Gut-Derived Metabolites and Their Role in Immune Dysfunction in Chronic Kidney Disease



Abstract
1. Introduction
2. The Gut Immune Homeostasis
3. Gut Dysbiosis in CKD
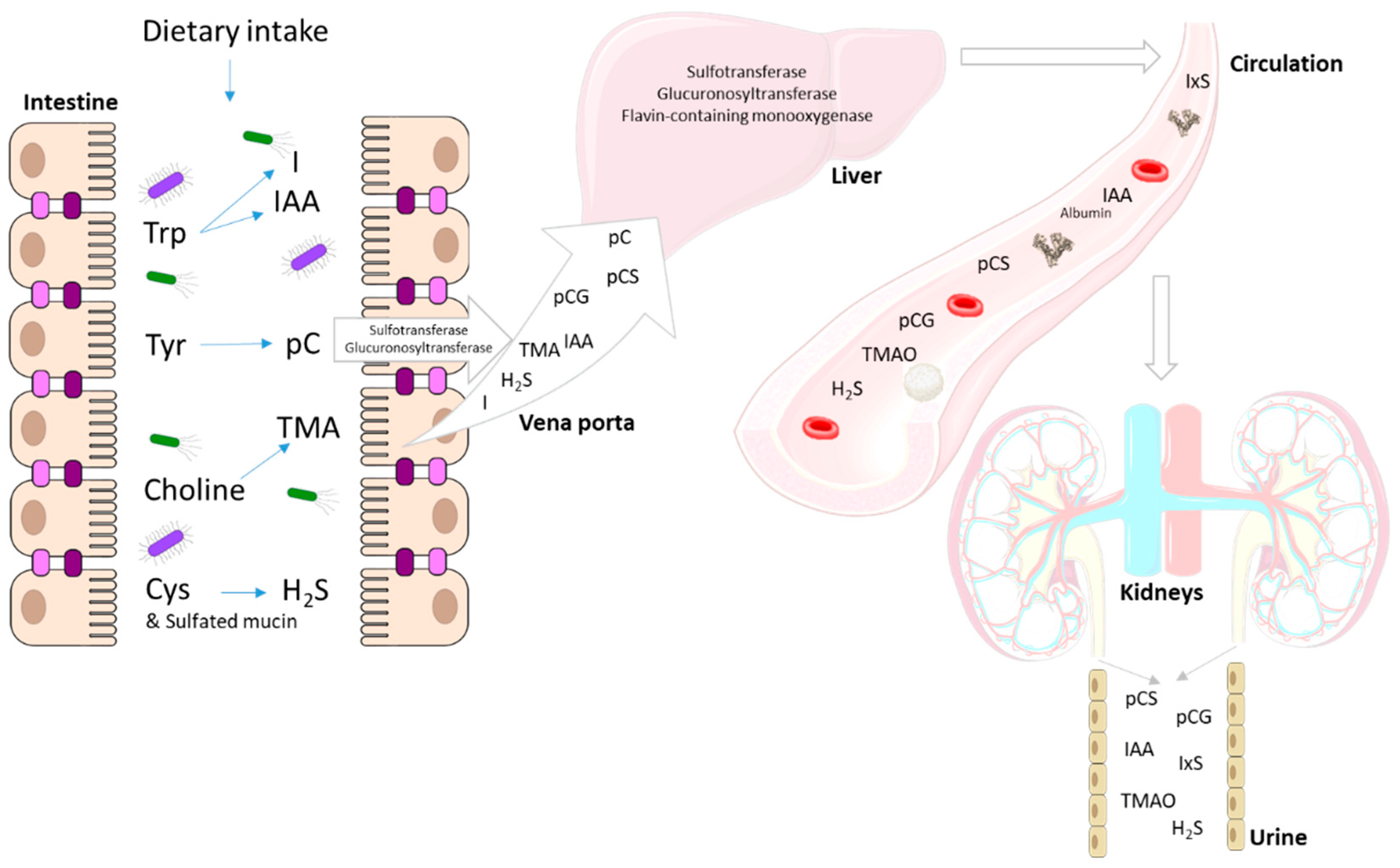
4. End Metabolites of Intestinal Bacterial Metabolism
4.1. p-Cresol
4.2. Indole and Indole-3-Acetic Acid
4.3. Trimethylamine
4.4. Sulfur Compounds
5. Immunomodulatory Effects of Uremic Toxins from Colonic Origin
5.1. p-Cresyl Sulfate
5.2. Indoxyl Sulfate
5.3. Indole-3-Acetic Acid
5.4. Trimethylamine N-Oxide
5.5. Sulfur Compounds
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Zewinger, S.; Schumann, T.; Fliser, D.; Speer, T. Innate immunity in CKD-associated vascular diseases. Nephrol. Dial. Transplant. 2016, 31, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Zhao, Y.Y.; Pahl, M.V. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol. Dial. Transplant. 2016, 31, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Aronov, P.A.; Luo, F.J.; Plummer, N.S.; Quan, Z.; Holmes, S.; Hostetter, T.H.; Meyer, T.W. Colonic contribution to uremic solutes. J. Am. Soc. Nephrol. JASN 2011, 22, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Mair, R.D.; Sirich, T.L.; Plummer, N.S.; Meyer, T.W. Characteristics of Colon-Derived Uremic Solutes. Clin. J. Am. Soc. Nephrol. CJASN 2018, 13, 1398–1404. [Google Scholar] [CrossRef]
- Dou, L.; Sallee, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. JASN 2015, 26, 876–887. [Google Scholar] [CrossRef]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef]
- Liabeuf, S.; Glorieux, G.; Lenglet, A.; Diouf, M.; Schepers, E.; Desjardins, L.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Does p-cresylglucuronide have the same impact on mortality as other protein-bound uremic toxins? PLoS ONE 2013, 8, e67168. [Google Scholar] [CrossRef]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef]
- Perna, A.F.; Glorieux, G.; Zacchia, M.; Trepiccione, F.; Capolongo, G.; Vigorito, C.; Anishchenko, E.; Ingrosso, D. The role of the intestinal microbiota in uremic solute accumulation: A focus on sulfur compounds. J. Nephrol. 2019, 32, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Maynard, C.L.; Elson, C.O.; Hatton, R.D.; Weaver, C.T. Reciprocal interactions of the intestinal microbiota and immune system. Nature 2012, 489, 231–241. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- McDermott, A.J.; Huffnagle, G.B. The microbiome and regulation of mucosal immunity. Immunology 2014, 142, 24–31. [Google Scholar] [CrossRef]
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef]
- Simenhoff, M.L.; Saukkonen, J.J.; Burke, J.F.; Wesson, L.G., Jr.; Schaedler, R.W.; Gordon, S.J. Bacterial populations of the small intestine in uremia. Nephron 1978, 22, 63–68. [Google Scholar] [CrossRef]
- Strid, H.; Simren, M.; Stotzer, P.O.; Ringstrom, G.; Abrahamsson, H.; Bjornsson, E.S. Patients with chronic renal failure have abnormal small intestinal motility and a high prevalence of small intestinal bacterial overgrowth. Digestion 2003, 67, 129–137. [Google Scholar] [CrossRef]
- Gryp, T.; Huys, G.R.B.; Joossens, M.; Van Biesen, W.; Glorieux, G.; Vaneechoutte, M. Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2020, 21, 1986. [Google Scholar] [CrossRef]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; Zuo, L.I.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Ehlting, C.; Haussinger, D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal. 2012, 24, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Feroze, U.; Kalantar-Zadeh, K.; Sterling, K.A.; Molnar, M.Z.; Noori, N.; Benner, D.; Shah, V.; Dwivedi, R.; Becker, K.; Kovesdy, C.P.; et al. Examining associations of circulating endotoxin with nutritional status, inflammation, and mortality in hemodialysis patients. J. Ren. Nutr. 2012, 22, 317–326. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.W.; Harrison, L.E.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating endotoxemia: A novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin. J. Am. Soc. Nephrol. CJASN 2011, 6, 133–141. [Google Scholar] [CrossRef]
- Raj, D.S.; Carrero, J.J.; Shah, V.O.; Qureshi, A.R.; Barany, P.; Heimburger, O.; Lindholm, B.; Ferguson, J.; Moseley, P.L.; Stenvinkel, P. Soluble CD14 levels, interleukin 6, and mortality among prevalent hemodialysis patients. Am. J. Kidney Dis. 2009, 54, 1072–1080. [Google Scholar] [CrossRef]
- Vince, A.; Dawson, A.M.; Park, N.; O’Grady, F. Ammonia production by intestinal bacteria. Gut 1973, 14, 171–177. [Google Scholar] [CrossRef]
- Wu, M.J.; Chang, C.S.; Cheng, C.H.; Chen, C.H.; Lee, W.C.; Hsu, Y.H.; Shu, K.H.; Tang, M.J. Colonic transit time in long-term dialysis patients. Am. J. Kidney Dis. 2004, 44, 322–327. [Google Scholar] [CrossRef]
- Gryp, T.; De Paepe, K.; Vanholder, R.; Kerckhof, F.M.; Van Biesen, W.; Van de Wiele, T.; Verbeke, F.; Speeckaert, M.; Joossens, M.; Couttenye, M.M.; et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. 2020. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.; De Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B. The Influence of CKD on Colonic Microbial Metabolism. J. Am. Soc. Nephrol. JASN 2016, 27, 1389–1399. [Google Scholar] [CrossRef]
- Kawakami, K.; Kojima, K.; Makino, I.; Kato, I.; Onoue, M. Fasting enhances p-Cresol production in the rat intestinal tract. Exp. Anim. 2007, 56, 301–307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wong, X.; Carrasco-Pozo, C.; Escobar, E.; Navarrete, P.; Blachier, F.; Andriamihaja, M.; Lan, A.; Tome, D.; Cires, M.J.; Pastene, E.; et al. Deleterious Effect of p-Cresol on Human Colonic Epithelial Cells Prevented by Proanthocyanidin-Containing Polyphenol Extracts from Fruits and Proanthocyanidin Bacterial Metabolites. J. Agric. Food Chem. 2016, 64, 3574–3583. [Google Scholar] [CrossRef] [PubMed]
- Al Hinai, E.A.; Kullamethee, P.; Rowland, I.R.; Swann, J.; Walton, G.E.; Commane, D.M. Modelling the role of microbial p-cresol in colorectal genotoxicity. Gut Microbes 2019, 10, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Makino, I.; Kato, I.; Uchida, K.; Onoue, M. p-Cresol inhibits IL-12 production by murine macrophages stimulated with bacterial immunostimulant. Immunopharmacol. Immunotoxicol. 2009, 31, 304–309. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Waterloos, M.A.; Van Landschoot, N.; Vogeleere, P.; Hoste, E.; Ringoir, S. Mechanisms of uremic inhibition of phagocyte reactive species production: Characterization of the role of p-cresol. Kidney Int. 1995, 47, 510–517. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci.USA 2010, 107, 228–233. [Google Scholar] [CrossRef]
- Hendrikx, T.; Duan, Y.; Wang, Y.; Oh, J.H.; Alexander, L.M.; Huang, W.; Starkel, P.; Ho, S.B.; Gao, B.; Fiehn, O.; et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut 2019, 68, 1504–1515. [Google Scholar] [CrossRef]
- Brieger, H.; Hodes, W.A. Toxic effects of exposure to vapors of aliphatic amines. AMA Arch. Ind. Hyg. Occup. Med. 1951, 3, 287–291. [Google Scholar] [PubMed]
- Fluhr, J.W.; Kelterer, D.; Fuchs, S.; Kaatz, M.; Grieshaber, R.; Kleesz, P.; Elsner, P. Additive impairment of the barrier function and irritation by biogenic amines and sodium lauryl sulphate: A controlled in vivo tandem irritation study. Skin Pharm. Physiol. 2005, 18, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Minkler, P.; Grove, D.; Wang, R.; Willard, B.; Dweik, R.; Hine, C. Non-enzymatic hydrogen sulfide production from cysteine in blood is catalyzed by iron and vitamin B6. Commun. Biol. 2019, 2, 194. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.; Bhattacharya, R.; Mukherjee, P. Hydrogen sulfide signaling in mitochondria and disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 13098–13125. [Google Scholar] [CrossRef]
- Koike, S.; Nishimoto, S.; Ogasawara, Y. Cysteine persulfides and polysulfides produced by exchange reactions with H2S protect SH-SY5Y cells from methylglyoxal-induced toxicity through Nrf2 activation. Redox Biol. 2017, 12, 530–539. [Google Scholar] [CrossRef]
- Koike, S.; Ogasawara, Y. Sulfur Atom in its Bound State Is a Unique Element Involved in Physiological Functions in Mammals. Molecules 2016, 21, 1753. [Google Scholar] [CrossRef]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.J.; Sakamoto, N.; Fearon, E.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 2019, 294, 12077–12090. [Google Scholar] [CrossRef]
- Shen, X.; Pattillo, C.B.; Pardue, S.; Bir, S.C.; Wang, R.; Kevil, C.G. Measurement of plasma hydrogen sulfide in vivo and in vitro. Free Radic. Biol. Med. 2011, 50, 1021–1031. [Google Scholar] [CrossRef]
- Tomasova, L.; Konopelski, P.; Ufnal, M. Gut Bacteria and Hydrogen Sulfide: The New Old Players in Circulatory System Homeostasis. Molecules 2016, 21, 1558. [Google Scholar] [CrossRef]
- Barton, L.L.; Ritz, N.L.; Fauque, G.D.; Lin, H.C. Sulfur Cycling and the Intestinal Microbiome. Dig. Dis. Sci. 2017, 62, 2241–2257. [Google Scholar] [CrossRef]
- Magee, E.A.; Richardson, C.J.; Hughes, R.; Cummings, J.H. Contribution of dietary protein to sulfide production in the large intestine: An in vitro and a controlled feeding study in humans. Am. J. Clin. Nutr. 2000, 72, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Maseda, C.; Hayakawa, A.; Okuda, K.; Asari, M.; Tanaka, H.; Yamada, H.; Jin, S.; Horioka, K.; Matoba, K.; Shiono, H.; et al. Liquid chromatography-tandem mass spectrometry method for the determination of thiosulfate in human blood and urine as an indicator of hydrogen sulfide poisoning. Leg. Med. (Tokyo, Japan) 2017, 24, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Shatalin, K.; Shatalina, E.; Mironov, A.; Nudler, E. H2S: A universal defense against antibiotics in bacteria. Science 2011, 334, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Mironov, A.; Seregina, T.; Nagornykh, M.; Luhachack, L.G.; Korolkova, N.; Lopes, L.E.; Kotova, V.; Zavilgelsky, G.; Shakulov, R.; Shatalin, K.; et al. Mechanism of H2S-mediated protection against oxidative stress in Escherichia coli. Proc. Natl. Acad. Sci. USA 2017, 114, 6022–6027. [Google Scholar] [CrossRef] [PubMed]
- Toliver-Kinsky, T.; Cui, W.; Toro, G.; Lee, S.J.; Shatalin, K.; Nudler, E.; Szabo, C. H2S, a Bacterial Defense Mechanism against the Host Immune Response. Infect. Immun. 2019, 87, e00272-18. [Google Scholar] [CrossRef]
- Roediger, W.E.; Duncan, A.; Kapaniris, O.; Millard, S. Reducing sulfur compounds of the colon impair colonocyte nutrition: Implications for ulcerative colitis. Gastroenterology 1993, 104, 802–809. [Google Scholar] [CrossRef]
- Shen, X.; Carlstrom, M.; Borniquel, S.; Jadert, C.; Kevil, C.G.; Lundberg, J.O. Microbial regulation of host hydrogen sulfide bioavailability and metabolism. Free Radic. Biol. Med. 2013, 60, 195–200. [Google Scholar] [CrossRef]
- Perna, A.F.; Di Nunzio, A.; Amoresano, A.; Pane, F.; Fontanarosa, C.; Pucci, P.; Vigorito, C.; Cirillo, G.; Zacchia, M.; Trepiccione, F.; et al. Divergent behavior of hydrogen sulfide pools and of the sulfur metabolite lanthionine, a novel uremic toxin, in dialysis patients. Biochimie 2016, 126, 97–107. [Google Scholar] [CrossRef]
- Jankowski, J.; Westhof, T.; Vaziri, N.D.; Ingrosso, D.; Perna, A.F. Gases as uremic toxins: Is there something in the air? Semin. Nephrol. 2014, 34, 135–150. [Google Scholar] [CrossRef]
- Perna, A.F.; Anishchenko, E.; Vigorito, C.; Zacchia, M.; Trepiccione, F.; D’Aniello, S.; Ingrosso, D. Zebrafish, a Novel Model System to Study Uremic Toxins: The Case for the Sulfur Amino Acid Lanthionine. Int. J. Mol. Sci. 2018, 19, 1323. [Google Scholar] [CrossRef]
- Perna, A.F.; Zacchia, M.; Trepiccione, F.; Ingrosso, D. The Sulfur Metabolite Lanthionine: Evidence for a Role as a Novel Uremic Toxin. Toxins 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Vigorito, C.; Anishchenko, E.; Mele, L.; Capolongo, G.; Trepiccione, F.; Zacchia, M.; Lombari, P.; Capasso, R.; Ingrosso, D.; Perna, A.F. Uremic Toxin Lanthionine Interferes with the Transsulfuration Pathway, Angiogenetic Signaling and Increases Intracellular Calcium. Int. J. Mol. Sci. 2019, 20, 2269. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, B.S.; Gee, D.; Weiss, A.; Pannall, P.; Roberts-Thomson, I.C.; Roediger, W.E. Estimation of phenolic conjugation by colonic mucosa. J. Clin. Pathol. 1989, 42, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Schepers, E.; Glorieux, G.; Vanholder, R. The gut: The forgotten organ in uremia? Blood Purif. 2010, 29, 130–136. [Google Scholar] [CrossRef]
- De Loor, H.; Bammens, B.; Evenepoel, P.; De Preter, V.; Verbeke, K. Gas chromatographic-mass spectrometric analysis for measurement of p-cresol and its conjugated metabolites in uremic and normal serum. Clin. Chem. 2005, 51, 1535–1538. [Google Scholar] [CrossRef]
- Poesen, R.; Evenepoel, P.; de Loor, H.; Kuypers, D.; Augustijns, P.; Meijers, B. Metabolism, Protein Binding, and Renal Clearance of Microbiota-Derived p-Cresol in Patients with CKD. Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 1136–1144. [Google Scholar] [CrossRef]
- Meyer, T.W.; Hostetter, T.H. Uremic solutes from colon microbes. Kidney Int. 2012, 81, 949–954. [Google Scholar] [CrossRef]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef]
- Meert, N.; Schepers, E.; Glorieux, G.; Van Landschoot, M.; Goeman, J.L.; Waterloos, M.A.; Dhondt, A.; Van der Eycken, J.; Vanholder, R. Novel method for simultaneous determination of p-cresylsulphate and p-cresylglucuronide: Clinical data and pathophysiological implications. Nephrol. Dial. Transplant. 2012, 27, 2388–2396. [Google Scholar] [CrossRef]
- Pletinck, A.; Glorieux, G.; Schepers, E.; Cohen, G.; Gondouin, B.; Van Landschoot, M.; Eloot, S.; Rops, A.; Van de Voorde, J.; De Vriese, A.; et al. Protein-bound uremic toxins stimulate crosstalk between leukocytes and vessel wall. J. Am. Soc. Nephrol. JASN 2013, 24, 1981–1994. [Google Scholar] [CrossRef]
- Han, H.; Chen, Y.; Zhu, J.; Ni, J.; Sun, J.; Zhang, R. Atorvastatin attenuates pcresyl sulfateinduced atherogenesis and plaque instability in ApoE knockout mice. Mol. Med. Rep. 2016, 14, 3122–3128. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Viaene, L.; Evenepoel, P.; Meijers, B.; Vanderschueren, D.; Overbergh, L.; Mathieu, C. Uremia suppresses immune signal-induced CYP27B1 expression in human monocytes. Am. J. Nephrol 2012, 36, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Shiba, T.; Kawakami, K.; Sasaki, T.; Makino, I.; Kato, I.; Kobayashi, T.; Uchida, K.; Kaneko, K. Effects of intestinal bacteria-derived p-cresyl sulfate on Th1-type immune response in vivo and in vitro. Toxicol. Appl. Pharmacol. 2014, 274, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Shiba, T.; Makino, I.; Kawakami, K.; Kato, I.; Kobayashi, T.; Kaneko, K. p-Cresyl sulfate suppresses lipopolysaccharide-induced anti-bacterial immune responses in murine macrophages in vitro. Toxicol. Lett. 2016, 245, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Shiba, T.; Makino, I.; Sasaki, T.; Fukuhara, Y.; Kawakami, K.; Kato, I.; Kobayashi, T. p-Cresyl sulfate decreases peripheral B cells in mice with adenine-induced renal dysfunction. Toxicol. Appl. Pharm. 2018, 342, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, M.L.; Bonan, N.B.; Dias, G.; Brehm, F.; Steiner, T.M.; Souza, W.M.; Stinghen, A.E.; Barreto, F.C.; Elifio-Esposito, S.; Pecoits-Filho, R.; et al. p-Cresyl sulfate affects the oxidative burst, phagocytosis process, and antigen presentation of monocyte-derived macrophages. Toxicol. Lett. 2016, 263, 1–5. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Shu, K.H.; Yang, F.J.; Chou, T.Y.; Chen, P.M.; Lay, F.Y.; Pan, S.Y.; Lin, C.J.; Litjens, N.H.R.; Betjes, M.G.H.; et al. A comprehensive characterization of aggravated aging-related changes in T lymphocytes and monocytes in end-stage renal disease: The iESRD study. Immun. Ageing 2018, 15, 27. [Google Scholar] [CrossRef]
- Borges Bonan, N.; Schepers, E.; Pecoits-Filho, R.; Dhondt, A.; Pletinck, A.; De Somer, F.; Vanholder, R.; Van Biesen, W.; Moreno-Amaral, A.; Glorieux, G. Contribution of the uremic milieu to an increased pro-inflammatory monocytic phenotype in chronic kidney disease. Sci. Rep. 2019, 9, 10236. [Google Scholar] [CrossRef]
- Hwang, W.B.; Kim, D.J.; Oh, G.S.; Park, J.H. Aryl Hydrocarbon Receptor Ligands Indoxyl 3-sulfate and Indole-3-carbinol Inhibit FMS-like Tyrosine Kinase 3 Ligand-induced Bone Marrow-derived plasmacytoid Dendritic Cell Differentiation. Immune Netw. 2018, 18, e35. [Google Scholar] [CrossRef]
- Ghimire, S.; Matos, C.; Caioni, M.; Weber, D.; Peter, K.; Holler, E.; Kreutz, M.; Renner, K. Indoxyl 3-sulfate inhibits maturation and activation of human monocyte-derived dendritic cells. Immunobiology 2018, 223, 239–245. [Google Scholar] [CrossRef]
- Kim, H.Y.; Yoo, T.H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.Y.; Lee, C.J.; et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Higuchi, Y.; Yagi, Y.; Nishijima, F.; Yamato, H.; Ishii, H.; Osaka, M.; Yoshida, M. Reduction of indoxyl sulfate by AST-120 attenuates monocyte inflammation related to chronic kidney disease. J. Leukoc. Biol. 2013, 93, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Yoo, T.H.; Cho, J.Y.; Kim, H.C.; Lee, W.W. Indoxyl sulfate-induced TNF-alpha is regulated by crosstalk between the aryl hydrocarbon receptor, NF-kappaB, and SOCS2 in human macrophages. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 10844–10858. [Google Scholar] [CrossRef]
- Adesso, S.; Popolo, A.; Bianco, G.; Sorrentino, R.; Pinto, A.; Autore, G.; Marzocco, S. The uremic toxin indoxyl sulphate enhances macrophage response to LPS. PLoS ONE 2013, 8, e76778. [Google Scholar] [CrossRef] [PubMed]
- Stockler-Pinto, M.B.; Soulage, C.O.; Borges, N.A.; Cardozo, L.; Dolenga, C.J.; Nakao, L.S.; Pecoits-Filho, R.; Fouque, D.; Mafra, D. From bench to the hemodialysis clinic: Protein-bound uremic toxins modulate NF-kappaB/Nrf2 expression. Int. Urol. Nephrol. 2018, 50, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef]
- Pires de Melo, M.; Curi, T.C.; Miyasaka, C.K.; Palanch, A.C.; Curi, R. Effect of indole acetic acid on oxygen metabolism in cultured rat neutrophil. Gen. Pharm. 1998, 31, 573–578. [Google Scholar] [CrossRef]
- De Melo, M.P.; Pithon-Curi, T.C.; Curi, R. Indole-3-acetic acid increases glutamine utilization by high peroxidase activity-presenting leukocytes. Life Sci. 2004, 75, 1713–1725. [Google Scholar] [CrossRef]
- De Melo, M.P.; Curi, T.C.; Curi, R.; Di Mascio, P.; Cilento, G. Peroxidase activity may play a role in the cytotoxic effect of indole acetic acid. Photochem. Photobiol. 1997, 65, 338–341. [Google Scholar] [CrossRef]
- De Melo, M.P.; de Lima, T.M.; Pithon-Curi, T.C.; Curi, R. The mechanism of indole acetic acid cytotoxicity. Toxicol. Lett. 2004, 148, 103–111. [Google Scholar] [CrossRef]
- Lins, P.G.; Valle, C.R.; Pugine, S.M.; Oliveira, D.L.; Ferreira, M.S.; Costa, E.J.; De Melo, M.P. Effect of indole acetic acid administration on the neutrophil functions and oxidative stress from neutrophil, mesenteric lymph node and liver. Life Sci. 2006, 78, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Salopek-Sondi, B.; Piljac-Zegarac, J.; Magnus, V.; Kopjar, N. Free radical-scavenging activity and DNA damaging potential of auxins IAA and 2-methyl-IAA evaluated in human neutrophils by the alkaline comet assay. J. Biochem. Mol. Toxicol. 2010, 24, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Cashman, J.R.; Zhang, J. Human flavin-containing monooxygenases. Ann. Rev. Pharm. Toxicol. 2006, 46, 65–100. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 2017, 37, BSR20160244. [Google Scholar] [CrossRef]
- Haghikia, A.; Li, X.S.; Liman, T.G.; Bledau, N.; Schmidt, D.; Zimmermann, F.; Krankel, N.; Widera, C.; Sonnenschein, K.; Haghikia, A.; et al. Gut Microbiota-Dependent Trimethylamine N-Oxide Predicts Risk of Cardiovascular Events in Patients with Stroke and Is Related to Proinflammatory Monocytes. Arter. Thromb. Vasc. Biol. 2018, 38, 2225–2235. [Google Scholar] [CrossRef]
- Li, L.; Bhatia, M.; Zhu, Y.Z.; Zhu, Y.C.; Ramnath, R.D.; Wang, Z.J.; Anuar, F.B.; Whiteman, M.; Salto-Tellez, M.; Moore, P.K. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1196–1198. [Google Scholar] [CrossRef]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 2118–2120. [Google Scholar] [CrossRef]
- Szabo, C. A timeline of hydrogen sulfide (H2S) research: From environmental toxin to biological mediator. Biochem. Pharm. 2018, 149, 5–19. [Google Scholar] [CrossRef]
- Rodrigues, C.; Percival, S.S. Immunomodulatory Effects of Glutathione, Garlic Derivatives, and Hydrogen Sulfide. Nutrients 2019, 11, 295. [Google Scholar] [CrossRef]
- Palinkas, Z.; Furtmuller, P.G.; Nagy, A.; Jakopitsch, C.; Pirker, K.F.; Magierowski, M.; Jasnos, K.; Wallace, J.L.; Obinger, C.; Nagy, P. Interactions of hydrogen sulfide with myeloperoxidase. Br. J. Pharmacol. 2015, 172, 1516–1532. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Sepe, I.; Lanza, D.; Capasso, R.; Zappavigna, S.; Capasso, G.; Caraglia, M.; Ingrosso, D. Hydrogen sulfide reduces cell adhesion and relevant inflammatory triggering by preventing ADAM17-dependent TNF-alpha activation. J. Cell Biochem. 2013, 114, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Fang, X.; Yang, X.; Mitsui, T.; Huang, Y.; Mao, Z.; Huang, Y.; Takeda, M.; Yao, J. Hydrogen sulfide donor NaHS alters antibody structure and function via sulfhydration. Int. Immunopharmacol. 2019, 73, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Luciano, M.G.; Ingrosso, D.; Pulzella, P.; Sepe, I.; Lanza, D.; Violetti, E.; Capasso, R.; Lombardi, C.; De Santo, N.G. Hydrogen sulphide-generating pathways in haemodialysis patients: A study on relevant metabolites and transcriptional regulation of genes encoding for key enzymes. Nephrol. Dial. Transplant. 2009, 24, 3756–3763. [Google Scholar] [CrossRef] [PubMed]
- Ling, W.H.; Hanninen, O. Shifting from a conventional diet to an uncooked vegan diet reversibly alters fecal hydrolytic activities in humans. J. Nutr. 1992, 122, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; De Preter, V.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-Cresyl sulfate serum concentrations in haemodialysis patients are reduced by the prebiotic oligofructose-enriched inulin. Nephrol. Dial. Transplant. 2010, 25, 219–224. [Google Scholar] [CrossRef]
- De Preter, V.; Vanhoutte, T.; Huys, G.; Swings, J.; De Vuyst, L.; Rutgeerts, P.; Verbeke, K. Effects of Lactobacillus casei Shirota, Bifidobacterium breve, and oligofructose-enriched inulin on colonic nitrogen-protein metabolism in healthy humans. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G358–G368. [Google Scholar] [CrossRef]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.C.; McWhinney, B.C.; Ungerer, J.P.; Campbell, K.L. Synbiotics Easing Renal Failure by Improving Gut Microbiology (SYNERGY): A Randomized Trial. Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 223–231. [Google Scholar] [CrossRef]


{kind=link}
| Metabolites | Side of Origin | (Patho) Physiological Effects | Ref. |
|---|---|---|---|
| Metabolites Generated by Gut Bacteria | |||
| p-Cresol | Colon | Intestinal epithelial cells: | |
| ↑LDH leakage | [32] | ||
| ↓ATP content | |||
| ↓TEER | |||
| ↑Paracellular transport | |||
| Genotoxicity | [33] | ||
| Leukocytes/Macrophages: | |||
| ↓IL-12 p40 production | [34] | ||
| ↓Respiratory burst activity | [35] | ||
| Indole | Colon | Intestinal epithelial cells: | |
| ↑Mucin2 expression | [36,37] | ||
| Regulation of gut homeostasis | [38] | ||
| ↓TNF-α mediated NF-κB activation | [39] | ||
| ↓IL-8 expression | |||
| ↑IL-10 expression | |||
| ↑TER | |||
| Indole-3-acetic acid | Colon | Intestinal epithelial cells: | |
| ↑IL-22 expression | [40] | ||
| ↑Antimicrobial C-type lectin REG3G | |||
| Trimethylamine | Colon | ND | |
| H2S | Colon | Intestinal bacteria: | |
| ↑Antibiotic resistance | [53] | ||
| Protection against ROS | [54] | ||
| Protection against immune cells | [55] | ||
| Maintenance of anaerobic conditions | |||
| ↓Butyrate oxidation | [56] | ||
| Circulating Uremic Toxins | |||
| p-Cresyl sulfate | Intestinal epithelial cells and liver | Leukocytes: | |
| ↑Baseline ROS and ↓ROS after stimulation | [68] | ||
| ↑Rolling | [70] | ||
| ↑Plaque growth and instability | [71] | ||
| ↓IFNγ-producing Th1 cells | [73] | ||
| ↓Anti-bacterial immune response | [74] | ||
| ↓Proliferation of CD43(+) B cell progenitors | [75] | ||
| ↑Macrophage activation | |||
| ↓Antigen processing | [76] | ||
| Premature aging of immune cells | [77] | ||
| p-Cresyl glucuronide | Intestinal epithelial cells and liver | Leukocytes: | |
| Synergistic to pCS: ↑ROS, impaired blood | [69] | ||
| flow; vascular leakage | [70] | ||
| Indoxyl sulfate | Liver | Anti-inflammatory and tolerizing effect on | [80] |
| DCs | |||
| Monocytes: | |||
| ↑TNF-α→↑HUVEC CX3CL1 | [81] | ||
| ↑ROS | [82] | ||
| ↑Leukocyte-endothelial cell adhesion | [82,86] | ||
| Macrophages: | |||
| ↑TNF-α | [83] | ||
| ↑NF-κB, ROS, mitochondrial Ca2+ | [84] | ||
| overload | |||
| ↓Nrf2 | [85] | ||
| Indole-3-acetic acid | Colon | Neutrophils: | |
| ↑peroxidase activity | [87] | ||
| ↑Glucose and glutamine metabolism | [88] | ||
| ↑Oxygen consumption | [88] | ||
| ↑Structural changes and cell death | [87,89] | ||
| ↑Phagocytic activity | [91] | ||
| Genotoxicity | [92] | ||
| Trimethylamine-oxide | Liver, kidney, and other | ↑VCAM-1 | [94] |
| ↑Monocyte-endothelial | |||
| adhesion→↑inflammatory gene expression | |||
| Activates PKC and p-NF-κB | |||
| H2S | Brain, vascular tissue, liver, kidney, RBC, and other | Leukocytes: | |
| ↓Leukocyte-endothelial adhesion | [97,98] | ||
| ↓Leukocyte infiltration | |||
| Regulation of post-translational | [100] | ||
| modification of NF-κB pathway | |||
| Macrophages: | |||
| ↓Pro-inflammatory cytokine production | [44] | ||
| ↓COX-2 and NO production | |||
| ↓Macrophage motitlity | |||
| ↓MPO activity | [101] | ||
| ↓Inflammation | [102] | ||
| ↓Antigen-binding | [103] | ||
| ↓Cell lysis (glomerular mesangial cells and T-lymphocytes) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glorieux, G.; Gryp, T.; Perna, A. Gut-Derived Metabolites and Their Role in Immune Dysfunction in Chronic Kidney Disease. Toxins 2020, 12, 245. https://doi.org/10.3390/toxins12040245
Glorieux G, Gryp T, Perna A. Gut-Derived Metabolites and Their Role in Immune Dysfunction in Chronic Kidney Disease. Toxins. 2020; 12(4):245. https://doi.org/10.3390/toxins12040245
Chicago/Turabian StyleGlorieux, Griet, Tessa Gryp, and Alessandra Perna. 2020. "Gut-Derived Metabolites and Their Role in Immune Dysfunction in Chronic Kidney Disease" Toxins 12, no. 4: 245. https://doi.org/10.3390/toxins12040245
APA StyleGlorieux, G., Gryp, T., & Perna, A. (2020). Gut-Derived Metabolites and Their Role in Immune Dysfunction in Chronic Kidney Disease. Toxins, 12(4), 245. https://doi.org/10.3390/toxins12040245