Infectious RNA: Human Immunodeficiency Virus (HIV) Biology, Therapeutic Intervention, and the Quest for a Vaccine


Abstract
:1. Introduction
2. HIV-1 Structure and Replication Cycle
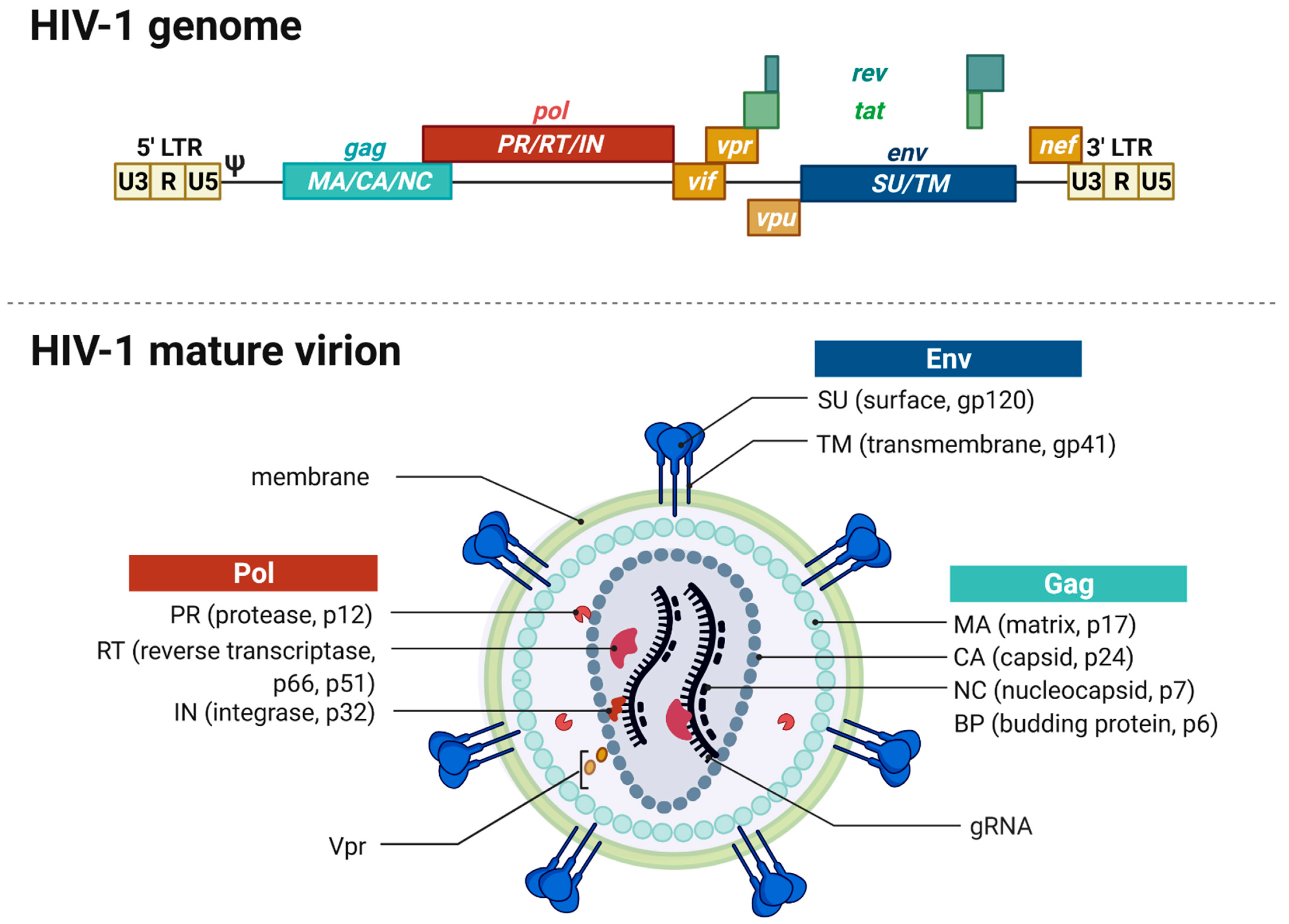
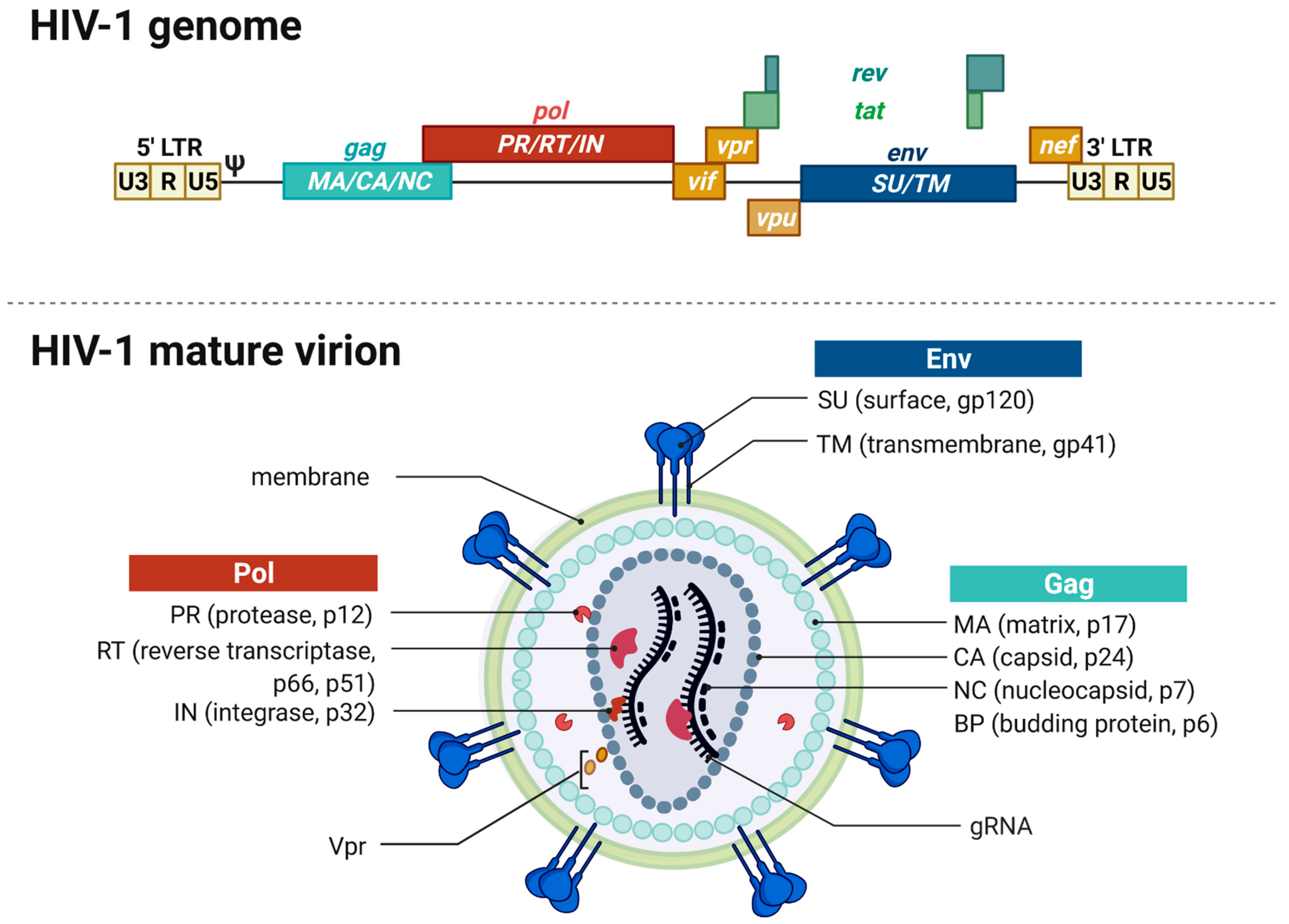
2.1. Genome and Virion Structure
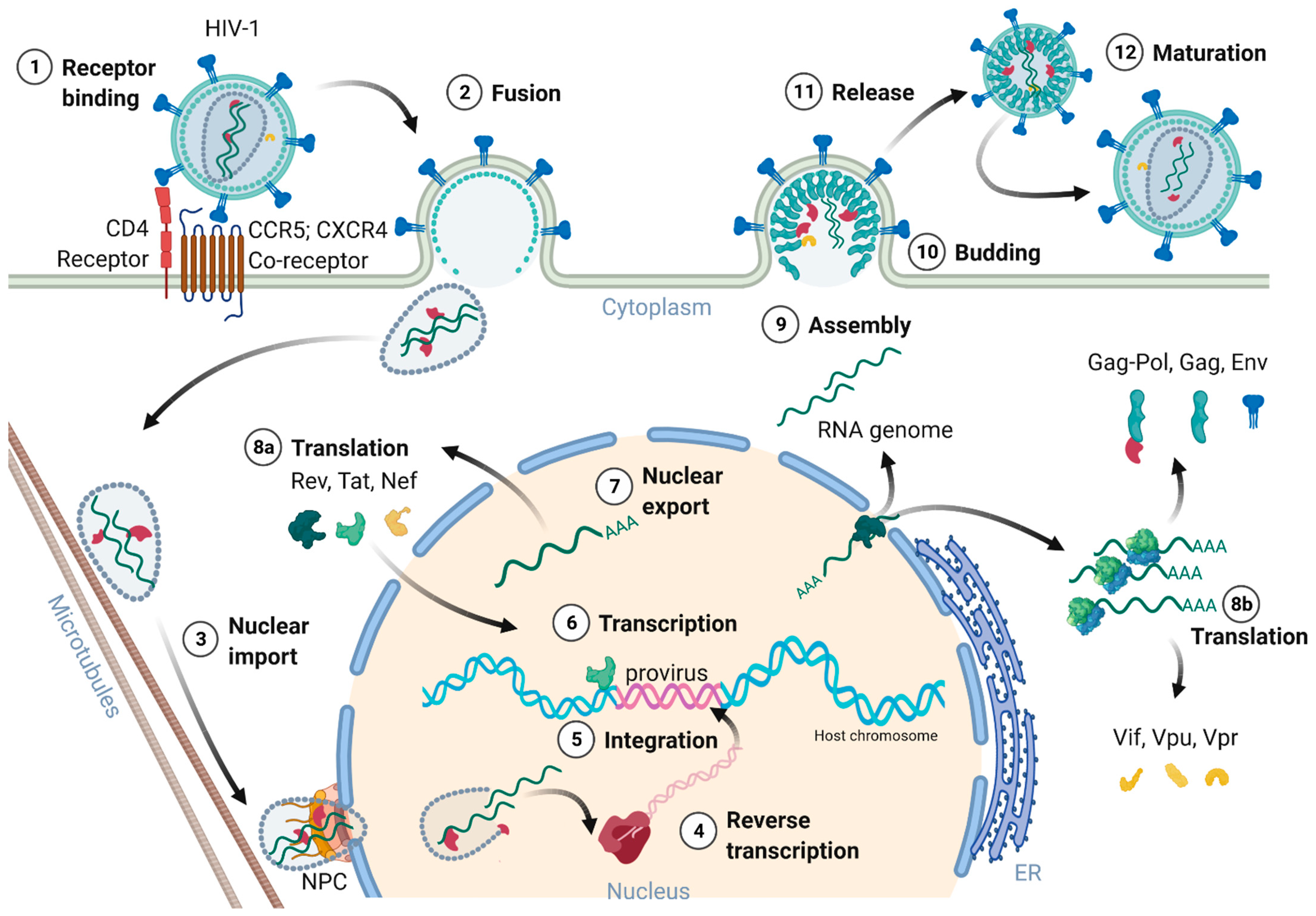
2.2. Receptors and Cell Entry
2.3. Nuclear Entry, Reverse Transcription, and Uncoating
2.4. Genome Integration
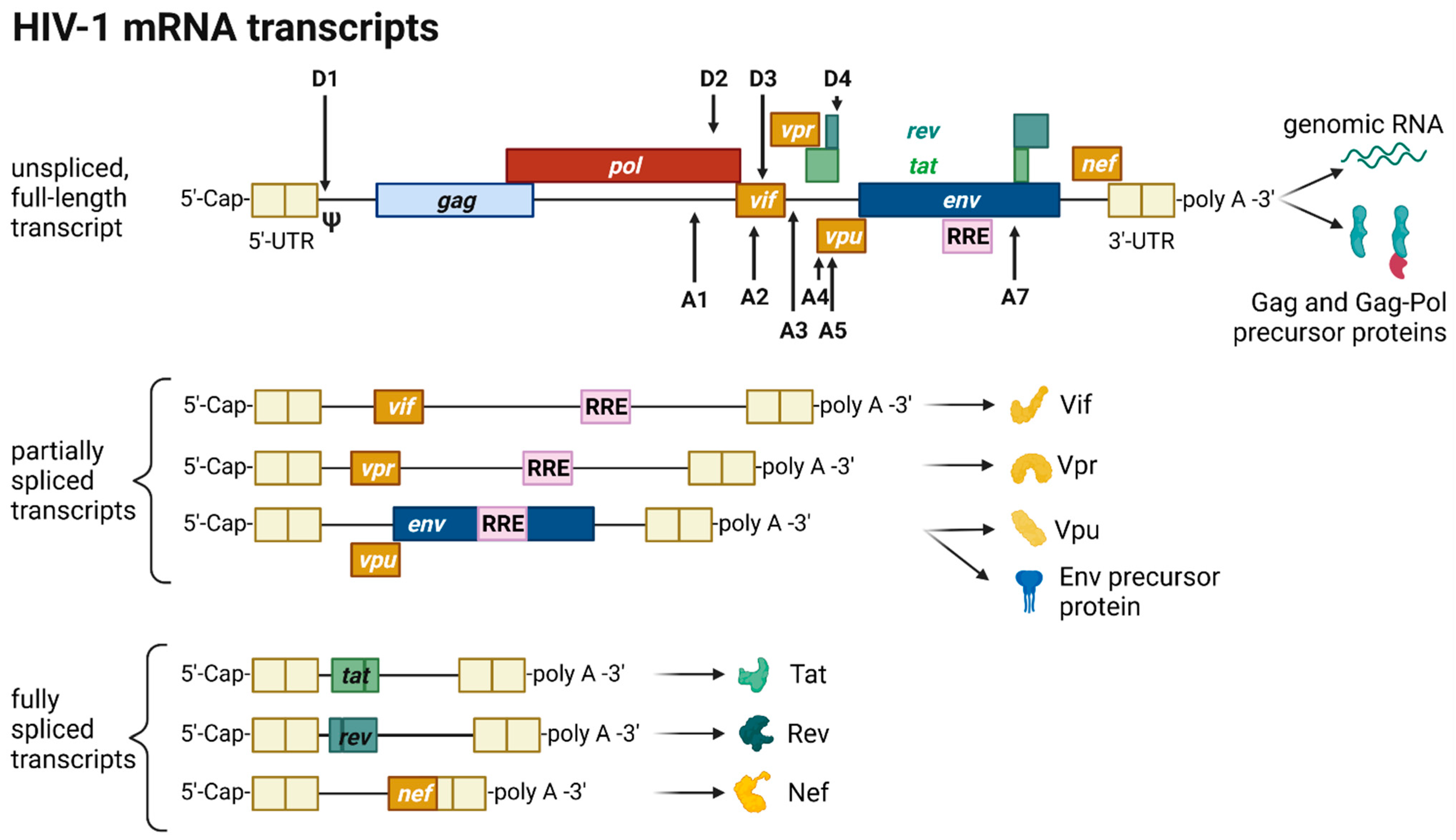
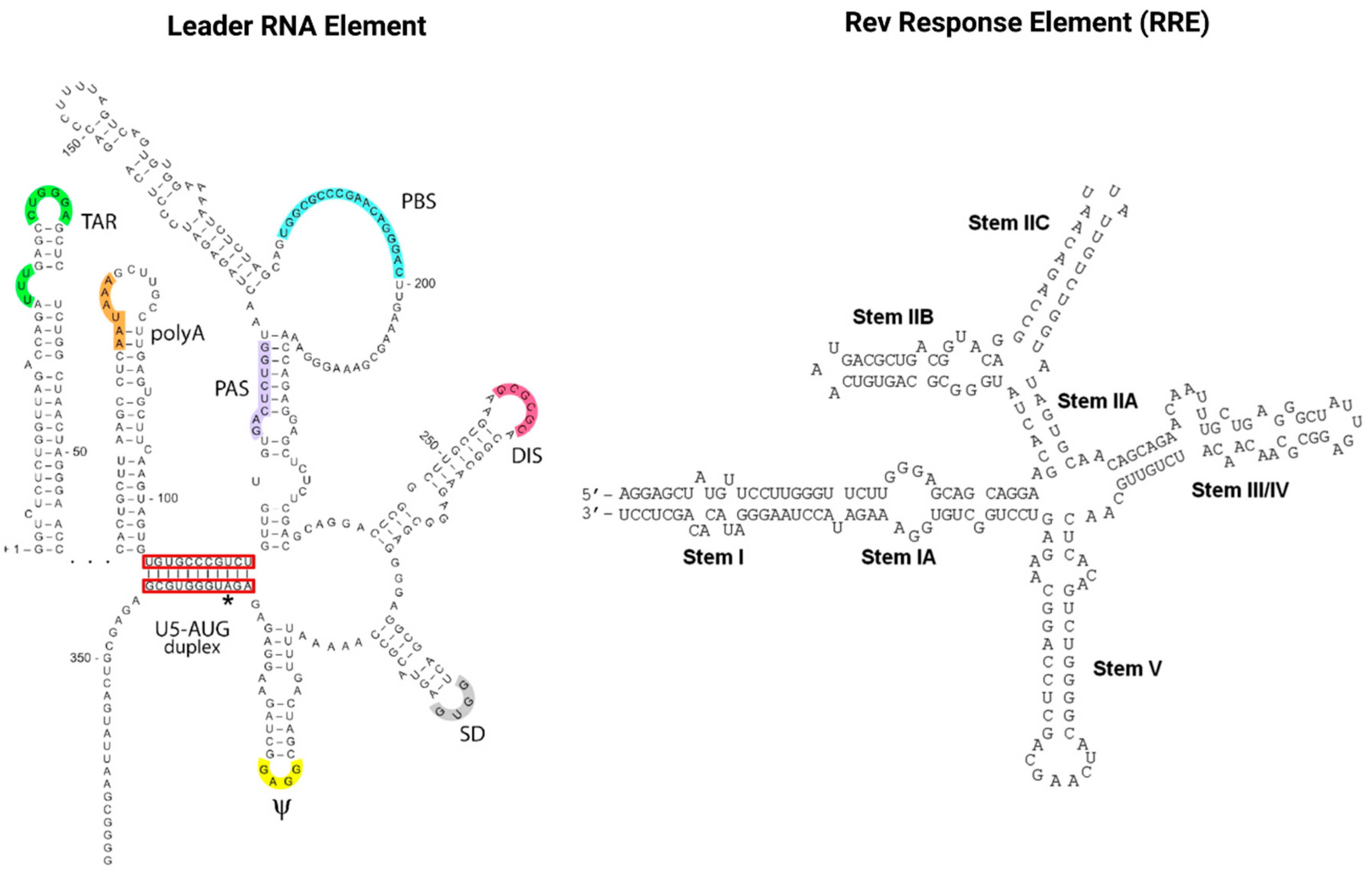
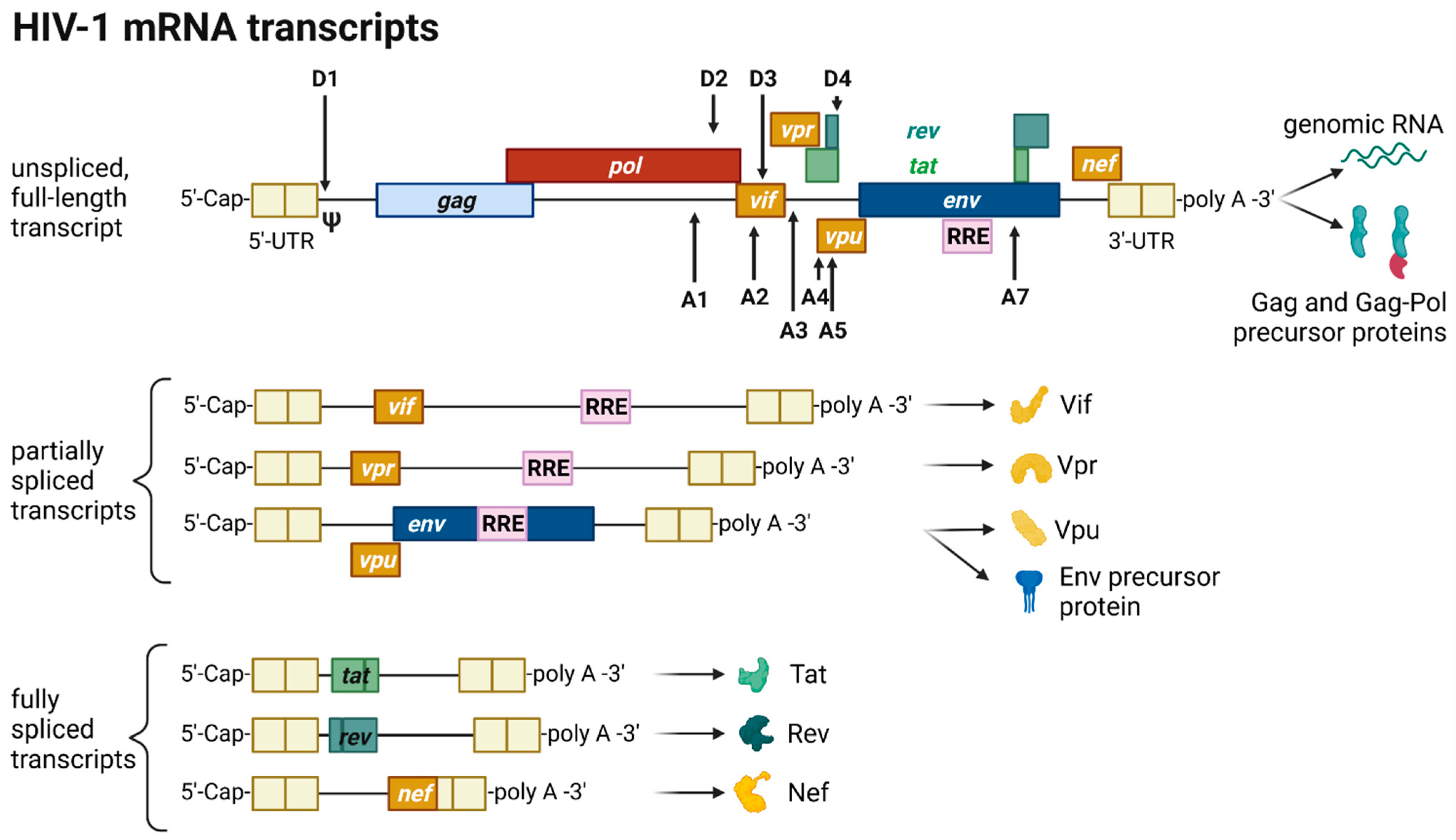
2.5. Transcription, Splicing, and Protein Expression
2.6. Assembly, Budding, and Virion Maturation
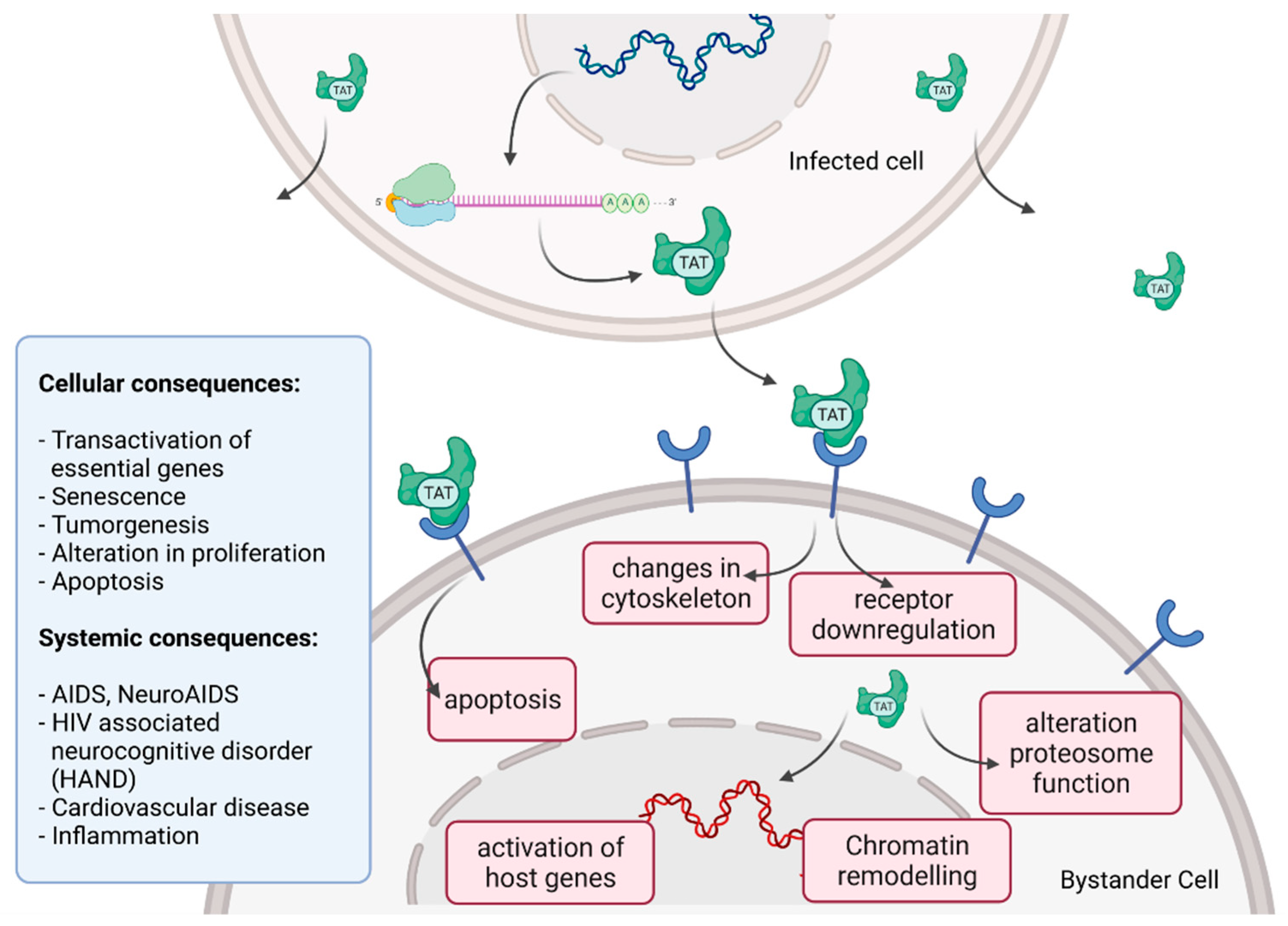
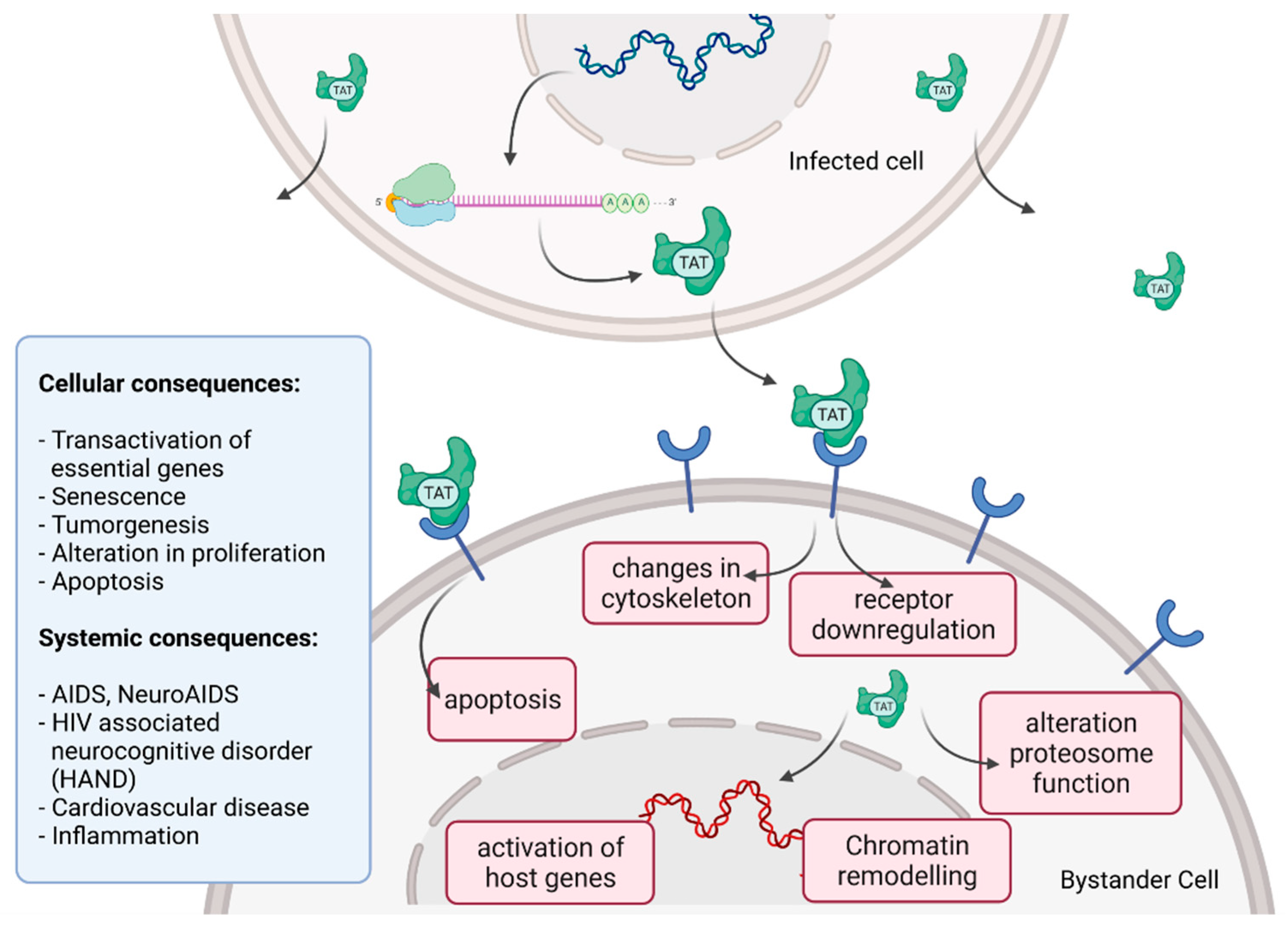
2.7. Cytoxicity of HIV Infection
3. Antiretroviral Therapy (ART)
4. Vaccines
4.1. Interplay of HIV and Immune Response—Implications for Vaccine Development
4.2. HIV Clinical Vaccine Trials
5. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woolhouse, M.E.J.; Brierley, L. Epidemiological characteristics of human-infective RNA viruses. Sci. Data 2018, 5, 180017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chase-Topping, M.; Guo, C.G.; van Bunnik, B.A.D.; Brierley, L.; Woolhouse, M.E.J. Global discovery of human-infective RNA viruses: A modelling analysis. PLoS Pathog. 2020, 16, 1009079. [Google Scholar] [CrossRef] [PubMed]
- Tarantola, A. Four thousand years of concepts relating to rabies in animals and humans, its prevention and its cure. Trop. Med. Infect. Dis. 2017, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galassi, F.M.; Habicht, M.E.; Rühli, F.J. Poliomyelitis in Ancient Egypt? Neurol. Sci. 2017, 38, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, O.B.; San Martín, J.L.; Montoya, R.H.; Del Diego, J.; Zambrano, B.; Dayan, G.H. Review: The history of dengue outbreaks in the Americas. Am. J. Trop. Med. Hyg. 2012, 87, 584–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology, 5th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1990; Volume 113, ISBN 9781451105636. [Google Scholar]
- van Doom, R.H. The epidemiology of emerging infectious diseases and pandemics. Medicine 2021, 49, 659–662. [Google Scholar] [CrossRef]
- Gallo RC, M.L. The chronology of AIDS research. Nature 1987, 326, 435–436. [Google Scholar] [CrossRef]
- Barré-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-Lymphotropic Retrovirus from a Patient at Risk for Acquired Immune Deficiency Syndrome (AIDS). Science. 1983, 220, 868–871. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.; Weaver, C. Das erworbene Immunschwächesyndrom (AIDS). In Janeway Immunologie; Springer-Spektrum: Berlin/Heidelberg, Germany, 2018; pp. 743–749. ISBN 978-3-662-56003-7. [Google Scholar]
- WHO HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 19 October 2021).
- Shaw, G.M.; Hunter, E. HIV transmission. Cold Spring Harb. Perspect. Med. 2012, 2, a006965. [Google Scholar] [CrossRef]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- Cassan, E.; Arigon-Chifolleau, A.M.; Mesnard, J.-M.; Gross, A.; Gascuel, O. Concomitant emergence of the antisense protein gene of HIV-1 and of the pandemic. Proc. Natl. Acad. Sci. USA 2016, 113, 11537–11542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sertznig, H.; Hillebrand, F.; Erkelenz, S.; Schaal, H.; Widera, M. Behind the scenes of HIV-1 replication: Alternative splicing as the dependency factor on the quiet. Virology 2018, 516, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H. Human Immunodeficiency Virus May Encode a Novel Protein on the Genomic DNA Plus Strand. Science 1988, 239, 1420–1422. [Google Scholar] [CrossRef]
- Churchill, M.J.; Cowley, D.J.; Wesselingh, S.L.; Gorry, P.R.; Gray, L.R. HIV-1 transcriptional regulation in the central nervous system and implications for HIV cure research. J. Neurovirol. 2015, 21, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Barboric, M.; Matija Peterlin, B. A new paradigm in eukaryotic biology: HIV Tat and the control of transcriptional elongation. PLoS Biol. 2005, 3, e76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbondji-Wonje, C.; Dong, M.; Zhao, J.; Wang, X.; Nanfack, A.; Ragupathy, V.; Sanchez, A.M.; Denny, T.N.; Hewlett, I. Genetic variability of the U5 and downstream sequence of major HIV-1 subtypes and circulating recombinant forms. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Kirchhoff, F. HIV Life Cycle: Overview. In Encyclopedia of AIDS; Springer: New York, NY, USA, 2013; pp. 1–9. [Google Scholar] [CrossRef]
- Li, L.; Li, H.S.; Pauza, C.D.; Bukrinsky, M.; Zhao, R.Y. Roles of HIV-1 auxiliary proteins in viral pathogenesis and host-pathogen interactions. Cell Res. 2005, 15, 923–934. [Google Scholar] [CrossRef]
- Das, A.T.; Harwig, A.; Vrolijk, M.M.; Berkhout, B. The TAR Hairpin of Human Immunodeficiency Virus Type 1 Can Be Deleted When Not Required for Tat-Mediated Activation of Transcription. J. Virol. 2007, 81, 7742–7748. [Google Scholar] [CrossRef] [Green Version]
- Seitz, R. Human Immunodeficiency Virus (HIV). Transfus. Med. Hemother. 2016, 43, 203–222. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S.; Hultquist, J.F.; Evans, D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012, 287, 40875–40883. [Google Scholar] [CrossRef] [Green Version]
- Malim, M.H.; Bieniasz, P.D. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Fu, R.M.; Liang, C.; Sloan, R.D. IFITM proteins inhibit HIV-1 protein synthesis. Sci. Rep. 2018, 8, 14451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramdas, P.; Sahu, A.K.; Mishra, T.; Bhardwaj, V.; Chande, A. From Entry to Egress: Strategic Exploitation of the Cellular Processes by HIV-1. Front. Microbiol. 2020, 11, 559792. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Sandrin, V.; Sundquist, W.I.; Bieniasz, P.D. An Interferon-α-Induced Tethering Mechanism Inhibits HIV-1 and Ebola Virus Particle Release but Is Counteracted by the HIV-1 Vpu Protein. Cell Host Microbe 2007, 2, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Mcnatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu Binds Directly to Tetherin and Displaces It from Nascent Virions. PLoS Pathog. 2013, 9, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [Green Version]
- Turner, B.G.; Summers, M.F. Structural biology of HIV 1 1Edited by P. E. Wright. J. Mol. Biol. 1999, 285, 1–32. [Google Scholar] [CrossRef]
- Burnie, J.; Guzzo, C. The incorporation of host proteins into the external HIV-1 envelope. Viruses 2019, 11, 85. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Liu, J.; Bess, J.; Chertova, E.; Lifson, J.D.; Grisé, H.; Ofek, G.A.; Taylor, K.A.; Roux, K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 2006, 441, 847–852. [Google Scholar] [CrossRef]
- Briggs, J.A.G.; Simon, M.N.; Gross, I.; Kräusslich, H.G.; Fuller, S.D.; Vogt, V.M.; Johnson, M.C. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 2004, 11, 672–675. [Google Scholar] [CrossRef]
- Goodsell, D.S. Viral Zone Expasy. Available online: https://viralzone.expasy.org/5182 (accessed on 29 September 2021).
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, R.; Xu, K.; Zhou, T.; Acharya, P.; Lemmin, T.; Liu, K.; Ozorowski, G.; Soto, C.; Taft, J.D.; Bailer, R.T.; et al. Fusion peptide of HIV-1 as a site of vulnerability to neutralizing antibody. Science 2016, 352, 828–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp 120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, S.A.; Finnegan, C.M.; Viard, M.; Raviv, Y.; Dimitrov, A.; Rawat, S.S.; Puri, A.; Durell, S.; Blumenthal, R. The HIV Env-mediated fusion reaction. Biochim. et Biophys. Acta-Biomembr. 2003, 1614, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Zila, V.; Margiotta, E.; Turoňová, B.; Müller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Börner, K.; Rada, J.; Müller, B.; et al. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. Cell 2021, 184, 1032–1046.e18. [Google Scholar] [CrossRef]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 5, 1088–1095. [Google Scholar] [CrossRef]
- Burdick, R.C.; Li, C.; Munshi, M.H.; Rawson, J.M.O.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef]
- Müller, T.G.; Zila, V.; Peters, K.; Schifferdecker, S.; Stanic, M.; Lucic, B.; Laketa, V.; Lusic, M.; Müller, B.; Kräusslich, H.G. Hiv-1 uncoating by release of viral cdna from capsid-like structures in the nucleus of infected cells. Elife 2021, 10, e64776. [Google Scholar] [CrossRef]
- Davis, A.J.; Carr, J.M.; Bagley, C.J.; Powell, J.; Warrilow, D.; Harrich, D.; Burrell, C.J.; Li, P. Human immunodeficiency virus type-1 reverse transcriptase exists as post-translationally modified forms in virions and cells. Retrovirology 2008, 5, 115. [Google Scholar] [CrossRef] [Green Version]
- Onafuwa-Nuga, A.; Telesnitsky, A. The Remarkable Frequency of Human Immunodeficiency Virus Type 1 Genetic Recombination. Microbiol. Mol. Biol. Rev. 2009, 73, 451–480. [Google Scholar] [CrossRef] [Green Version]
- Ruelas, D.S.; Greene, W.C. An Integrated Overview of HIV-1 Latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craigie, R.; Bushman, F.D. HIV DNA integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, T.; Davies, D. Structure and Function of HIV-1 Integrase. Curr. Top. Med. Chem. 2004, 4, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Pilon, A.A.; Bajaj, K.; Mazumder, A.; Neamati, N. HIV-1 integrase as a target for antiviral drugs. Antivir. Chem. Chemother. 1997, 8, 463–483. [Google Scholar] [CrossRef]
- Karn, J.; Stoltzfus, C.M. Transcriptional and Posttranscriptional Regulation of HIV-1 Gene Expression. Cold Spring Harb. Perspect. Med. 2012, 2, a006916. [Google Scholar] [CrossRef] [PubMed]
- Dutilleul, A.; Rodari, A.; Van Lint, C. Depicting HIV-1 Transcriptional Mechanisms: A Summary of What We Know. Viruses 2020, 12, 1385. [Google Scholar] [CrossRef]
- Verdikt, R.; Hernalsteens, O.; Van Lint, C. Epigenetic Mechanisms of HIV-1 Persistence. Vaccines 2021, 9, 514. [Google Scholar] [CrossRef]
- Shukla, A.; Ramirez, N.G.P.; D’Orso, I. HIV-1 Proviral Transcription and Latency in the New Era. Viruses 2020, 12, 555. [Google Scholar] [CrossRef]
- Hokello, J.; Lakhikumar Sharma, A.; Tyagi, M. AP-1 and NF-κB synergize to transcriptionally activate latent HIV upon T-cell receptor activation. FEBS Lett. 2021, 595, 577–594. [Google Scholar] [CrossRef]
- Kharytonchyk, S.; Monti, S.; Smaldino, P.J.; Van, V.; Bolden, N.C.; Brown, J.D.; Russo, E.; Swanson, C.; Shuey, A.; Telesnitsky, A.; et al. Transcriptional start site heterogeneity modulates the structure and function of the HIV-1 genome. Proc. Natl. Acad. Sci. USA 2016, 113, 13378–13383. [Google Scholar] [CrossRef] [Green Version]
- Schier, A.C.; Taatjes, D.J. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev. 2020, 34, 465–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Jiménez, A.; Campos, A.; Navarro, F.; Clemente-Blanco, A.; Calvo, O. Regulation of Eukaryotic RNAPs Activities by Phosphorylation. Front. Mol. Biosci. 2021, 8, 681865. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, A.J.C.; Bugai, A.; Barboric, M. Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res. 2016, 44, 7527–7539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengal, E.; Aloni, Y. Transcriptional elongation by purified RNA polymerase II is blocked at the trans-activation-responsive region of human immunodeficiency virus type 1 in vitro. J. Virol. 1991, 65, 4910–4918. [Google Scholar] [CrossRef] [Green Version]
- Kao, S.-Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- McNamara, R.P.; McCann, J.L.; Gudipaty, S.A.; D’Orso, I. Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep. 2013, 5, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, M.; Chen, L.F.; Chen, R. P-TEFb: Finding its ways to release promoter-proximally paused RNA polymerase II. Transcription 2018, 9, 88–94. [Google Scholar] [CrossRef]
- Barboric, M.; Yik, J.H.N.; Czudnochowski, N.; Yang, Z.; Chen, R.; Contreras, X.; Geyer, M.; Peterlin, B.M.; Zhou, Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007, 35, 2003–2012. [Google Scholar] [CrossRef]
- Tantale, K.; Garcia-Oliver, E.; Robert, M.C.; L’Hostis, A.; Yang, Y.; Tsanov, N.; Topno, R.; Gostan, T.; Kozulic-Pirher, A.; Basu-Shrivastava, M.; et al. Stochastic pausing at latent HIV-1 promoters generates transcriptional bursting. Nat. Commun. 2021, 12, 4508. [Google Scholar] [CrossRef]
- Nguyen Quang, N.; Goudey, S.; Ségéral, E.; Mohammad, A.; Lemoine, S.; Blugeon, C.; Versapuech, M.; Paillart, J.C.; Berlioz-Torrent, C.; Emiliani, S.; et al. Dynamic nanopore long-read sequencing analysis of HIV-1 splicing events during the early steps of infection. Retrovirology 2020, 17, 25. [Google Scholar] [CrossRef]
- Emery, A.; Swanstrom, R. HIV-1: To Splice or Not to Splice, That Is the Question. Viruses 2021, 13, 181. [Google Scholar] [CrossRef] [PubMed]
- Bohne, J.; Wodrich, H.; Kräusslich, H.G. Splicing of human immunodeficiency virus RNA is position-dependent suggesting sequential removal of introns from the 5′ end. Nucleic Acids Res. 2005, 33, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, V.; Summers, M.F. How retroviruses select their genomes. Nat. Rev. Microbiol. 2005, 3, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Obayashi, C.M.; Shinohara, Y.; Masuda, T.; Kawai, G. Influence of the 5′-terminal sequences on the 5′-UTR structure of HIV-1 genomic RNA. Sci. Rep. 2021, 11, 10920. [Google Scholar] [CrossRef] [PubMed]
- Esquiaqui, J.M.; Kharytonchyk, S.; Drucker, D.; Telesnitsky, A. HIV-1 spliced RNAs display transcription start site bias. RNA 2020, 26, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Toro-Ascuy, D.; Rojas-Araya, B.; Valiente-Echeverría, F.; Soto-Rifo, R. Interactions between the HIV-1 unspliced mRNA and host mRNA decay machineries. Viruses 2016, 8, 320. [Google Scholar] [CrossRef]
- Bresson, S.; Tollervey, D. Surveillance-ready transcription: Nuclear RNA decay as a default fate. Open Biol. 2018, 8, 170270. [Google Scholar] [CrossRef] [Green Version]
- Nawroth, I.; Mueller, F.; Basyuk, E.; Beerens, N.; Rahbek, U.L.; Darzacq, X.; Bertrand, E.; Kjems, J.; Schmidt, U. Stable assembly of HIV-1 export complexes occurs cotranscriptionally. RNA 2014, 20, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Malim, M.H.; Emerman, M. HIV-1 Accessory Proteins-Ensuring Viral Survival in a Hostile Environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Faust, T.B.; Binning, J.M.; Gross, J.D.; Frankel, A.D. Making Sense of Multifunctional Proteins: Human Immunodeficiency Virus Type 1 Accessory and Regulatory Proteins and Connections to Transcription. Annu. Rev. Virol. 2017, 4, 241–260. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; De Clercq, E. HIV Genome-Wide Protein Associations: A Review of 30 Years of Research. Microbiol. Mol. Biol. Rev. 2016, 80, 679–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacks, T.; Powert, M.D.; Masiarz, F.R.; Luciw, P.A.; Barr, P.J.; Varmus, H.E. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 1988, 331, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.E.; Saad, J.S. The interplay between HIV-1 Gag binding to the plasma membrane and Env incorporation. Viruses 2020, 12, 548. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Kharytonchyk, S.; Chaudry, I.; Iyer, A.S.; Carter, H.; Becker, G.; Desai, Y.; Glang, L.; Choi, S.H.; Singh, K.; et al. Structural basis for transcriptional start site control of HIV-1 RNA fate. Science 2020, 368, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.; Kharytonchyk, S.; Kuo, N.; Cannistraci, E.; Flores, H.; Chaudhary, R.; Sarkar, M.; Dong, X.; Telesnitsky, A.; Summers, M.F. 5′-Cap sequestration is an essential determinant of HIV-1 genome packaging. Proc. Natl. Acad. Sci. USA 2021, 118, e2112475118. [Google Scholar] [CrossRef]
- Mouhand, A.; Pasi, M.; Catala, M.; Zargarian, L.; Belfetmi, A.; Barraud, P.; Mauffret, O.; Tisné, C. Overview of the nucleic-acid binding properties of the HIV-1 nucleocapsid protein in its different maturation states. Viruses 2020, 12, 1109. [Google Scholar] [CrossRef]
- Zhao, H.; Datta, S.A.K.; Kim, S.H.; To, S.C.; Chaturvedi, S.K.; Rein, A.; Schuck, P. Nucleic acid–induced dimerization of HIV-1 Gag protein. J. Biol. Chem. 2019, 294, 16480–16493. [Google Scholar] [CrossRef] [Green Version]
- Sarni, S.; Biswas, B.; Liu, S.; Olson, E.D.; Kitzrow, J.P.; Rein, A.; Wysocki, V.H.; Musier-Forsyth, K. HIV-1 Gag protein with or without p6 specifically dimerizes on the viral RNA packaging signal. J. Biol. Chem. 2020, 295, 14391–14401. [Google Scholar] [CrossRef]
- Murray, P.S.; Li, Z.; Wang, J.; Tang, C.L.; Honig, B.; Murray, D. Retroviral matrix domains share electrostatic homology: Models for membrane binding function throughout the viral life cycle. Structure 2005, 13, 1521–1531. [Google Scholar] [CrossRef]
- Alfadhli, A.; Still, A.; Barklis, E. Analysis of Human Immunodeficiency Virus Type 1 Matrix Binding to Membranes and Nucleic Acids. J. Virol. 2009, 83, 12196–12203. [Google Scholar] [CrossRef] [Green Version]
- Gaines, C.R.; Tkacik, E.; Rivera-Oven, A.; Somani, P.; Achimovich, A.; Alabi, T.; Zhu, A.; Getachew, N.; Yang, A.L.; McDonough, M.; et al. HIV-1 Matrix Protein Interactions with tRNA: Implications for Membrane Targeting. J. Mol. Biol. 2018, 430, 2113–2127. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.K.; Heinrich, F.; Raghunandan, S.; Krueger, S.; Curtis, J.E.; Rein, A.; Nanda, H. HIV-1 Gag extension: Conformational changes require simultaneous interaction with membrane and nucleic acid. J. Mol. Biol. 2011, 406, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Xu, J.; Liu, M.; Rao, A.L.N.; Zandi, R.; Gill, S.S.; Mohideen, U. Investigation of HIV-1 Gag binding with RNAs and lipids using Atomic Force Microscopy. PLoS ONE 2020, 15, e0228036. [Google Scholar] [CrossRef] [PubMed]
- Mateu, M.G. The capsid protein of human immunodeficiency virus: Intersubunit interactions during virus assembly. FEBS J. 2009, 276, 6098–6109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, E.; Mammano, F.; Cohen, E.A.; Göttlinger, H.G. The p6gag domain of human immunodeficiency virus type 1 is sufficient for the incorporation of Vpr into heterologous viral particles. J. Virol. 1995, 69, 2759–2764. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Cada, A.K. Inside job: How the ESCRTs release HIV-1 from infected cells. Biochem. Soc. Trans. 2018, 46, 1029–1036. [Google Scholar] [CrossRef]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 maturation: Lessons learned from inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef]
- Pornillos, O.; Ganser-Pornillos, B.K. Maturation of retroviruses. Curr. Opin. Virol. 2019, 36, 47–55. [Google Scholar] [CrossRef]
- Blakemore, R.J.; Burnett, C.; Swanson, C.; Kharytonchyk, S.; Telesnitsky, A.; Munro, J.B. Stability and conformation of the dimeric HIV-1 genomic RNA 5′UTR. Biophys. J. 2021, 120, 4874–4890. [Google Scholar] [CrossRef]
- Murakami, T.; Ablan, S.; Freed, E.O.; Tanaka, Y. Regulation of Human Immunodeficiency Virus Type 1 Env-Mediated Membrane Fusion by Viral Protease Activity. J. Virol. 2004, 78, 1026–1031. [Google Scholar] [CrossRef] [Green Version]
- Elcheva, I.A.; Spiegelman, V.S. The role of cis-and trans-acting rna regulatory elements in leukemia. Cancers 2020, 12, 3854. [Google Scholar] [CrossRef] [PubMed]
- Ranum, L.P.W.; Cooper, T.A. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 2006, 29, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. Expandable DNA repeats and human disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Heinz, A.; Nabariya, D.K.; Krauss, S. Huntingtin and Its Role in Mechanisms of RNA-Mediated Toxicity. Toxins 2021, 13, 487. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, D.Y.; Lee, K.H. Rna-binding proteins and the complex pathophysiology of als. Int. J. Mol. Sci. 2021, 22, 2598. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Moreno, M.; Noerenberg, M.; Ni, S.; Järvelin, A.I.; González-Almela, E.; Lenz, C.E.; Bach-Pages, M.; Cox, V.; Avolio, R.; Davis, T.; et al. System-wide Profiling of RNA-Binding Proteins Uncovers Key Regulators of Virus Infection. Mol. Cell 2019, 74, 196–211.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.T.; Vrolijk, M.M.; Harwig, A.; Berkhout, B. Opening of the TAR hairpin in the HIV-1 genome causes aberrant RNA dimerization and packaging. Retrovirology 2012, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.D.; Jayaraman, B.; Frankel, A.D. The HIV-1 Rev response element. RNA Biol. 2012, 9, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: Role in Bystander Toxicity. Front. Cell. Infect. Microbiol. 2020, 10, 61. [Google Scholar] [CrossRef]
- Fujii, R.; Okamoto, M.; Aratani, S.; Oishi, T.; Ohshima, T.; Taira, K.; Baba, M.; Fukamizu, A.; Nakajima, T. A Role of RNA Helicase A in cis-Acting Transactivation Response Element-mediated Transcriptional Regulation of Human Immunodeficiency Virus Type 1. J. Biol. Chem. 2001, 276, 5445–5451. [Google Scholar] [CrossRef] [Green Version]
- Gerena, Y.; Menéndez-Delmestre, R.; Delgado-Nieves, A.; Vélez, J.; Méndez-Álvarez, J.; Sierra-Pagan, J.E.; Skolasky, R.L.; Henderson, L.; Nath, A.; Wojna, V. Release of Soluble Insulin Receptor From Neurons by Cerebrospinal Fluid From Patients With Neurocognitive Dysfunction and HIV Infection. Front. Neurol. 2019, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Moir, S.; Chun, T.-W.; Fauci, A.S. Pathogenic Mechanisms of HIV Disease. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 223–248. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Prim. 2015, 1, 15035. [Google Scholar] [CrossRef]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Prim. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Justiz Vaillant, A.A.; Naik, R. HIV-1 Associated Opportunistic Infections. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30969609 (accessed on 21 October 2021).
- U.S. Department of Health & Human Services Global Statistics. Available online: https://www.hiv.gov/hiv-basics/overview/data-and-trends/global-statistics (accessed on 21 October 2021).
- Maenza, J.; Flexner, C. Combination antiretroviral therapy for HIV infection. Am. Fam. Physician 1998, 57, 2789–2798. [Google Scholar] [PubMed]
- Warnke, D.; Barreto, J.; Temesgen, Z. Therapeutic review: Antiretroviral drugs. J. Clin. Pharmacol. 2007, 47, 1570–1579. [Google Scholar] [CrossRef]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. In Dep. Heal. Hum. Serv. Available online: https://clinicalinfo.hiv.gov/sites/default/files/guidelines/archive/AdultandAdolescentGL_2021_08_16.pdf (accessed on 25 October 2021).
- U.S. Department of Health & Human Services FDA-Approved HIV Medicines. Available online: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines (accessed on 21 October 2021).
- Qi, B.; Fang, Q.; Liu, S.; Hou, W.; Li, J.; Huang, Y.; Shi, J. Advances of CCR5 antagonists: From small molecules to macromolecules. Eur. J. Med. Chem. 2020, 208, 112819. [Google Scholar] [CrossRef]
- Jamjian, M.C.; McNicholl, I.R. Enfuvirtide: First fusion inhibitor for treatment of HIV infection. Am. J. Health Pharm. 2004, 61, 1242–1247. [Google Scholar] [CrossRef]
- Meanwell, N.A.; Krystal, M.R.; Nowicka-Sans, B.; Langley, D.R.; Conlon, D.A.; Eastgate, M.D.; Grasela, D.M.; Timmins, P.; Wang, T.; Kadow, J.F. Inhibitors of HIV-1 Attachment: The Discovery and Development of Temsavir and its Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 62–80. [Google Scholar] [CrossRef]
- Beccari, M.V.; Mogle, B.T.; Sidman, E.F.; Mastro, K.A.; Asiago-Reddy, E.; Kufel, W.D. Ibalizumab, a Novel Monoclonal Antibody for the Management of Multidrug-Resistant HIV-1 Infection. Antimicrob. Agents Chemother. 2019, 63, e00110–e00119. [Google Scholar] [CrossRef] [Green Version]
- Kemnic, T.R.; Gulick, P.G. HIV Antiretroviral Therapy. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30020680 (accessed on 21 October 2021).
- Ghosh, R.K.; Ghosh, S.M.; Chawla, S. Recent advances in antiretroviral drugs. Expert Opin. Pharmacother. 2011, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.S. Protease-Mediated Maturation of HIV: Inhibitors of Protease and the Maturation Process. Mol. Biol. Int. 2012, 2012, 604261. [Google Scholar] [CrossRef] [PubMed]
- Palella, F.J.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining Morbidity and Mortality among Patients with Advanced Human Immunodeficiency Virus Infection. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.D.; Chaisson, R.E. Natural history of HIV infection in the era of combination antiretroviral therapy. Aids 1999, 13, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H.S.; et al. Prevention of HIV-1 Infection with Early Antiretroviral Therapy. N. Engl. J. Med. 2011, 365, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H.S.; et al. Antiretroviral Therapy for the Prevention of HIV-1 Transmission. N. Engl. J. Med. 2016, 375, 830–839. [Google Scholar] [CrossRef]
- Thomas, R.; Galanakis, C.; Vézina, S.; Longpré, D.; Boissonnault, M.; Huchet, E.; Charest, L.; Murphy, D.; Trottier, B.; Machouf, N. Adherence to Post-Exposure Prophylaxis (PEP) and Incidence of HIV Seroconversion in a Major North American Cohort. PLoS ONE 2015, 10, e0142534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, D. Post-exposure prophylaxis for HIV infection. Expert Rev. Anti. Infect. Ther. 2011, 9, 431–442. [Google Scholar] [CrossRef]
- Mohri, H.; Singh, M.K.; Ching, W.T.W.; Ho, D.D. Quantitation of zidovudine-resistant human immunodeficiency virus type 1 in the blood of treated and untreated patients. Proc. Natl. Acad. Sci. USA 1993, 90, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, D.E.; Webb, G.F. Understanding drug resistance for monotherapy treatment of HIV infection. Bull. Math. Biol. 1997, 59, 763–785. [Google Scholar] [CrossRef]
- Pennings, P.S. HIV drug resistance: Problems and perspectives. Infect. Dis. Rep. 2013, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richman, D.D. HIV drug resistance. Annu. Rev. Pharmacol. Toxicol. 1993, 33, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Dau, B.; Holodniy, M. Novel targets for antiretroviral therapy: Clinical progress to date. Drugs 2009, 69, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.F.; Simões, S.; Carvalheiro, M.; Pereira, J.M.A.; Costa, Q.; Ascenso, A. Novel Antiretroviral Therapeutic Strategies for HIV. Molecules 2021, 26, 5305. [Google Scholar] [CrossRef] [PubMed]
- Cutrell, A.G.; Schapiro, J.M.; Perno, C.F.; Kuritzkes, D.R.; Quercia, R.; Patel, P.; Polli, J.W.; Dorey, D.; Wang, Y.; Wu, S.; et al. Exploring predictors of HIV-1 virologic failure to long-acting cabotegravir and rilpivirine: A multivariable analysis. AIDS 2021, 35, 1333–1342. [Google Scholar] [CrossRef]
- Rizzardini, G.; Overton, E.T.; Orkin, C.; Swindells, S.; Arasteh, K.; Górgolas Hernández-Mora, M.; Pokrovsky, V.; Girard, P.-M.; Oka, S.; Andrade-Villanueva, J.F.; et al. Long-Acting Injectable Cabotegravir + Rilpivirine for HIV Maintenance Therapy: Week 48 Pooled Analysis of Phase 3 ATLAS and FLAIR Trials. JAIDS J. Acquir. Immune Defic. Syndr. 2020, 85, 498–506. [Google Scholar] [CrossRef]
- Li, X.-D.; Liu, L.; Cheng, L. Identification of thienopyridine carboxamides as selective binders of HIV-1 trans Activation Response (TAR) and Rev Response Element (RRE) RNAs. Org. Biomol. Chem. 2018, 16, 9191–9196. [Google Scholar] [CrossRef]
- Chavali, S.S.; Mali, S.M.; Jenkins, J.L.; Fasan, R.; Wedekind, J.E. Co-crystal structures of HIV TAR RNA bound to lab-evolved proteins show key roles for arginine relevant to the design of cyclic peptide TAR inhibitors. J. Biol. Chem. 2020, 295, 16470–16486. [Google Scholar] [CrossRef]
- Melidis, L.; Styles, I.B.; Hannon, M.J. Targeting structural features of viral genomes with a nano-sized supramolecular drug. Chem. Sci. 2021, 12, 7174–7184. [Google Scholar] [CrossRef]
- Sosic, A.; Olivato, G.; Carraro, C.; Göttlich, R.; Fabris, D.; Gatto, B. Bis-3-Chloropiperidines Targeting TAR RNA as A Novel Strategy to Impair the HIV-1 Nucleocapsid Protein. Molecules 2021, 26, 1874. [Google Scholar] [CrossRef]
- Dai, Y.; Wynn, J.E.; Peralta, A.N.; Sherpa, C.; Jayaraman, B.; Li, H.; Verma, A.; Frankel, A.D.; Le Grice, S.F.; Santos, W.L. Discovery of a Branched Peptide That Recognizes the Rev Response Element (RRE) RNA and Blocks HIV-1 Replication. J. Med. Chem. 2018, 61, 9611–9620. [Google Scholar] [CrossRef] [PubMed]
- Medina-Trillo, C.; Sedgwick, D.M.; Herrera, L.; Beltrán, M.; Moreno, Á.; Barrio, P.; Bedoya, L.M.; Alcamí, J.; Fustero, S.; Gallego, J. Nucleic acid recognition and antiviral activity of 1,4-substituted terphenyl compounds mimicking all faces of the HIV-1 Rev protein positively-charged α-helix. Sci. Rep. 2020, 10, 7190. [Google Scholar] [CrossRef] [PubMed]
- Dietz, J.; Koch, J.; Kaur, A.; Raja, C.; Stein, S.; Grez, M.; Pustowka, A.; Mensch, S.; Ferner, J.; Möller, L.; et al. Inhibition of HIV-1 by a Peptide Ligand of the Genomic RNA Packaging Signal Ψ. ChemMedChem 2008, 3, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Warui, D.M.; Baranger, A.M. Identification of specific small molecule ligands for stem loop 3 ribonucleic acid of the packaging signal Ψ of human immunodeficiency virus-1. J. Med. Chem. 2009, 52, 5462–5473. [Google Scholar] [CrossRef] [PubMed]
- Ingemarsdotter, C.K.; Zeng, J.; Long, Z.; Lever, A.M.L.; Kenyon, J.C. An RNA-binding compound that stabilizes the HIV-1 gRNA packaging signal structure and specifically blocks HIV-1 RNA encapsidation. Retrovirology 2018, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Eakle, R.; Venter, F.; Rees, H. Pre-exposure prophylaxis (PrEP) in an era of stalled HIV prevention: Can it change the game? Retrovirology 2018, 15, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, T.D.; Carter, A.; Jahagirdar, D.; Biehl, M.H.; Douwes-Schultz, D.; Larson, S.L.; Arora, M.; Dwyer-Lindgren, L.; Steuben, K.M.; Abbastabar, H.; et al. Global, regional, and national incidence, prevalence, and mortality of HIV, 1980–2017, and forecasts to 2030, for 195 countries and territories: A systematic analysis for the Global Burden of Diseases, Injuries, and Risk Factors Study 2017. Lancet HIV 2019, 6, e831–e859. [Google Scholar] [CrossRef] [Green Version]
- Assessing Global HIV Targets in PEPFAR Countries: A Dashboard. Available online: https://www.kff.org/global-health-policy/issue-brief/assessing-global-hiv-targets-in-pepfar-countries-a-dashboard/# (accessed on 21 October 2021).
- UNAIDS Joint United Nations; Programme on HIV/AIDS 90-90-90 An ambitious treatment target to help end the AIDS epidemic. United Nations 2014, 40.
- Joulaei, H.; Shooshtarian, S.; Dianatinasab, M. Is UNAIDS 90-90-90 Target a Dream or a Reality for Middle East and North Africa Region on Ending the AIDS Epidemic? A Review Study. Aids Rev. 2019, 20, 83–93. [Google Scholar] [CrossRef]
- Porter, K.; Gourlay, A.; Attawell, K.; Hales, D.; Supervie, V.; Touloumi, G.; Rosinska, M.; Vourli, G.; van Sighem, A.; Pharris, A.; et al. Substantial Heterogeneity in Progress Toward Reaching the 90-90-90 HIV Target in the WHO European Region. JAIDS J. Acquir. Immune Defic. Syndr. 2018, 79, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Schackman, B.R.; Fleishman, J.A.; Su, A.E.; Berkowitz, B.K.; Moore, R.D.; Walensky, R.P.; Becker, J.E.; Voss, C.; Paltiel, A.D.; Weinstein, M.C.; et al. The lifetime medical cost savings from preventing HIV in the United States. Med. Care 2015, 53, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, N.C.; Horn, T.H.; Hyle, E.P.; Walensky, R.P. HIV Antiretroviral Therapy Costs in the United States, 2012–2018. JAMA Intern. Med. 2020, 180, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.T.; Bhat, N.; Yoder, C.; Chun, T.-W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [Green Version]
- Hamers, R.L.; Rinke de Wit, T.F.; Holmes, C.B. HIV drug resistance in low-income and middle-income countries. Lancet HIV 2018, 5, e588–e596. [Google Scholar] [CrossRef]
- Bandera, A.; Gori, A.; Clerici, M.; Sironi, M. Phylogenies in ART: HIV reservoirs, HIV latency and drug resistance. Curr. Opin. Pharmacol. 2019, 48, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Saleem, K.; Lim, M.; Chow, E.P.F.; Fairley, C.K.; Terris-Prestholt, F.; Ong, J.J. Global estimates for the lifetime cost of managing HIV. AIDS 2021, 35, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.L. HIV/AIDS Vaccines: 2018. Clin. Pharmacol. Ther. 2018, 104, 1062–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Douek, D.C.; Kwong, P.D.; Nabel, G.J. The rational design of an AIDS vaccine. Cell 2006, 124, 677–681. [Google Scholar] [CrossRef] [Green Version]
- Mona Sadat, L.; Seyed Mehdi, S.; Amitis, R. HIV-1 Immune evasion: The main obstacle toward a successful vaccine. Arch. Asthma, Allergy Immunol. 2018, 2, 13–15. [Google Scholar] [CrossRef] [Green Version]
- Rolland, M. HIV-1 immune evasion—A threat to effective vaccines? Nat. Med. 2016, 22, 580–581. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.L.; Milush, J.M.; Buggert, M.; Eriksson, E.M.; Larsen, M.V.; Liegler, T.; Hartogensis, W.; Bacchetti, P.; Lund, O.; Hecht, F.M.; et al. Targeting of Conserved Gag-Epitopes in Early HIV Infection Is Associated with Lower Plasma Viral Load and Slower CD4 + T Cell Depletion. AIDS Res. Hum. Retroviruses 2013, 29, 602–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiepiela, P.; Ngumbela, K.; Thobakgale, C.; Ramduth, D.; Honeyborne, I.; Moodley, E.; Reddy, S.; De Pierres, C.; Mncube, Z.; Mkhwanazi, N.; et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 2007, 13, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Guha, D.; Ayyavoo, V. Innate Immune Evasion Strategies by Human Immunodeficiency Virus Type 1. ISRN AIDS 2013, 2013, 954806. [Google Scholar] [CrossRef] [Green Version]
- Blankson, J.N.; Persaud, D.; Siliciano, R.F. The Challenge of Viral Reservoirs in HIV-1 Infection. Annu. Rev. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef]
- Tomaras, G.D.; Haynes, B.F. HIV-1-specific antibody responses during acute and chronic HIV-1 infection. Curr. Opin. HIV AIDS 2009, 4, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Overbaugh, J.; Morris, L. The Antibody Response against HIV-1. Cold Spring Harb. Perspect. Med. 2012, 2, a007039. [Google Scholar] [CrossRef]
- Wyatt, R.; Kwong, P.D.; Desjardins, E.; Sweet, R.W.; Robinson, J.; Hendrickson, W.A.; Sodroski, J.G. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 1998, 393, 705–711. [Google Scholar] [CrossRef]
- Ward, A.B.; Wilson, I.A. The HIV-1 envelope glycoprotein structure: Nailing down a moving target. Immunol. Rev. 2017, 275, 21–32. [Google Scholar] [CrossRef]
- Harada, S.; Yoshimura, K. Driving HIV-1 into a Vulnerable corner by taking advantage of viral adaptation and evolution. Front. Microbiol. 2017, 8, 390. [Google Scholar] [CrossRef] [Green Version]
- Hraber, P.; Seaman, M.S.; Bailer, R.T.; Mascola, J.R.; Montefiori, D.C.; Korber, B.T. Prevalence of broadly neutralizing antibody responses during chronic HIV-1 infection. AIDS 2014, 28, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Doores, K.J. The HIV glycan shield as a target for broadly neutralizing antibodies. FEBS J. 2015, 282, 4679–4691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sok, D.; Burton, D.R. Recent progress in broadly neutralizing antibodies to HIV. Nat. Immunol. 2018, 19, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Binley, J.M.; Wrin, T.; Korber, B.; Zwick, M.B.; Wang, M.; Chappey, C.; Stiegler, G.; Kunert, R.; Zolla-Pazner, S.; Katinger, H.; et al. Comprehensive Cross-Clade Neutralization Analysis of a Panel of Anti-Human Immunodeficiency Virus Type 1 Monoclonal Antibodies. J. Virol. 2004, 78, 13232–13252. [Google Scholar] [CrossRef] [Green Version]
- Gray, E.S.; Moore, P.L.; Choge, I.A.; Decker, J.M.; Bibollet-Ruche, F.; Li, H.; Leseka, N.; Treurnicht, F.; Mlisana, K.; Shaw, G.M.; et al. Neutralizing Antibody Responses in Acute Human Immunodeficiency Virus Type 1 Subtype C Infection. J. Virol. 2007, 81, 6187–6196. [Google Scholar] [CrossRef] [Green Version]
- Gorny, M.K.; Conley, A.J.; Karwowska, S.; Buchbinder, A.; Xu, J.Y.; Emini, E.A.; Koenig, S.; Zolla-Pazner, S. Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J. Virol. 1992, 66, 7538–7542. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.-Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; et al. Broad and Potent Neutralizing Antibodies from an African Donor Reveal a New HIV-1 Vaccine Target. Science 2009, 326, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Woldemeskel, B.A.; Kwaa, A.K.; Blankson, J.N. Viral reservoirs in elite controllers of HIV-1 infection: Implications for HIV cure strategies. EBioMedicine 2020, 62, 103118. [Google Scholar] [CrossRef]
- Deeks, S.G.; Walker, B.D. Human Immunodeficiency Virus Controllers: Mechanisms of Durable Virus Control in the Absence of Antiretroviral Therapy. Immunity 2007, 27, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.D.; Yu, X.G. Unravelling the mechanisms of durable control of HIV-1. Nat. Rev. Immunol. 2013, 13, 487–498. [Google Scholar] [CrossRef]
- Baker, B.; Block, B.; Rothchild, A.; Walker, B. Elite control of HIV infection: Implications for vaccine design. Expert Opin. Biol. Ther. 2009, 9, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Lian, X.; Gao, C.; Sun, X.; Einkauf, K.B.; Chevalier, J.M.; Chen, S.M.Y.; Hua, S.; Rhee, B.; Chang, K.; et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 2020, 585, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H. Challenges in the development of an HIV-1 vaccine. Nature 2008, 455, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.W.; Frahm, N. Current views on the potential for development of a HIV vaccine. Expert Opin. Biol. Ther. 2017, 17, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Zolla-Pazner, S. Identifying epitopes of HIV-1 that induce protective antibodies. Nat. Rev. Immunol. 2004, 4, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Pantophlet, R.; Burton, D.R. GP120: Target for Neutralizing HIV-1 Antibodies. Annu. Rev. Immunol. 2006, 24, 739–769. [Google Scholar] [CrossRef]
- Mascola, J.R.; Montefiori, D.C. The Role of Antibodies in HIV Vaccines. Annu. Rev. Immunol. 2010, 28, 413–444. [Google Scholar] [CrossRef]
- Burton, D.R.; Hangartner, L. Broadly Neutralizing Antibodies to HIV and Their Role in Vaccine Design. Annu. Rev. Immunol. 2016, 34, 635–659. [Google Scholar] [CrossRef]
- Saphire, E.O.; Parren, P.W.H.I.; Pantophlet, R.; Zwick, M.B.; Morris, G.M.; Rudd, P.M.; Dwek, R.A.; Stanfield, R.L.; Burton, D.R.; Wilson, I.A. Crystal Structure of a Neutralizing Human IgG Against HIV-1: A Template for Vaccine Design. Science 2001, 293, 1155–1159. [Google Scholar] [CrossRef]
- Ghosn, J.; Taiwo, B.; Seedat, S.; Autran, B.; Katlama, C. HIV. Lancet 2018, 392, 685–697. [Google Scholar] [CrossRef]
- Zagury, D.; Léonard, R.; Fouchard, M.; Réveil, B.; Bernard, J.; Ittelé, D.; Cattan, A.; Zirimwabagabo, L.; Kalumbu, M.; Justin, W.; et al. Immunization against AIDS in humans. Nature 1987, 326, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Hatziioannou, T.; Evans, D.T. Animal models for HIV/AIDS research. Nat Rev Microbiol. 2015, 10, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Policicchio, B.B.; Pandrea, I.; Apetrei, C. Animal Models for HIV Cure Research. Front. Immunol. 2016, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rompay, K.K.A. Tackling HIV and AIDS: Contributions by non-human primate models. Lab Anim. 2017, 46, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Ng’uni, T.; Chasara, C.; Ndhlovu, Z.M. Major Scientific Hurdles in HIV Vaccine Development: Historical Perspective and Future Directions. Front. Immunol. 2020, 11, 590780. [Google Scholar] [CrossRef]
- Esparza, J. A brief history of the global effort to develop a preventive HIV vaccine. Vaccine 2013, 31, 3502–3518. [Google Scholar] [CrossRef] [Green Version]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to Prevent HIV-1 Infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef]
- Pitisuttithum, P.; Rerks-Ngarm, S.; Bussaratid, V.; Dhitavat, J.; Maekanantawat, W.; Pungpak, S.; Suntharasamai, P.; Vanijanonta, S.; Nitayapan, S.; Kaewkungwal, J.; et al. Safety and Reactogenicity of Canarypox ALVAC-HIV (vCP1521) and HIV-1 gp120 AIDSVAX B/E Vaccination in an Efficacy Trial in Thailand. PLoS ONE 2011, 6, e27837. [Google Scholar] [CrossRef] [Green Version]
- Zolla-Pazner, S.; DeCamp, A.; Gilbert, P.B.; Williams, C.; Yates, N.L.; Williams, W.T.; Howington, R.; Fong, Y.; Morris, D.E.; Soderberg, K.A.; et al. Vaccine-Induced IgG Antibodies to V1V2 Regions of Multiple HIV-1 Subtypes Correlate with Decreased Risk of HIV-1 Infection. PLoS ONE 2014, 9, e87572. [Google Scholar] [CrossRef]
- Karasavvas, N.; Billings, E.; Rao, M.; Williams, C.; Zolla-Pazner, S.; Bailer, R.T.; Koup, R.A.; Madnote, S.; Arworn, D.; Shen, X.; et al. The Thai Phase III HIV Type 1 Vaccine Trial (RV144) Regimen Induces Antibodies That Target Conserved Regions Within the V2 Loop of gp120. AIDS Res. Hum. Retrovir. 2012, 28, 1444–1457. [Google Scholar] [CrossRef]
- Lin, L.; Finak, G.; Ushey, K.; Seshadri, C.; Hawn, T.R.; Frahm, N.; Scriba, T.J.; Mahomed, H.; Hanekom, W.; Bart, P.A.; et al. COMPASS identifies T-cell subsets correlated with clinical outcomes. Nat. Biotechnol. 2015, 33, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Gilbert, P.B.; McElrath, M.J.; Zolla-Pazner, S.; Tomaras, G.D.; Alam, S.M.; Evans, D.T.; Montefiori, D.C.; Karnasuta, C.; Sutthent, R.; et al. Immune-Correlates Analysis of an HIV-1 Vaccine Efficacy Trial. N. Engl. J. Med. 2012, 366, 1275–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Excler, J.-L.; Nitayaphan, S.; Kaewkungwal, J.; Premsri, N.; Kunasol, P.; Karasavvas, N.; Schuetz, A.; Ngauy, V.; et al. Randomized, Double-Blind Evaluation of Late Boost Strategies for HIV-Uninfected Vaccine Recipients in the RV144 HIV Vaccine Efficacy Trial. J. Infect. Dis. 2017, 215, 1255–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitisuttithum, P.; Nitayaphan, S.; Chariyalertsak, S.; Kaewkungwal, J.; Dawson, P.; Dhitavat, J.; Phonrat, B.; Akapirat, S.; Karasavvas, N.; Wieczorek, L.; et al. Late boosting of the RV144 regimen with AIDSVAX B/E and ALVAC-HIV in HIV-uninfected Thai volunteers: A double-blind, randomised controlled trial. Lancet HIV 2020, 7, e238–e248. [Google Scholar] [CrossRef]
- Gray, G.E.; Bekker, L.-G.; Laher, F.; Malahleha, M.; Allen, M.; Moodie, Z.; Grunenberg, N.; Huang, Y.; Grove, D.; Prigmore, B.; et al. Vaccine Efficacy of ALVAC-HIV and Bivalent Subtype C gp120–MF59 in Adults. N. Engl. J. Med. 2021, 384, 1089–1100. [Google Scholar] [CrossRef]
- Seaman, M.S.; Janes, H.; Hawkins, N.; Grandpre, L.E.; Devoy, C.; Giri, A.; Coffey, R.T.; Harris, L.; Wood, B.; Daniels, M.G.; et al. Tiered Categorization of a Diverse Panel of HIV-1 Env Pseudoviruses for Assessment of Neutralizing Antibodies. J. Virol. 2010, 84, 1439–1452. [Google Scholar] [CrossRef] [Green Version]
- Fischer, W.; Perkins, S.; Theiler, J.; Bhattacharya, T.; Yusim, K.; Funkhouser, R.; Kuiken, C.; Haynes, B.; Letvin, N.L.; Walker, B.D.; et al. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat. Med. 2007, 13, 100–106. [Google Scholar] [CrossRef]
- Corey, L.; McElrath, M.J. HIV vaccines: Mosaic approach to virus diversity. Nat. Med. 2010, 16, 268–270. [Google Scholar] [CrossRef]
- Barouch, D.H.; O’Brien, K.L.; Simmons, N.L.; King, S.L.; Abbink, P.; Maxfield, L.F.; Sun, Y.H.; La Porte, A.; Riggs, A.M.; Lynch, D.M.; et al. Mosaic HIV-1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat. Med. 2010, 16, 319–323. [Google Scholar] [CrossRef] [Green Version]
- Santra, S.; Liao, H.X.; Zhang, R.; Muldoon, M.; Watson, S.; Fischer, W.; Theiler, J.; Szinger, J.; Balachandran, H.; Buzby, A.; et al. Mosaic vaccines elicit CD8+ T lymphocyte responses that confer enhanced immune coverage of diverse HIV strains in monkeys. Nat. Med. 2010, 16, 324–328. [Google Scholar] [CrossRef]
- Barouch, D.H.; Tomaka, F.L.; Wegmann, F.; Stieh, D.J.; Alter, G.; Robb, M.L.; Michael, N.L.; Peter, L.; Nkolola, J.P.; Borducchi, E.N.; et al. Evaluation of a mosaic HIV-1 vaccine in a multicentre, randomised, double-blind, placebo-controlled, phase 1/2a clinical trial (APPROACH) and in rhesus monkeys (NHP 13-19). Lancet 2018, 392, 232–243. [Google Scholar] [CrossRef]
- Sanders, R.W.; Derking, R.; Cupo, A.; Julien, J.-P.; Yasmeen, A.; de Val, N.; Kim, H.J.; Blattner, C.; de la Peña, A.T.; Korzun, J.; et al. A Next-Generation Cleaved, Soluble HIV-1 Env Trimer, BG505 SOSIP.664 gp140, Expresses Multiple Epitopes for Broadly Neutralizing but Not Non-Neutralizing Antibodies. PLoS Pathog. 2013, 9, e1003618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, R.W.; Van Gils, M.J.; Derking, R.; Sok, D.; Ketas, T.J.; Burger, J.A.; Ozorowski, G.; Cupo, A.; Simonich, C.; Goo, L.; et al. HIV-1 neutralizing antibodies induced by native-like envelope trimers. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Study to Assess the Efficacy of a Heterologous Prime/Boost Vaccine Regimen of Ad26.Mos4.HIV and Aluminum Phosphate-Adjuvanted Clade C gp140 in Preventing Human Immunodeficiency Virus (HIV)-1 Infection in Women in Sub-Saharan Africa [NCT03060629]. Available online: https://clinicaltrials.gov/ct2/show/NCT03060629 (accessed on 21 October 2021).
- Johnson & Johnson and Global Partners Announce Results from Phase 2b Imbokodo HIV Vaccine Clinical Trial in Young Women in Sub-Saharan Africa. Available online: https://www.janssen.com/johnson-johnson-and-global-partners-announce-results-phase-2b-imbokodo-hiv-vaccine-clinical-trial (accessed on 21 October 2021).
- A Study of Heterologous Vaccine Regimen of Adenovirus Serotype 26 Mosaic4 Human Immunodeficiency Virus(Ad26.Mos4.HIV), Adjuvanted Clade C gp140 and Mosaic gp140 to Prevent HIV-1 Infection Among Cis-gender Men and Transgender Individuals Who Have Sex With. Available online: https://clinicaltrials.gov/ct2/show/NCT03964415 (accessed on 29 October 2021).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Restriction Factor (RF) | RF Mechanism of Action | HIV Counter Reaction | References |
|---|---|---|---|
| APOBEC3G | Encapsidated into virion, induces G-to-A hypermutation during reverse transcription | Vif | [24,25] |
| IFITMs/IFI16 | Excludes viral mRNA from polysome processing, inhibits the protein synthesis | Nef | [26] |
| SAMHD1 | Deoxynucleoside triphosphate triphosphohydrolase 1 activity, prevents reverse transcription | Vpx (only HIV-2) | [25] |
| SerinC3/5 | Incorporated into the virion, inhibits membrane fusion | Nef | [27] |
| Tetherin/BST-2 | Anchors virions on the cell surface of infected cells, inhibits virion release | Vpu | [28,29] |
| TRIM5α/TRIMCyp/TRIM22 | Binds directly to HIV-1 capsids, accelerates uncoating and inhibits reverse transcription | p24-CA variation | [30] |
| Antiretroviral Drug Class | Mechanism of Action | Generic Name, Examples | FDA Approval Year |
|---|---|---|---|
| Nucleoside reverse transcriptase inhibitors (NRTIs) | Incorporation of nucleoside or nucleotide analogues by the reverse transcriptase leads to chain-termination of proviral DNA synthesis [13,120,121] | abacir | 1998 |
| emtricitabine | 2003 | ||
| lamivudine | 1995 | ||
| tenofovir disoproxil fumarate | 2001 | ||
| zidovudine | 1987 | ||
| Non-nucleoside reverse transcriptase inhibitors (NNRTIs) | NNRTIs bind the substrate pocket of the reverse transcriptase, hence reducing polymerase activity and impeding proviral DNA synthesis [13,120,121] | doravirine | 2018 |
| efavirenz | 1998 | ||
| etravirine | 2008 | ||
| nevirapine | 1996, 2001 | ||
| rilpivirine | 2011 | ||
| Protease inhibitors (PIs) | Blocking the active site of the viral protease inhibits the processing of the Gag-Pol polyprotein precursor [13,120,121,122] | atazanavir | 2003 |
| darunavir | 2006 | ||
| fosamprenavir | 2003 | ||
| ritonavir | 1996 | ||
| saquinavir | 1995 | ||
| tipranavir | 2005 | ||
| Integrase inhibitors (IIs) | Blocking the active site of the viral integrase inhibits insertion of the proviral DNA into host cell genome [13,120,121] | cabotegravir | 2021 |
| dolutegravir | 2013 | ||
| raltegravir | 2007, 2017 | ||
| (Post-)Attachment inhibitors (AIs) | Viral entry is either prevented by binding to gp120-SU (AIs) or by binding the CD4 receptor (post-AIs) [118,119] | fostemsavir | 2020 |
| ibalizumab-uiyk | 2018 | ||
| CCR5 antagonists | Blocking of the co-receptor CCR5 impedes viral entry [116] | maraviroc | 2007 |
| Fusion inhibitors (FIs) | Binding to gp41-TM inhibits viral entry [117] | enfuvirtide | 2003 |
| Vaccine Trial | Study ID | Start Year | Target Site | Vaccine Regimen | Outcome |
|---|---|---|---|---|---|
| HVTN 702 | NCT02968849 | 2016 | South Africa | IM administration of ALVAC-HIV (vCP2438) at months 0 and 1 followed by IM injection of ALVAC-HIV (vCP2438) and bivalent gp120-MF59 adjuvant at a total dose of 200 µg at months 3, 6, and 12 | Safe, no serious adverse events were observed, no sufficient protection |
| Mosaico | NCT03964415 | 2019 | Argentina Brazil Mexico Peru Italy Poland Spain USA | Priming (IM) with Ad26.Mos4.HIV (Ad26.Mos.1.Env, Ad26.Mos.2S.Env, Ad26.Mos1.Gag-Pol, Ad26.Mos2.Gag-Pol) at months 0 and 3 followed by boosting (IM) with Ad26.Mos4.HIV vaccine and adjuvanted bivalent clade C and mosaic gp140 at months 6 and 12 | Results not yet available |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Heuvel, Y.; Schatz, S.; Rosengarten, J.F.; Stitz, J. Infectious RNA: Human Immunodeficiency Virus (HIV) Biology, Therapeutic Intervention, and the Quest for a Vaccine. Toxins 2022, 14, 138. https://doi.org/10.3390/toxins14020138
van Heuvel Y, Schatz S, Rosengarten JF, Stitz J. Infectious RNA: Human Immunodeficiency Virus (HIV) Biology, Therapeutic Intervention, and the Quest for a Vaccine. Toxins. 2022; 14(2):138. https://doi.org/10.3390/toxins14020138
Chicago/Turabian Stylevan Heuvel, Yasemin, Stefanie Schatz, Jamila Franca Rosengarten, and Jörn Stitz. 2022. "Infectious RNA: Human Immunodeficiency Virus (HIV) Biology, Therapeutic Intervention, and the Quest for a Vaccine" Toxins 14, no. 2: 138. https://doi.org/10.3390/toxins14020138
APA Stylevan Heuvel, Y., Schatz, S., Rosengarten, J. F., & Stitz, J. (2022). Infectious RNA: Human Immunodeficiency Virus (HIV) Biology, Therapeutic Intervention, and the Quest for a Vaccine. Toxins, 14(2), 138. https://doi.org/10.3390/toxins14020138