Comparative Analysis of Microcystin Prevalence in Michigan Lakes by Online Concentration LC/MS/MS and ELISA


Abstract
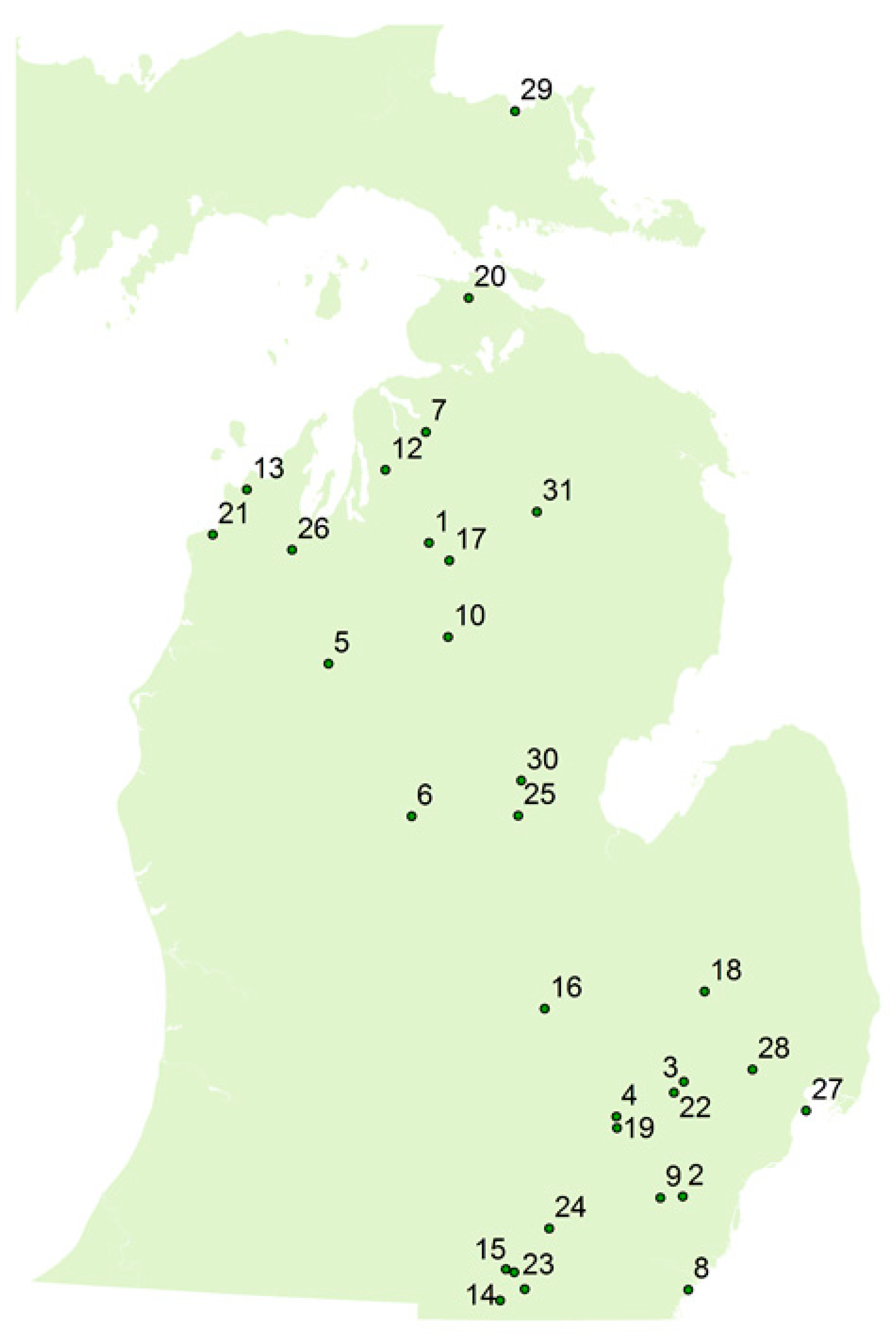
1. Introduction
2. Results and Discussion
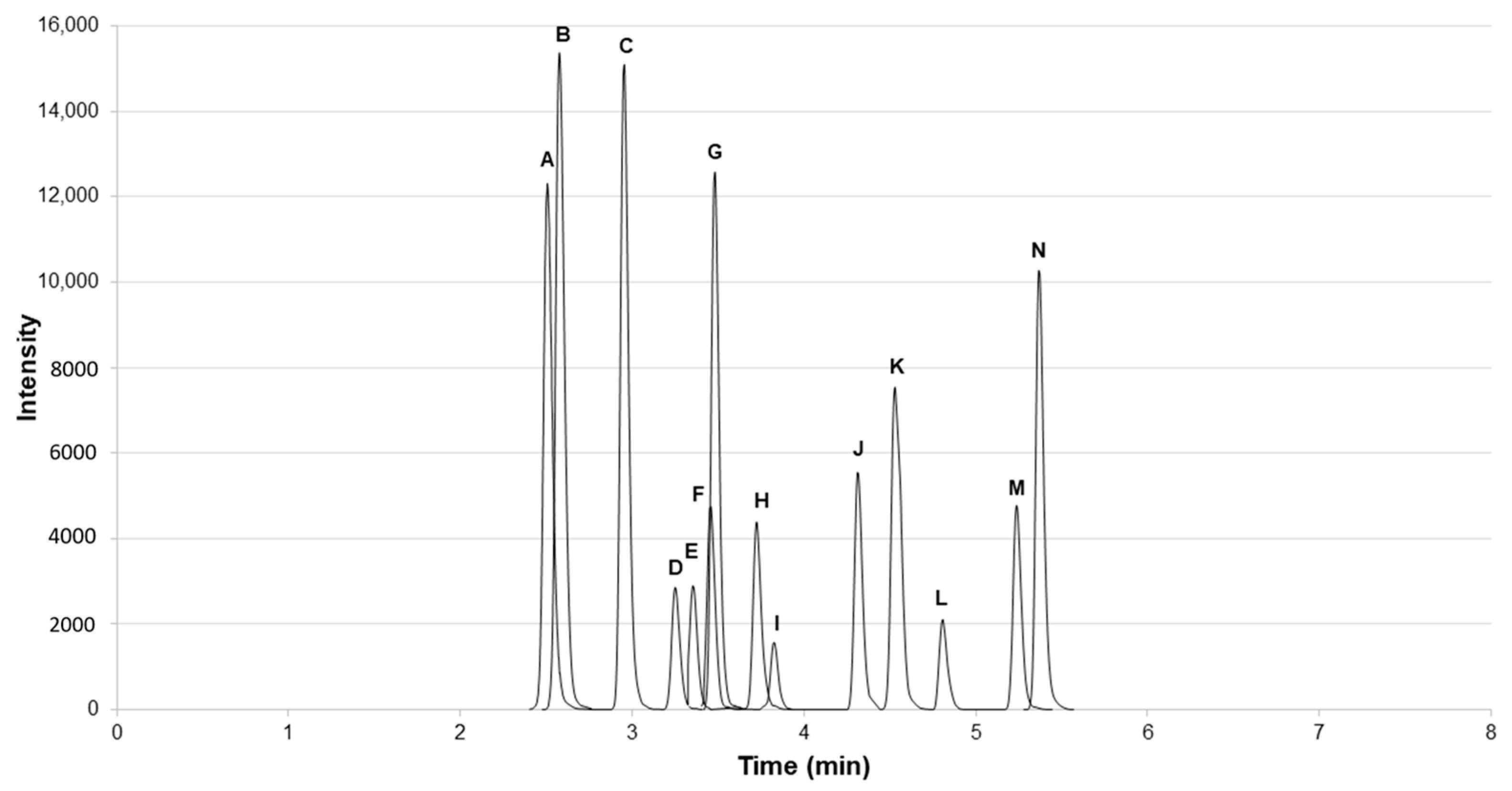
2.1. Online Concentration and LC/MS/MS Methodology
2.2. LC/MS/MS Method Validation
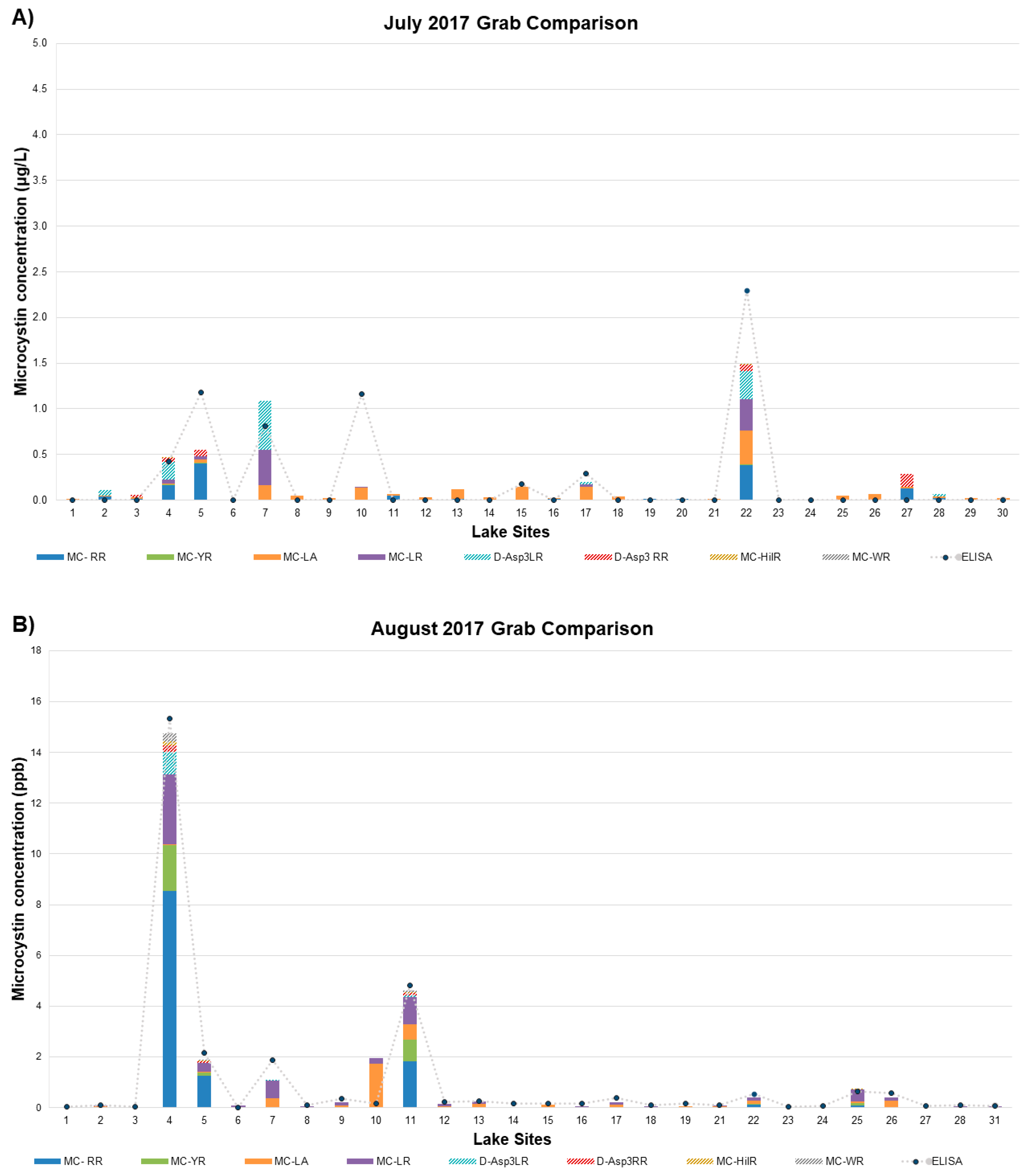
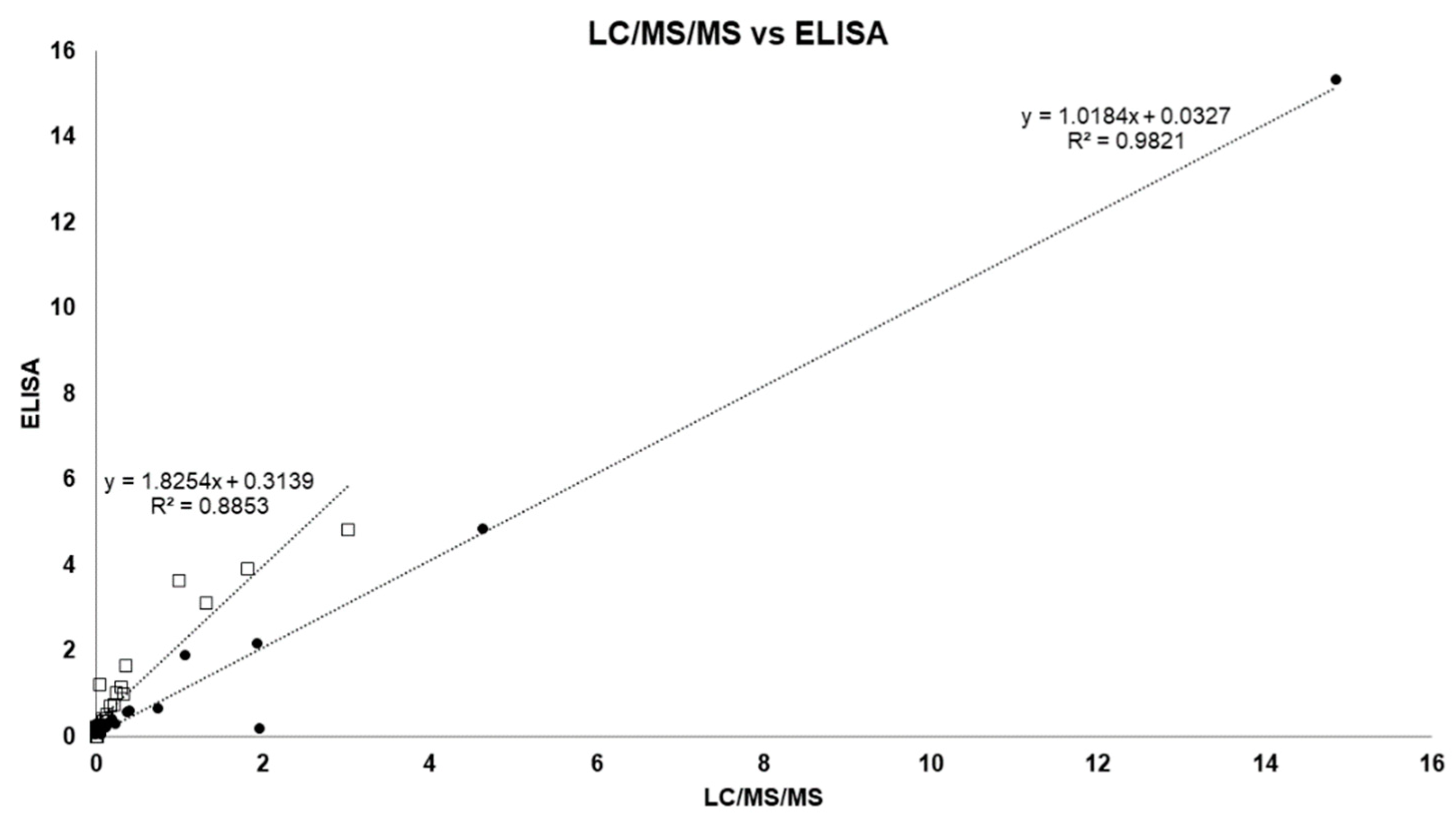
2.3. LC/MS/MS and ELISA Method Results and Comparison
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. ELISA
4.3. Liquid Chromatography Mass Spectrometry
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chorus, I.; Bartram, J. Toxic Cyanobacteria in Water: A Guide to Their Public Health Consequences, Monitoring and Management; CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar]
- Sanseverino, I.; Conduto, D.; Pozzoli, L.; Dobricic, S.; Lettieri, T. Algal Bloom and Its Economic Impact; Report no. EUR 27905; Publications Office of the European Union: Ispra VA, Italy, 2016. [Google Scholar]
- Pham, T.-L.; Dang, T.N. Microcystins in Freshwater Ecosystems: Occurrence, Distribution, and Current Treatment Approaches. In Water and Wastewater Treatment Technologies; Bui, X., Chiemchaisri, C., Fujioka, T., Varjani, S., Eds.; Springer: Gateway East, Singapore, 2019; pp. 15–36. [Google Scholar]
- Ground Water and Drinking Water: Drinking Water Health Advisory Documents for Cyanobacterial Toxins. Available online: https://www.epa.gov/ground-water-and-drinking-water/drinking-water-health-advisory-documents-cyanobacterial-toxins (accessed on 27 November 2018).
- Health Canada. Cyanobacterial Toxins in Drinking Water: Document for Public Consultation Water, Federal-Provincial-Territorial Committee on Drinking Water; Health Canada: Ottawa, Canada, 2016; pp. 1–177.
- Massey, I.Y.; Yang, F.; Ding, Z.; Yang, S.; Guo, J.; Tezi, C.; Al-Osman, M.; Kamegni, R.B.; Zeng, W. Exposure routes and health effects of microcystins on animals and humans: A mini-review. Toxicon 2018, 151, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, W. A world overview—One-hundred-twenty-seven years of research on toxic cyanobacteria—Where do we go from here? In Cyanobacterial Harmful Algal Blooms: State of the Science and Research Needs; Springer: New York, NY, USA, 2008; pp. 105–125. [Google Scholar]
- Puddick, J.; Prinsep, M.R.; Wood, S.A.; Kaufononga, S.A.; Cary, S.C.; Hamilton, D.P. High levels of structural diversity observed in microcystins from Microcystis CAWBG11 and characterization of six new microcystin congeners. Mar. Drugs 2014, 12, 5372–5395. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guidelines for Safe Recreational Water Environments: Coastal and Fresh Waters; WHO: Geneva, Switzerland, 2003; Volume 1, pp. 1–253. [Google Scholar]
- U.S. EPA (United States Environmental Protection Agency). Human Health Recreational Ambient Water Quality Criteria or Swimming Advisories for Microcystins and Cylindrospermopsin Draft; Division, Office of Water Health and Ecological Criteria Division: Washington, DC, USA, 2016; pp. 1–185.
- Shoemaker, J.A.; Tettenhorst, D.R.; de la Cruz, A. Method 544: Determination of Microcystins and Nodularin in Drinking Water by Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry (LC/MS/MS); U.S. EPA (United States Environmental Protection Agency), National Exposure Research Laboratory: Cincinnati, OH, USA, 2015; pp. 1–70.
- Zaffiro, A.; Rosenblum, L.; Wendelken, S.C. Method 546: Determination of Total Microcystins and Nodularins in Drinking Water and Ambient Water by Adda Enzyme-Linked Immunosorbent Assay; U.S. EPA (United States Environmental Protection Agency), Standards and Risk Management Division: Cincinnati, OH, USA, 2016; pp. 1–21.
- Appa, R.; Mhaisalkar, V.A.; Bafana, A.; Devi, S.S.; Krishnamurthi, K.; Chakrabarti, T.; Naoghare, P.K. Simultaneous quantitative monitoring of four indicator contaminants of emerging concern (CEC) in different water sources of Central India using SPE/LC-(ESI)MS-MS. Environ. Monit. Assess. 2018, 190, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Kuroda, K.; Tominaga, Y.; Naito, T.; Sueyoshi, K.; Hosoya, K.; Otsuka, K. Effective determination of a pharmaceutical, sulpiride, in river water by online SPE-LC-MS using a molecularly imprinted polymer as a preconcentration medium. J. Pharmaceut. Biomed. 2014, 89, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Carlson, K. Quantification of human and veterinary antibiotics in water and sediment using SPE/LC/MS/MS. Anal. Bioanal. Chem. 2007, 387, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.E.; Wang, C.T.; Gardinali, P.R. Fully automated trace level determination of parent and alkylated PAHs in environmental waters by online SPE-LC-APPI-MS/MS. Anal. Bioanal. Chem. 2014, 406, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Beltran, E.; Ibanez, M.; Sancho, J.V.; Hernandez, F. Determination of six microcystins and nodularin in surface and drinking waters by on-line solid phase extraction-ultra high pressure liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2012, 1266, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Fayad, P.B.; Roy-Lachapelle, A.; Duy, S.V.; Prévost, M.; Sauvé, S. On-line solid-phase extraction coupled to liquid chromatography tandem mass spectrometry for the analysis of cyanotoxins in algal blooms. Toxicon 2015, 108, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Munoz, G.; Duy, S.V.; Roy-Lachapelle, A.; Husk, B.; Sauve, S. Analysis of individual and total microcystins in surface water by on-line preconcentration and desalting coupled to liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2017, 1516, 9–20. [Google Scholar] [CrossRef]
- Balest, L.; Murgolo, S.; Sciancalepore, L.; Montemurro, P.; Abis, P.P.; Pastore, C.; Mascolo, G. Ultra-trace levels analysis of microcystins and nodularin in surface water by on-line solid-phase extraction with high-performance liquid chromatography tandem mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 4063–4071. [Google Scholar] [CrossRef]
- Ortiz, X.; Korenkova, E.; Jobst, K.J.; MacPherson, K.A.; Reiner, E.J. A high throughput targeted and non-targeted method for the analysis of microcystins and anatoxin—A using on-line solid phase extraction coupled to liquid chromatography-quadrupole time-of-flight high resolution mass spectrometry. Anal. Bioanal. Chem. 2017, 409, 4959–4969. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Pant, S.C.; Vijayaraghavan, R.; Rao, P.V.L. Comparative toxicity evaluation of cyanobacterial cyclic peptide toxin microcystin variants (LR, RR, YR) in mice. Toxicology 2003, 188, 285–296. [Google Scholar] [CrossRef]
- Foss, A.J.; Aubel, M.T. Using the MMPB technique to confirm microcystin concentrations in water measured by ELISA and HPLC (UV, MS, MS/MS). Toxicon 2015, 104, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Teta, R.; Della Sala, G.; Glukhov, E.; Gerwick, L.; Gerwick, W.H.; Mangoni, A.; Costantino, V. Combined LC-MS/MS and Molecular Networking Approach Reveals New Cyanotoxins from the 2014 Cyanobacterial Bloom in Green Lake, Seattle. Environ. Sci. Technol. 2015, 49, 14301–14310. [Google Scholar] [CrossRef] [PubMed]
- Loftin, K.A.; Graham, J.L.; Hilborn, E.D.; Lehmann, S.C.; Meyer, M.T.; Dietze, J.E.; Griffith, C.B. Cyanotoxins in inland lakes of the United States: Occurrence and potential recreational health risks in the EPA National Lakes Assessment 2007. Harmful Algae 2016, 56, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Namikoshi, M.; Rinehart, K.L.; Sakai, R.; Stotts, R.R.; Dahlem, A.M.; Beasley, V.R.; Carmichael, W.W.; Evans, W.R. Identification of 12 Hepatotoxins from a Homer Lake Bloom of the Cyanobacteria Microcystis-Aeruginosa, Microcystis-Viridis, and Microcystis-Wesenbergii—9 New Microcystins. J. Org. Chem. 1992, 57, 866–872. [Google Scholar] [CrossRef]
- Fuller, L.M.; Brennan, A.K.; Fogarty, L.R.; Loftin, K.A.; Johnson, H.E.; VanderMeulen, D.D.; Lafrancois, B.M. Detection of Microcystin and Other Cyanotoxins in Lakes at Isle Royale National Park, Pictured Rocks National Lakeshore, and Sleeping Bear Dunes National Lakeshore, Northern Michigan, 2012–13. Scientific Investigations Report; U.S. Geological Survey: Reston, VA, USA, 2017; pp. 1–44. Available online: https://dx.doi.org/10.3133/sir20175122 (accessed on 1 October 2018).
- Graham, J.L.; Loftin, K.A.; Meyer, M.T.; Ziegler, A.C. Cyanotoxin mixtures and taste-and-odor compounds in cyanobacterial blooms from the Midwestern United States. Environ. Sci. Technol. 2010, 44, 7361–7368. [Google Scholar] [CrossRef]
- Kann, J.; Corum, S.; Fetcho, K. Microcystin Bioaccumulation in Klamath River Freshwater Mussel Tissue: 2009 Results; Aquatic Ecosystem Sciences, LLC: Ashland, OR, USA, 2010; pp. 1–37. [Google Scholar]
- Lehman, P.W.; Boyer, G.; Hall, C.; Waller, S.; Gehrts, K. Distribution and toxicity of a new colonial Microcystis aeruginosa bloom in the San Francisco Bay Estuary, California. Hydrobiologia 2005, 541, 87–99. [Google Scholar] [CrossRef]
- Miller, M.A.; Kudela, R.M.; Mekebri, A.; Crane, D.; Oates, S.C.; Tinker, M.T.; Staedler, M.; Miller, W.A.; Toy-Choutka, S.; Dominik, C. Evidence for a Novel Marine Harmful Algal Bloom: Cyanotoxin (Microcystin) Transfer from Land to Sea Otters. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Holden, S. Are Harmful Algae Blooms (HABs) a Problem in Michigan’s Inland Lakes? Available online: https://www.mi-wea.org/docs/4B%20Sarah%20Holden%20MWEA%202016.pdf (accessed on 1 October 2018).
- Beaver, J.R.; Manis, E.E.; Loftin, K.A.; Graham, J.L.; Pollard, A.I.; Mitchell, R.M. Land use patterns, ecoregion, and microcystin relationships in U.S. lakes and reservoirs: A preliminary evaluation. Harmful Algae 2014, 36, 57–62. [Google Scholar] [CrossRef]
- Sarnelle, O.; Wandell, H. Monitoring and Predicting Concentrations of Cyanobacterial Toxins in Michigan Lakes; Michigan State University Department of Fisheries and Wildlife: East Lansing, MI, USA, 2008; pp. 1–41. [Google Scholar]
- Dreelin, E.; McNinch, R.; Rose, J.B. Waterborne Pathogens: Where Michigan Stands Now and Recommendations for Our Future; Center for Water Sciences Pathogen Workshop Series; Michigan State University: East Lansing, MI, USA, 2007; pp. 1–35. [Google Scholar]
- Xie, L.; Rediske, R.R.; Hong, Y.; O’Keefe, J.; Gillett, N.D.; Dyble, J.; Steinman, A.D. The role of environmental parameters in the structure of phytoplankton assemblages and cyanobacteria toxins in two hypereutrophic lakes. Hydrobiologia 2012, 691, 255–268. [Google Scholar] [CrossRef]
- Backer, L.C.; Carmichael, W.; Kirkpatrick, B.; Williams, C.; Irvin, M.; Zhou, Y.; Johnson, T.B.; Nierenberg, K.; Hill, V.R.; Kieszak, S.M.; et al. Recreational exposure to low concentrations of microcystins during an algal bloom in a small lake. Mar. Drugs 2008, 6, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Raikow, D.F.; Sarnelle, O.; Wilson, A.E.; Hamilton, S.K. Dominance of the noxious cyanobacterium Microcystis aeruginosa in low-nutrient lakes is associated with exotic zebra mussels. Limnol. Oceanogr. 2004, 49, 482–487. [Google Scholar] [CrossRef]
- Michalak, A.M.; Anderson, E.J.; Beletsky, D.; Boland, S.; Bosch, N.S.; Bridgeman, T.B.; Chaffin, J.D.; Cho, K.; Confesor, R.; Daloglu, I.; et al. Record-setting algal bloom in Lake Erie caused by agricultural and meteorological trends consistent with expected future conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6448–6452. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Glibert, P.M.; Burkholder, J.M. Harmful algal blooms and eutrophication: Nutrient sources, composition, and consequences. Estuaries 2002, 25, 704–726. [Google Scholar] [CrossRef]
- Guo, Y.C.B.; Lee, A.K.; Yates, R.S.; Liang, S.; Rochelle, P.A. Analysis of Microcystins in Drinking Water by ELISA and LC/MS/MS. J. Am. Water Works Assoc. 2017, 109, 13–25. [Google Scholar] [CrossRef]
- Fischer, W.J.; Garthwaite, I.; Miles, C.O.; Ross, K.M.; Aggen, J.B.; Chamberlin, A.R.; Towers, N.R.; Dietrich, D.R. Congener-independent immunoassay for microcystins and nodularins. Environ. Sci. Technol. 2001, 35, 4849–4856. [Google Scholar] [CrossRef]
- Lawton, L.A.; Chambers, H.; Edwards, C.; Nwaopara, A.A.; Healy, M. Rapid detection of microcystins in cells and water. Toxicon 2010, 55, 973–978. [Google Scholar] [CrossRef]
- Li, J.M.; Li, R.H.; Li, J. Current research scenario for microcystins biodegradation—A review on fundamental knowledge, application prospects and challenges. Sci. Total Environ. 2017, 595, 615–632. [Google Scholar] [CrossRef]
- Dziga, D.; Wasylewski, M.; Wladyka, B.; Nybom, S.; Meriluoto, J. Microbial degradation of microcystins. Chem. Res. Toxicol. 2013, 26, 841–852. [Google Scholar] [CrossRef]
- Bourne, D.G.; Jones, G.J.; Blakeley, R.L.; Jones, A.; Negri, A.P.; Riddles, P. Enzymatic pathway for the bacterial degradation of the cyanobacterial cyclic peptide toxin microcystin LR. Appl. Environ. Microbiol. 1996, 62, 4086–4094. [Google Scholar] [PubMed]
- Thees, A.; Atari, E.; Birbeck, J.; Westrick, J.A.; Huntley, J. Isolation and Characterization of Lake Erie Bacteria that Degrade the Cyanobacterial Microcystin Toxin MC-LR. J. Great Lakes Res. 2018. [Google Scholar] [CrossRef]
- Liu, I.; Lawton, L.A.; Robertson, P.K.J. Mechanistic studies of the photocatalytic oxidation of microcystin-LR: An investigation of byproducts of the decomposition process. Environ. Sci. Technol. 2003, 37, 3214–3219. [Google Scholar] [CrossRef] [PubMed]
- Lawton, L.A.; Robertson, P.K.J.; Cornish, B.J.P.A.; Jaspars, M. Detoxification of microcystins (cyanobacterial hepatotoxins) using TiO2 photocatalytic oxidation. Environ. Sci. Technol. 1999, 33, 771–775. [Google Scholar] [CrossRef]
- Edwards, C.; Graham, D.; Fowler, N.; Lawton, L.A. Biodegradation of microcystins and nodularin in freshwaters. Chemosphere 2008, 73, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- U.S. EPA (United States Environmental Protection Agency). Survey of the Nation’s Lakes. Field Operations Manual; U.S. EPA: Washington, DC, USA, 2007.
- Kubwabo, C.; Vais, N.; Benoit, F.M. Characterization of microcystins using in-source collision-induced dissociation. Rapid Commun. Mass Spectrom. 2005, 19, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, M.C.; Neilan, B.A. Characterization of the nodularin synthetase gene cluster and proposed theory of the evolution of cyanobacterial hepatotoxins. Appl. Environ. Microb. 2004, 70, 6353–6362. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Validation Procedure | Equation |
|---|---|
| Initial demonstration of precision: relative standard deviation (%RSD) | |
| Initial demonstration of accuracy: percent recovery (%Rec) | |
| MRL confirmation using the Half Rang for the prediction interval of results (HRPIR) | HRPIR = 3.963σ |
| Upper PIR limit | |
| Lower PIR limit | |
| Detection Limit (DL) determination |
| Compound | DL (ng/L) | EPA 544 DL (ng/L) [11] | MRL (ng/L) | Upper PIR | Lower PIR | r2 | %RSD | %Rec |
|---|---|---|---|---|---|---|---|---|
| D-Asp3-MC-RR | 0.6 | 5 | 122.46 | 98.32 | 0.9994 | 2.76 | 110 | |
| MC-RR | 0.6 | 1.2 | 5 | 118.66 | 93.98 | 0.9998 | 2.93 | 106 |
| Nodularin | 0.9 | 1.8 | 10 | 104.89 | 85.45 | 0.9999 | 2.58 | 95 |
| MC-YR | 3.6 | 4.6 | 10 | 147.37 | 70.86 | 0.9980 | 8.85 | 109 |
| MC-HtyR | 2.6 | 10 | 117.91 | 62.17 | 0.9996 | 7.81 | 90 | |
| MC-LR | 3.8 | 4.3 | 10 | 131.90 | 50.57 | 0.9988 | 11.25 | 91 |
| D-Asp3-MC-LR | 1.7 | 5 | 141.78 | 70.82 | 0.9996 | 8.42 | 106 | |
| MC-HilR | 3.3 | 5 | 139.94 | 68.82 | 0.9987 | 8.60 | 104 | |
| MC-WR | 2.0 | 5 | 144.79 | 74.78 | 0.9991 | 8.04 | 110 | |
| MC-LA | 3.3 | 4.0 | 10 | 135.37 | 65.77 | 0.9981 | 8.73 | 101 |
| MC-LY | 2.6 | 2.2 | 10 | 139.18 | 84.08 | 0.9986 | 6.23 | 111 |
| MC-LW | 3.2 | 10 | 137.61 | 68.33 | 0.9994 | 8.49 | 102 | |
| MC-LF | 1.2 | 3.4 | 5 | 138.41 | 88.53 | 0.9993 | 5.55 | 113 |
| Date | D-Asp3 MC-RR | MC-RR | MC-YR | MC-HtyR | MC-LR | D-Asp3-MC-LR | MC-HilR | MC-WR | MC-LA | MC-LY | MC-LW | MC-LF | MS SUM | ELISA MC-LR eq. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 07/17 | 5 | 14 | 0 | 0 | 5 | 6 | 2 | 0 | 24 | 0 | 0 | 0 | 56 | 7 |
| 08/17 | 4 | 11 | 6 | 2 | 21 | 5 | 5 | 4 | 21 | 1 | 1 | 1 | 82 | 16 |
| 09/17 | 2 | 8 | 6 | 1 | 22 | 7 | 4 | 2 | 19 | 0 | 1 | 0 | 72 | 17 |
| 10/17 | 0 | 7 | 5 | 1 | 20 | 7 | 2 | 4 | 14 | 1 | 0 | 0 | 61 | 27 |
| Total | 11 | 40 | 17 | 4 | 68 | 25 | 13 | 10 | 78 | 2 | 2 | 1 | 271 | 67 |
| Compound | Precursor (m/z) | Quant ions (m/z) | Qual ions (m/z) |
|---|---|---|---|
| D-Asp3-MC-RR | 512.861 | 135.071 | 375.054 |
| MC-RR * | 519.850 | 135.070 | 440.130 |
| Nodularin * | 825.383 | 135.111 | 389.111 |
| MC-LA * | 910.365 | 375.054 | 135.050 |
| D-Asp3-MC-LR | 981.430 | 135.111 | 375.111 |
| MC-LF * | 986.365 | 852.286 | 478.214 |
| MC-LR * | 995.378 | 135.039 | 213.050t |
| MC-LY * | 1002.304 | 494.214 | 868.286 |
| MC-HilR | 1009.461 | 135.111 | 213.054 |
| MC-LW | 1025.639 | 517.214 | 891.286 |
| C2D5 MC-LR * | 1028.743 | 135.097 | 163.083 |
| MC-YR * | 1045.639 | 213.032 | 136.222 |
| MC-HtyR | 1059.426 | 135.111 | 617.222 |
| MC-WR | 1068.452 | 135.097 | 626.183 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birbeck, J.A.; Westrick, J.A.; O’Neill, G.M.; Spies, B.; Szlag, D.C. Comparative Analysis of Microcystin Prevalence in Michigan Lakes by Online Concentration LC/MS/MS and ELISA. Toxins 2019, 11, 13. https://doi.org/10.3390/toxins11010013
Birbeck JA, Westrick JA, O’Neill GM, Spies B, Szlag DC. Comparative Analysis of Microcystin Prevalence in Michigan Lakes by Online Concentration LC/MS/MS and ELISA. Toxins. 2019; 11(1):13. https://doi.org/10.3390/toxins11010013
Chicago/Turabian StyleBirbeck, Johnna A., Judy A. Westrick, Grace M. O’Neill, Brian Spies, and David C. Szlag. 2019. "Comparative Analysis of Microcystin Prevalence in Michigan Lakes by Online Concentration LC/MS/MS and ELISA" Toxins 11, no. 1: 13. https://doi.org/10.3390/toxins11010013
APA StyleBirbeck, J. A., Westrick, J. A., O’Neill, G. M., Spies, B., & Szlag, D. C. (2019). Comparative Analysis of Microcystin Prevalence in Michigan Lakes by Online Concentration LC/MS/MS and ELISA. Toxins, 11(1), 13. https://doi.org/10.3390/toxins11010013