J. Sci. Food Agric. 2018, 98, 4003–4013. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.L.; Fernandes, J.O.; Cunha, S.C. Comparative assessment of three cleanup procedures after QuEChERS extraction for determination of trichothecenes (type A and type B) in processed cereal-based baby foods by GC–MS. Food Chem. 2015, 182, 143–149. [Google Scholar] [CrossRef]
- European Commission. Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, 50, 8–36. [Google Scholar]
- Foerster, C.; Monsalve, L.; Ríos-Gajardo, G. Mycotoxin Exposure in Children through Breakfast Cereal Consumption in Chile. Toxins 2022, 14, 324. [Google Scholar] [CrossRef]
- Mallmann, C.A.; Tyska, D.; Almeida, C.A.A.; Oliveira, M.S.; Gressler, L.T. Mycotoxicological monitoring of breakfast and infant cereals marketed in Brazil. Int. J. Food Microbiol. 2020, 331, 108628. [Google Scholar] [CrossRef] [PubMed]
- MycoKey: Integrated and Innovative Key Actions for Mycotoxin Management in the Food and Feed Chain. Available online: http://www.mycokey.eu/results/ (accessed on 4 February 2023).
- Capei, R.; Pettini, L.; Tacconi, F.M. Occurrence of Ochratoxin A in breakfast cereals and sweet snacks in Italy: Dietary exposure assessment. Ann. Ig 2019, 31, 130–139. [Google Scholar]
- Lo Magro, S.; Campaniello, M.; Nardiello, D.; Muscarella, M. Assessment of Fumonisins B-1 and B-2 Levels in Commercial Maize-Based Food Products by Liquid Chromatography with Fluorimetric Detection and Postcolumn Chemical Derivatization. J. Food Sci. 2011, 76, T1–T4. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, B.; Ferrari, M.; Bergamini, C. Simultaneous determination of deoxynivalenol, zearalenone, T-2 and HT-2 toxins in breakfast cereals and baby food by high-performance liquid chromatography and tandem mass spectrometry. J. Mass Spectrom. 2010, 45, 1075–1080. [Google Scholar] [CrossRef]
- EFSA CONTAM Panel. Appropriateness to set a group health-based guidance value for zearalenone and its modified forms. EFSA J. 2016, 14, e04425. [Google Scholar]
- Mahnine, N.; Meca, G.; Elabidi, A.; Fekhaoui, M.; Saoiabi, A.; Font, G.; Manes, J.; Zinedine, A. Further data on the levels of emerging Fusarium mycotoxins enniatins (A, A1, B, B1), beauvericin and fusaproliferin in breakfast and infant cereals from Morocco. Food Chem. 2011, 124, 481–485. [Google Scholar] [CrossRef]
- Malachova, A.; Dzuman, Z.; Veprikova, Z.; Vaclavikova, M.; Zachariasova, M.; Hajslova, J. Deoxynivalenol, Deoxynivalenol-3-glucoside, and Enniatins: The Major Mycotoxins Found in Cereal-Based Products on the Czech Market. J. Agric. Food Chem. 2011, 59, 12990–12997. [Google Scholar] [CrossRef]
- Stanciu, O.; Juan, C.; Miere, D.; Loghin, F.; Mañes, J. Analysis of enniatins and beauvericin by LC-MS/MS in wheat-based products. CYTA J. Food 2017, 15, 433–440. [Google Scholar] [CrossRef]
- Juan, C.; Covarelli, L.; Beccari, G.; Colasante, V.; Manes, J. Simultaneous analysis of twenty-six mycotoxins in durum wheat grain from Italy. Food Control 2016, 62, 322–329. [Google Scholar] [CrossRef]
- Juan, C.; Ritieni, A.; Manes, J. Occurrence of Fusarium mycotoxins in Italian cereal and cereal products from organic farming. Food Chem. 2013, 141, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.; Assunção, R.; Cunha, S.C.; Fernandes, J.O.; Jager, A.; Petta, T.; Oliveira, C.A.; Alvito, P. Assessment of multiple mycotoxins in breakfast cereals available in the Portuguese market. Food Chem. 2018, 239, 132–140. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Izzo, L.; Gaspari, A.; Graziani, G.; Mañes, J.; Ritieni, A. Urinary levels of enniatin B and its phase I metabolites: First human pilot biomonitoring study. Food Chem. Toxicol. 2018, 118, 454–459. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Narváez, A.; Izzo, L.; Gaspari, A.; Graziani, G.; Ritieni, A. Biomonitoring of Enniatin B1 and Its Phase I Metabolites in Human Urine: First Large-Scale Study. Toxins 2020, 12, 415. [Google Scholar] [CrossRef]
- Izzo, L.; Narváez, A.; Castaldo, L.; Gaspari, A.; Rodríguez-Carrasco, Y.; Grosso, M.; Ritieni, A. Multiclass and multi-residue screening of mycotoxins, pharmacologically active substances, and pesticides in infant milk formulas through ultra-high-performance liquid chromatography coupled with high-resolution mass spectrometry analysis. J. Dairy Sci. 2022, 105, 2948–2962. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Features | Method A | Method B | Method C |
|---|---|---|---|
| Amount of sample (g) | 2.5 | 2.5 | 2.5 |
| Dilution in water (mL) | - | 4 | 15 |
| Quantity of solvent (mL) | 20 | 16 | 10 |
| Acidification with formic acid (%) | 1.0 | 0.1 | 0.1 |
| Clean up | 200 mg C18 + 900 MgSO4 | - | - |
| Recovery (%) | Precision (%) [RSDr, (RSDR)] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Analyte | Linearity (R2) | SSE (%) | 50 µg/kg | 25 µg/kg | 10 µg/kg | 50 µg/kg | 25 µg/kg | 10 µg/kg | LOQ (µg/kg) |
| DON | 0.9969 | 107 | 107 | 115 | 117 | 12 (8) | 13 (9) | 13 (11) | 12.5 |
| NEO | 0.9948 | 81 | 105 | 108 | 118 | 10 (9) | 9 (14) | 7 (7) | 0.78 |
| HT-2 | 0.9981 | 96 | 111 | 112 | 91 | 6 (8) | 5 (4) | 10 (13) | 0.78 |
| T2 | 0.9982 | 85 | 102 | 115 | 104 | 10 (11) | 17 (11) | 15 (11) | 0.39 |
| BEA | 0.9986 | 85 | 106 | 120 | 100 | 14 (10) | 9 (7) | 14 (10) | 0.20 |
| ENNA | 0.9978 | 82 | 107 | 120 | 97 | 16 (13) | 5 (5) | 11 (11) | 0.20 |
| AFB1 | 0.9940 | 83 | 104 | 107 | 76 | 14 (11) | 10 (15) | 15 (12) | 1.56 |
| ENNA1 | 0.9980 | 99 | 108 | 106 | 119 | 14 (10) | 7 (6) | 15 (10) | 1.56 |
| ENNB | 0.9903 | 98 | 106 | 119 | 113 | 13 (10) | 10 (10) | 7 (9) | 0.39 |
| ENNB1 | 0.9903 | 99 | 107 | 112 | 116 | 6 (12) | 6 (4) | 12 (8) | 1.56 |
| AFG1 | 0.9924 | 96 | 100 | 109 | 97 | 14 (14) | 7 (9) | 11 (7) | 0.78 |
| AFG2 | 0.9921 | 95 | 113 | 115 | 119 | 6 (5) | 13 (12) | 14 (10) | 0.78 |
| AFB2 | 0.9960 | 90 | 101 | 119 | 119 | 17 (10) | 12 (19) | 5 (7) | 0.20 |
| OTA | 0.9932 | 87 | 109 | 115 | 115 | 6 (7) | 11 (7) | 9 (11) | 6.25 |
| FUS X | 0.9914 | 92 | 108 | 105 | 119 | 9 (9) | 17 (11) | 2 (4) | 12.5 |
| FB2 | 0.9962 | 82 | 105 | 98 | 112 | 6 (12) | 12 (11) | 13 (13) | 0.39 |
| FB1 | 0.9993 | 85 | 109 | 111 | 114 | 13 (17) | 11 (23) | 8 (9) | 3.12 |
| α-ZEL | 0.9947 | 94 | 105 | 105 | 114 | 7 (8) | 19 (16) | 9 (5) | 3.12 |
| α-ZAL | 0.9935 | 89 | 99 | 94 | 87 | 9 (11) | 10 (7) | 11 (14) | 1.56 |
| β-ZEL | 0.9972 | 84 | 107 | 108 | 117 | 6 (6) | 14 (11) | 13 (10) | 1.56 |
| β-ZAL | 0.9936 | 91 | 103 | 98 | 91 | 5 (8) | 9 (10) | 11 (13) | 1.56 |
| ZAN | 0.9930 | 86 | 97 | 111 | 103 | 5 (3) | 10 (11) | 5 (6) | 1.56 |
| ZEN | 0.9980 | 97 | 107 | 107 | 111 | 11 (10) | 16 (11) | 14 (16) | 1.56 |
| Mycotoxin | Prevalence (%) | Mean Concentration (µg/kg) | SD (µg/kg) | Maximum (µg/kg) |
|---|---|---|---|---|
| BEA | 86 | 6.7 | 7.74 | 30.66 |
| ENNA | 7 | 1.05 | - | 1.05 |
| ENNA1 | 21 | 6.34 | 7.35 | 14.79 |
| ENNB | 21 | 23.09 | 17.55 | 35.39 |
| ENNB1 | 14 | 15.03 | 17.57 | 27.45 |
| FB2 | 14 | 1.61 | 0.97 | 2.3 |
| FB1 | 36 | 22.18 | 20.4 | 55.51 |
| α-ZEL | 14 | 5.79 | 0.23 | 5.96 |
| β-ZEL | 7 | 4.39 | - | 4.39 |
| Analyte | Retention Time (min) | Elemental Composition | Adduct | Theoretical Mass (m/z) | Measured Mass (m/z) | Accuracy (Δ ppm) | Collision Energy (eV) | Product Ions (m/z) |
|---|---|---|---|---|---|---|---|---|
| Ion | ||||||||
| DON | 2.69 | C15H20O6 | [M+H]+ | 297.13326 | 297.13345 | 0.64 | 13 | 249.11194; 203.10648 |
| FUS-X | 3.58 | C17H22O8 | [M+Na]+ | 377.12073 | 377.12063 | −0.27 | 20 | 228.16002; 175.07550 |
| NEO | 3.76 | C19H26O8 | [M+NH4]+ | 400.19659 | 400.19632 | −0.67 | 10 | 305.13803; 141.0053 |
| HT-2 | 4.3 | C22H32O8 | [M+NH4]+ | 442.24354 | 442.24323 | −0.7 | 27 | 263.12744; 215.10641 |
| β-ZEL | 4.34 | C18H24O5 | [M-H]− | 319.1551 | 319.155 | −0.31 | 36 | 174.95604; 160.97665 |
| α-ZEL | 4.44 | C18H24O5 | [M-H]− | 319.1551 | 319.155 | −0.31 | 36 | 174.95604; 129.01947 |
| T-2 | 4.49 | C24H34O9 | [M+NH4]+ | 484.25411 | 484.2543 | 0.39 | 23 | 215.10603; 185.09561 |
| ZAN | 4.68 | C18H24O5 | [M-H]− | 319.1551 | 319.155 | −0.31 | 35 | 273.01187; 131.05020 |
| ZEN | 4.7 | C18H22O5 | [M-H]− | 317.13945 | 317.13928 | −0.54 | -32 | 175.03989; 131.05008 |
| AFG1 | 4.73 | C17H12O7 | [M+H]+ | 329.06558 | 329.06549 | −0.27 | 40 | 243.06467; 200.04640 |
| AFG2 | 5.03 | C17H14O7 | [M+H]+ | 331.08123 | 331.08078 | −1.36 | 37 | 313.07010; 245.08032 |
| ENNB | 5.15 | C33H57N3O9 | [M+NH4]+ | 657.44331 | 657.44348 | 0.26 | 50 | 214.1432; 196.1328 |
| AFB2 | 5.16 | C17H14O6 | [M+H]+ | 315.08631 | 315.08615 | −0.51 | 36 | 287.09064; 259.05945 |
| ENNB1 | 5.18 | C34H59N3O9 | [M+NH4]+ | 671.45986 | 671.45935 | −0.76 | 48 | 214.14343; 196.13295 |
| ENNA1 | 5.24 | C35H61N3O9 | [M+NH4]+ | 685.47461 | 685.47449 | −0.18 | 48 | 228.15900; 210.14847 |
| AFB1 | 5.26 | C17H12O6 | [M+H]+ | 313.07066 | 313.07053 | −0.42 | 36 | 285.07489; 269.04373 |
| ENNA | 5.28 | C36H63N3O9 | [M+NH4]+ | 699.49026 | 699.48987 | −0.56 | 43 | 228.15900; 210.14847 |
| BEA | 5.4 | C45H57N3O9 | [M+NH4]+ | 801.44331 | 801.44339 | 0.1 | 35 | 262.76715; 244.18239 |
| FB1 | 6.03 | C34H59NO15 | [M+H]+ | 722.39575 | 722.39539 | −0.5 | 48 | 352.32010; 334.30963 |
| OTA | 6.5 | C20H18NO6Cl | [M+H]+ | 404.08954 | 404.08931 | 0.57 | 16 | 358.08304; 341.05658 |
| FB2 | 6.78 | C34H59NO14 | [M+H]+ | 706.40083 | 706.40192 | −1.54 | 58 | 336.32547; 318.31488 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narváez, A.; Izzo, L.; Castaldo, L.; Lombardi, S.; Rodríguez-Carrasco, Y.; Ritieni, A. Multi-Mycotoxin Method Development Using Ultra-High Liquid Chromatography with Orbitrap High-Resolution Mass Spectrometry Detection in Breakfast Cereals from the Campania Region, Italy. Toxins 2023, 15, 148. https://doi.org/10.3390/toxins15020148
Narváez A, Izzo L, Castaldo L, Lombardi S, Rodríguez-Carrasco Y, Ritieni A. Multi-Mycotoxin Method Development Using Ultra-High Liquid Chromatography with Orbitrap High-Resolution Mass Spectrometry Detection in Breakfast Cereals from the Campania Region, Italy. Toxins. 2023; 15(2):148. https://doi.org/10.3390/toxins15020148
Chicago/Turabian StyleNarváez, Alfonso, Luana Izzo, Luigi Castaldo, Sonia Lombardi, Yelko Rodríguez-Carrasco, and Alberto Ritieni. 2023. "Multi-Mycotoxin Method Development Using Ultra-High Liquid Chromatography with Orbitrap High-Resolution Mass Spectrometry Detection in Breakfast Cereals from the Campania Region, Italy" Toxins 15, no. 2: 148. https://doi.org/10.3390/toxins15020148
APA StyleNarváez, A., Izzo, L., Castaldo, L., Lombardi, S., Rodríguez-Carrasco, Y., & Ritieni, A. (2023). Multi-Mycotoxin Method Development Using Ultra-High Liquid Chromatography with Orbitrap High-Resolution Mass Spectrometry Detection in Breakfast Cereals from the Campania Region, Italy. Toxins, 15(2), 148. https://doi.org/10.3390/toxins15020148