1. Introduction
Sugar alcohols such as sorbitol, mannitol, and xylitol are naturally present in some fruits, vegetables, and mushrooms but are classified as artificial sweeteners since they can be industrially produced by reducing saccharides such as glucose [
1,
2]. These sugar alcohols have a low caloric content due to poor absorption in the small intestine and are thus frequently used as sweeteners in “sugar-free” candy, chewing gum, and beverages [
3]. Because of their stability and resistance to heat, they are also used for a wide variety of other purposes, including as food moisturizing agents, preservatives, and excipients. However, ingestion of these poorly-absorbed sugar alcohols may lead to gastrointestinal symptoms in some people and have laxative effects. For example, it has been reported that the excessive use of chewing gum containing sorbitol may cause symptoms such as diarrhea, weight loss, and bloating [
4,
5]. The main cause of the diarrhea is thought to be the accumulation of poorly-absorbed sugar alcohols in the colon, where they increase colonic osmotic pressure and prevent water absorption [
6]. Interestingly, susceptibility to sugar alcohol–induced diarrhea is known to vary among individuals [
6]. However, the mechanisms underlying these effects remain unknown.
Poorly-absorbed carbohydrates can be a source of energy for some bacteria residing in the gut [
7]. The human gut contains approximately 40 trillion bacteria that form complex communities of which the composition varies among individuals due to differences in diet, lifestyle, and antibiotic use [
8,
9]. Consumption of poorly-absorbed carbohydrates is one factor that can alter the composition of the gut microbiota. Because the carbohydrates preferred by gut bacteria vary from species to species, the ingestion of poorly-absorbed carbohydrates with different chemical structures can promote the development of a distinctive gut flora [
10]. Additionally, sugar alcohols like sorbitol, which are a type of poorly-absorbed carbohydrate, are known to be degraded by certain bacteria [
11,
12]. However, the relationship between the consumption of poorly-absorbed sugar alcohols and their biological effects, such as osmotic diarrhea and gut bacteria makeup, remains poorly understood.
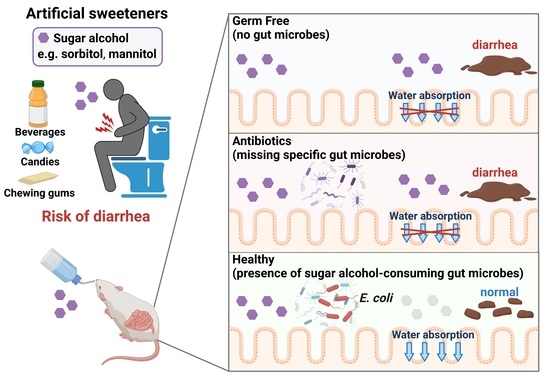
We hypothesized that sugar alcohol-degrading bacteria exist in the intestine and also that susceptibility to sugar alcohol-induced diarrhea depends on the amount of these bacteria being present. When the intestine contains low amounts of sugar alcohol–degrading bacteria, sugar alcohols accumulate, which leads to increased osmotic pressure and diarrhea. In the current study, we investigated the relationship between sugar alcohol-induced diarrhea and gut microbiota in an effort to identify the bacteria responsible for the prevention of diarrhea symptoms.
2. Materials and Methods
2.1. Mice
Male C57BL/6 mice were purchased from the Sankyo Labo Service Corporation (Tokyo, Japan) and were kept under conventional conditions. Male MCH (ICR) mice, germ-free (GF) male ICR mice, and ICR-derived inbred strain IQI mice were purchased from CLEA Japan, Inc. (Tokyo, Japan). GF male MCH and IQI mice were maintained in vinyl isolators. Male MCH mice were kept under specific pathogen-free (SPF) conditions. These mice were fed γ-ray-sterilized AIN-93G (Oriental Yeast, Tokyo, Japan). In order to evaluate the effect of sugar alcohol on diarrhea, mice were administered water drinking water containing 5% or 10% sorbitol, or 5% mannitol for 4 days. For experiments involving antibiotics, mice were administered drinking water containing ampicillin (1 g/L), streptomycin (5 g/L), erythromycin (200 mg/L), or vancomycin (250 mg/L). For gnotobiotic experiments, GF male ICR or IQI mice (obtained from CLEA Japan) were bred in vinyl isolators and fed γ-ray-sterilized CMF. During the experiment, GF mice were fed a γ-ray-sterilized AIN-93G.
All mice were housed under 21–22 °C with a 12 h alternating light–dark cycle at the animal facilities of Faculty of Pharmacy, Keio University (Tokyo, Japan). All animal experiments were performed according to the Institutional Guidelines for the Care and Use of Laboratory Animals in Research and were approved by the local ethics committees at Keio University.
2.2. Bacteria
Bacteria isolated from mouse feces and strains purchased from the Japan Collection of Microorganisms (JCM, Ibaraki, Japan) were used in the study. Wild-type (WT) and gene mutants (srlA-, srlB-, srlE-, or srlD) of E. coli K-12 were purchased from the National Bio Resource Project (NBRP, Shizuoka, Japan).
2.3. Colonization of Germ-Free Mice
Escherichia coli and Proteus mirabilis isolated from the stool of C57BL/6 mice, or WT and srlD mutant of E. coli K-12 were used for the experiments. GF male IQI or ICR mice were colonized with 200 µL of bacterial culture via oral gavage.
2.4. Evaluation of Diarrhea Symptoms
The severity of diarrhea was evaluated based on body weight change and fecal water content after the sugar alcohol treatment. Body weight change was recorded daily in the morning. The collected feces were weighed, dried overnight in an incubator, and re-weighed. The fecal water content was calculated from the weight before drying and the weight after drying.
2.5. 16S rRNA Gene Sequencing and Analysis
Approximately 200 mg of each stool sample was transferred into 2 mL tubes containing 0.1 mm zirconia/silica beads and 3.0 mm zirconia beads. The stool samples were homogenized for 10 min in a Shake Master Neo cell disruption device (Biomedical Sciences, Tokyo, Japan) after the addition of 540 µL of SLX–Mlus Buffer from an E.Z.N.A. Stool DNA Kit (Omega Bio-tek, Norcross, GA, USA). After homogenization, 60 µL of DS buffer and 20 µL of Proteinase K solution from the E.Z.N.A. Stool DNA Kit were added. The samples were then incubated at 70 °C for 10 min and at 95 °C for 5 min. SP2 buffer from the E.Z.N.A. Stool DNA Kit was then added and the samples were incubated on ice for 5 min. The samples were centrifuged at 13,000×
g for 5 min and DNA was extracted from 200 µL of the supernatant using a magLEAD 12 gC nucleic acid extraction instrument (Precision System Science, Chiba, Japan) with a MagLEAD Consumable Kit and MagDEA
®Dx SV. The extracted genomic DNA was resuspended at 5 ng/μL in 10 mM Tris-HCl buffer. The DNA was used to prepare 16S rRNA gene libraries in accordance with the protocol described in an Illumina technical note (
https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 9 May 2019). Briefly, each DNA sample was amplified by polymerase chain reaction (PCR) using KAPA HiFi HS ReadyMix (Nippon Genetics, Tokyo, Japan) and primers specific for variable regions 3 and 4 of the 16S rRNA gene. The PCR products were purified using AMPure XP Beads (Beckman Coulter, Brea, CA, USA) and adapters added by PCR using a Nextera XT Index Kit. The libraries were further purified using AMPure XP Beads, diluted to 10 nM with 10 mM Tris–HCl buffer, and pooled. The pooled samples were sequenced using a MiSeq System (Illumina, San Diego, CA, USA) with a 2 × 300-base-pair protocol. Sequences were analyzed using QIIME (v. 1.9.1) software (
http://qiime.org, accessed on 10 June 2021) [
13]. Reads with an average quality value < 20 were excluded using Trimmomatic (v. 0.36) software (
http://www.usadellab.org/cms/?page=trimmomatic, accessed on 10 June 2021) [
14]. Paired-end sequences were joined using a Fastq-join tool in the EA-Utils software package (
https://expressionanalysis.github.io/ea-utils/,
https://openbioinformaticsjournal.com/VOLUME/7/PAGE/1/, accessed on 10 June 2021). High-quality sequences (11,000) were randomly chosen from the quality filter-passed sequences that were obtained for each sample. After trimming both primer sequences using cutadapt (
https://doi.org/10.14806/ej.17.1.200, accessed on 10 June 2021), chimeras were detected using the USEARCH de novo method. The sequences were assigned to operational taxonomic units (OTUs) using the UCLUST algorithm with a sequence identity threshold of 96%. Taxonomic assignments for each OTU were made by similarity searching against publicly available 16S sequences (RDP v. 10.27 (The Center for Microbial Ecology, East Lansing, Michigan, US) and CORE update 2 September 2012) and NCBI genome databases using the blastn program available from the local Basic Local Alignment Search Tool (BLAST). The data were simplified to 5000–10,000 sequences per sample using rarefaction curves, and the relative abundances of the community members were determined using the rarefied data. The analysis was performed as described in a previous report [
15]. Principal coordinate analysis (PCoA) was used to analyze the beta diversity using an unweighted UniFrac metric calculated by QIIME. Permutational multivariate analysis of the variance (PERMANOVA) test was performed using the vegan package (v 2.7) (
https://github.com/vegandevs/vegan, accessed on 10 June 2021) in R.
2.6. DNA Extraction and Quantitative PCR (qPCR)
Bacterial DNA was isolated from 20 mg of fecal samples (or 200 µL of standard culture). Extraction and quantification of the bacteria DNA were conducted as described in a previous report [
16]. Briefly, a 20-fold diluted fecal sample (200 µL) was mixed with 300 μL of extraction buffer (100 mM Tris–HCl, 40 mM EDTA, 1.7% SDS, pH 9.0), 500 µL of buffer-saturated phenol, and 300 mg of glass beads. The mixture was vortexed vigorously for 15 s at 5000 rpm 3 times using a Precellys24 homogenizer (M&S Instruments Inc., Osaka, Japan). After centrifugation at 14,000×
g for 5 min, 400 µL of the supernatant was collected. Phenol–chloroform extractions were subsequently performed and 250 µL of the supernatant was subjected to isopropanol precipitation. Finally, the DNA was suspended in 1 mL of TE buffer.
The total number of bacteria in the feces was analyzed by qPCR using universal primers. PCR amplification and detection were performed using a CFX-96 Real-Time system (Bio-Rad, Hercules, CA, USA). Each reaction mixture (20 µL) consisted of 10 µL of SYBR premix Ex Taq II (Takara, Shiga, Japan), 0.4 µL of each primer (10 µM), 2 µL of DNA template, and 7.2 µL of distilled water. The amplification profile consisted of one cycle at 95 °C for 30 s followed by 35 cycles of 95 °C for 5 s, 60 °C for 30 s. The fluorescent signal of the amplified products was detected during the last step of each cycle. Melting curve analysis was performed after amplification to distinguish the targeted PCR product from non-targeted product. The melting curves were obtained by slow heating the samples from 65 °C to 95 °C at a rate 0.5 °C/s.
2.7. Isolation and Identification of Bacteria from Feces
Fecal samples were diluted with phosphate-buffered saline (PBS) and applied to Brain Heart Infusion agar (BD, Franklin Lakes, NJ, Japan) and modified Gifu Anaerobic Medium agar (Nissui, Tokyo, Japan) and cultured under anaerobic conditions. After 24 h to 72 h culture, some colonies were picked and the isolated colonies lysed in 100 µL of 50 mM NaOH. After incubation at 95 °C for 10 min, the lysed colonies were centrifuged at 3000× g for 10 min. Then, 20 µL of extracted solution was diluted with 100 µL Tris–HCl (pH 7.0-7.2). The diluted DNA was used as template for PCR amplification. The PCR reaction mixtures (25 µL) consisted of 18 µL of KOD FX Neo reagent solution (12.5 µL 2× buffer, 5 µL dNTPs, and 0.5 µL enzyme solution), 0.75 µL of each primer (10 µM), 2 µL of DNA template, and 3.5 µL of distilled water. The amplification profile consisted of one cycle at 94 °C for 2 min followed by 40 cycles of 98 °C for 10 s, 55 °C for 30 s, and 68 °C for 30 s with a final extension step of 72 °C for 5 min. After purification using ExoSAP-IT ExpressPCR Cleanup Reagents (ThermoFisher, Waltham, MA, Japan), the samples were sequenced using capillary sequence services from Hokkaido System Science Co., Ltd. (Sapporo, Hokkaido, Japan).
2.8. Evaluation of Sorbitol Degradation and Measurement of Sorbitol Concentration in Culture Supernatant
M9 medium [
17] was used as a control medium to culture Enterobacteriaceae isolates. The other bacteria were cultured using the Gifu Anaerobic Medium (GAM) Semisolid without Dextrose (Nissui), prepared in water and filtered to remove the agar. When sugar was added to the media, the concentration was adjusted to 10 mg/mL. Following incubation under anaerobic conditions, 10 µL of the bacterial culture broth from the previous day was placed in each medium. Bacterial growth was evaluated based on absorbance at 600 nm or 570 nm that was measured using an Infinite 200 Pro plate reader (Tecan, Seestrasse, Switzerland). Sorbitol concentrations in the culture supernatants were measured using an EnzyChrom Sorbitol Assay Kit (BioAssay Systems, Hayward, CA, USA).
2.9. Statistical Analysis
Statistical analyses were performed using R (version 3.6.3) (R Foundation, Viennam, Austria). For parametrical data when the variances were equal, we used Student’s t-test for two groups, and one-way analysis of variance (ANOVA) followed by Dunnett’s test (post-hoc test) for more than two groups, respectively. When the variances were unequal, we used Welch’s t-test for two groups, and ANOVA followed by Steel’s test (post-hoc test) for more than two groups, respectively. Differences with p-values < 0.05 were considered statistically significant.
4. Discussion
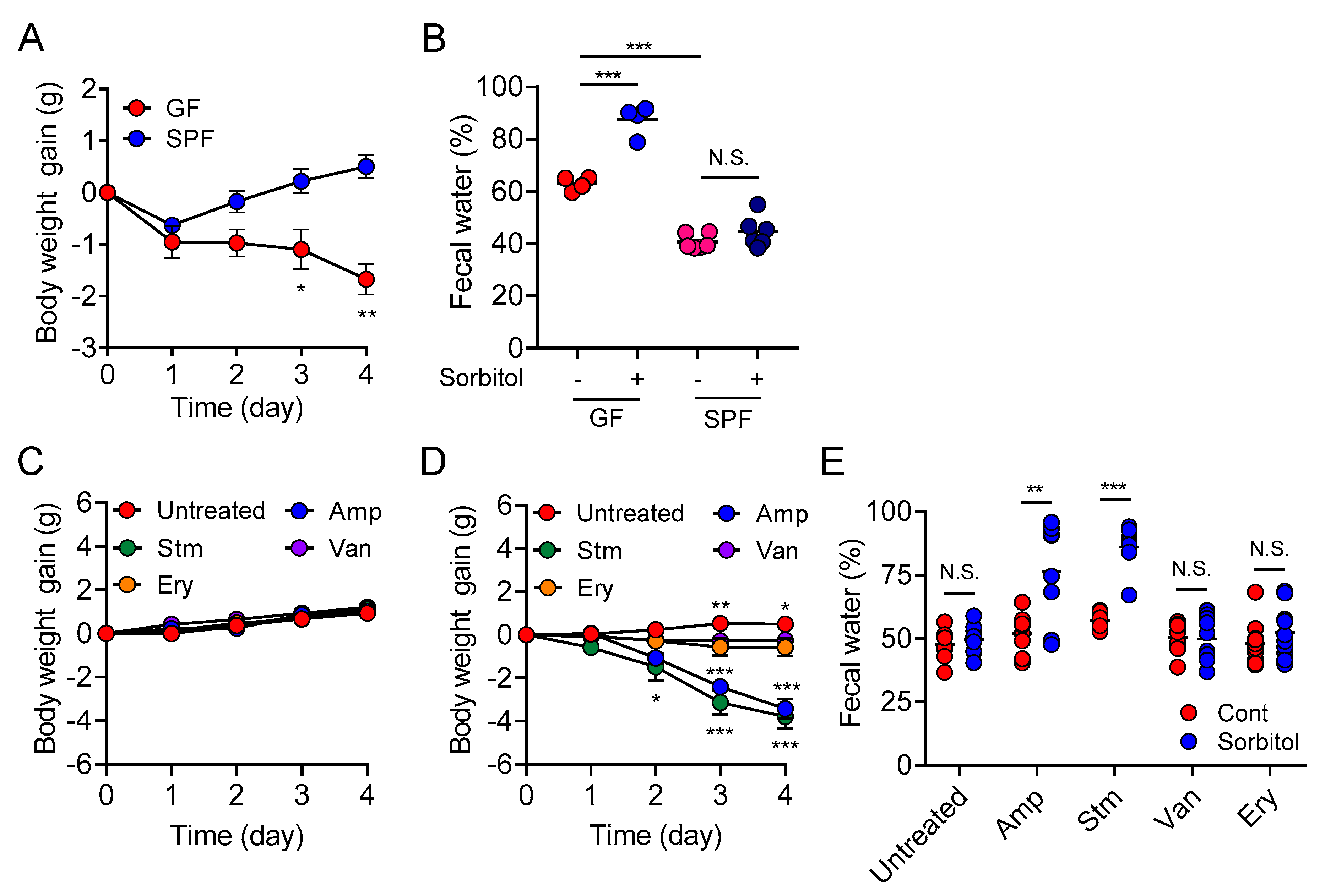
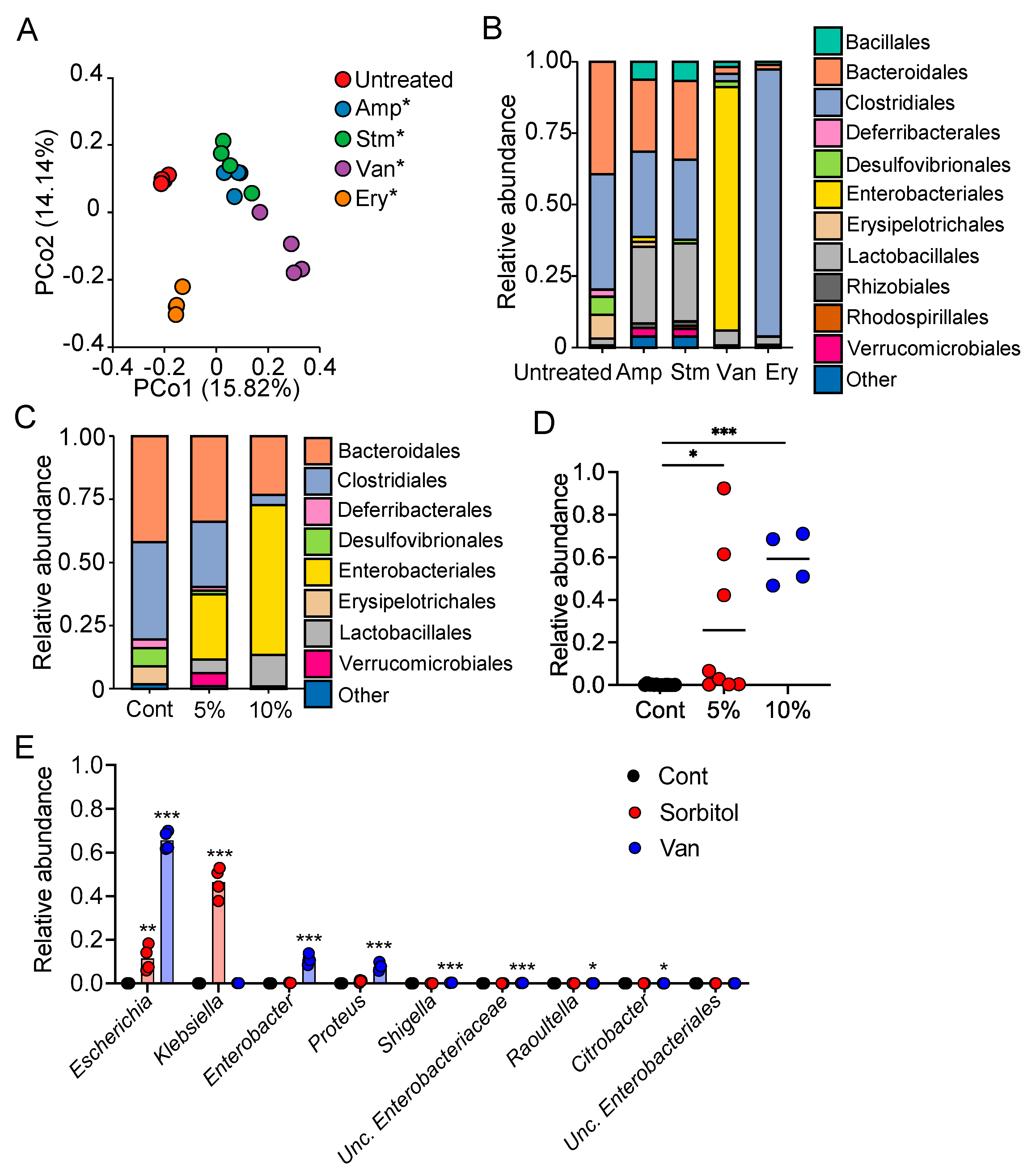
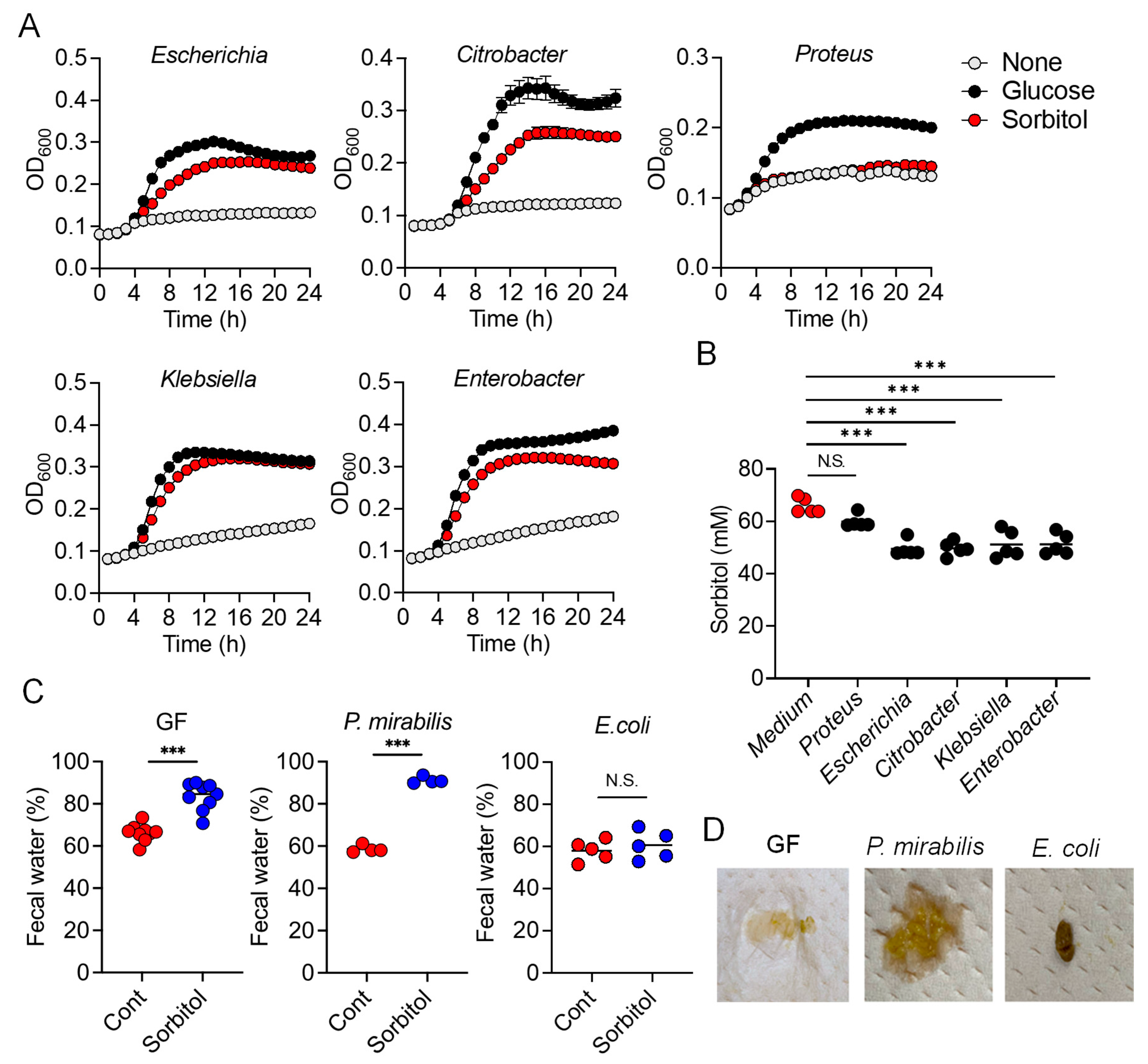
In the current study, we demonstrated the protective role of gut bacteria against sugar alcohol-induced diarrhea using both GF mice that lack gut bacteria and mice that had altered gut microbiota as a result treatment with antibiotics. When exposed to sorbitol at a dose that failed to induce diarrhea in SPF mice, GF mice exhibited severe diarrhea and significant weight loss. GF mice are known to have higher fecal water content than that of SPF mice under normal conditions, but while the fecal water content of the SPF mice in our study did not change with sorbitol treatment, the fecal water content of the GF mice increased with sorbitol treatment. Since antibiotics such as ampicillin, streptomycin, vancomycin, and erythromycin have different antimicrobial spectra, administration of those antibiotics changed the gut microbiota composition of mice to specific states that were dependent on their antimicrobial spectra. Mice with gut microbiota composition altered by ampicillin and streptomycin treatments exhibited increased susceptibility to sorbitol-induced diarrhea. In comparison, mice with gut microbiota composition that had increased proportions of Enterobacteriaceae and Lachnoclostridium as a result of vancomycin and erythromycin treatment, respectively, did not show an increase in their susceptibility to sorbitol-induced diarrhea. Interestingly, the abundance of Enterobacteriaceae increased in a concentration-dependent manner as sorbitol intake increased. This effect was particularly dramatic in mice that received 10% sorbitol. This sorbitol-induced growth of Enterobacteriaceae most likely contributed to the transient diarrhea and subsequent disappearance of diarrheal symptoms observed in these mice. Enterobacteriaceae expansion may also represent the mechanism underlying the acquisition of sorbitol tolerance observed in individuals after long-term intake.
At the species level, E. coli constituted approximately 70% of the gut Enterobacteriaceae population in vancomycin-treated mice with Enterobacter spp. and P. mirabilis accounting for the remaining 30%. As Enterobacter spp. and P. mirabilis were not able to utilize sorbitol as an energy source, E. coli appears to have been the main contributor to the suppression of sorbitol-induced diarrhea by Enterobacteriaceae. Indeed, the inoculation of GF mice with E. coli alone was sufficient to generate resistance to sorbitol-induced diarrhea.
While the pathways of sorbitol metabolism vary depending on the particular bacterial species, the sugar phosphotransferase system (PTS) is one of the major known pathways. Sorbitol is phosphorylated in this pathway to sorbitol-6-phosphate by a sugar-specific permease, which is a membrane-bound complex known as enzyme II. The sorbitol-6-phosphate is subsequently converted to fructose by PTS-dependent dehydrogenase and the fructose is used for glycolysis [
19,
20,
21]. An
E. coli mutant lacking
srlD, which encodes the PTS-dependent sorbitol-6-phosphate dehydrogenase, was unable to degrade sorbitol and did not protect mice against sorbitol-induced diarrhea. This indicates the sorbitol degradation by
E. coli was dependent on PTS. However, mutants of
srlA,
srlB, or
srlE, which are genes encoding components of the enzyme II complex, were able to utilize sorbitol as an energy source, suggesting that an enzyme II complex–independent pathway for the production of sorbitol-6-phosphate via phosphorylation of sorbitol may exist in
E. coli. On the other hand, we also showed that some
Clostridium species (re-classified as
Lachnoclostridium) were able to utilize sorbitol. However,
Clostridium is known to be able to directly metabolize sorbitol to fructose unlike
E. coli which metabolizes sorbitol to fructose via the PTS pathway [
22,
23].
Although some strains of
E. coli can cause infectious and inflammatory diseases in humans, many others live peacefully in human intestines, aiding in digestion and even defending their host against harmful microbes [
24]. We found that
E. coli was increased and suppressed sorbitol-induced diarrhea after the ingestion of sorbitol. In human adults,
E. coli is not the dominant bacteria in the intestine. However, these microbes may maintain a hospitable environment in the gut by preventing the detrimental effects of artificial sweeteners which are widely used in processed foods.
Our current study revealed the relationship between sugar alcohol-induced diarrhea and the gut microbiota, and identified specific gut bacteria that responded to and degraded the sorbitol. However, mannitol and xylitol (which like sorbitol, are poorly-absorbed sugar alcohols) are also widely used as artificial sweeteners in processed foods. As the metabolism of these sugar alcohols by the gut bacteria may differ from that of sorbitol, the species of gut bacteria that protect against diarrhea induced by these sugar alcohols may also be different. Further work is needed to clarify whether sugar alcohols other than sorbitol are degraded by different gut microbes.
The gut microbiota has been reported to be associated with various diseases, including some lifestyle-related diseases [
25,
26,
27]. Accordingly, manipulation of the gut microbiota has received attention for its potential exploitation in supporting human health, such as the use of stool transplantation or metabolites derived from gut microbiota [
28,
29]. Prebiotics are compounds in food that induce the growth or activity of beneficial microorganisms, such as by altering the composition of the gut microbiome [
30]. In contrast, dysbiosis is a term for microbial imbalance that may causes negative health symptoms [
31]. Dysbiosis can arise from diverse causes, including antibiotic use or consumption of an inappropriate diet [
32]. Our current findings regarding the effect of sorbitol on the gut microbiota suggest that artificial sweeteners in processed foods can act as either prebiotics that promote a healthy gut or as inappropriate dietary elements that may cause dysbiosis. We believe our findings provide helpful information elucidating the effects of artificial sweeteners in processed foods on human health via gut microbiota.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



