
Quality Risk Management in Pharmaceutical Manufacturing Operations: Case Study for Sterile Product Filling and Final Product Handling Stage


Abstract
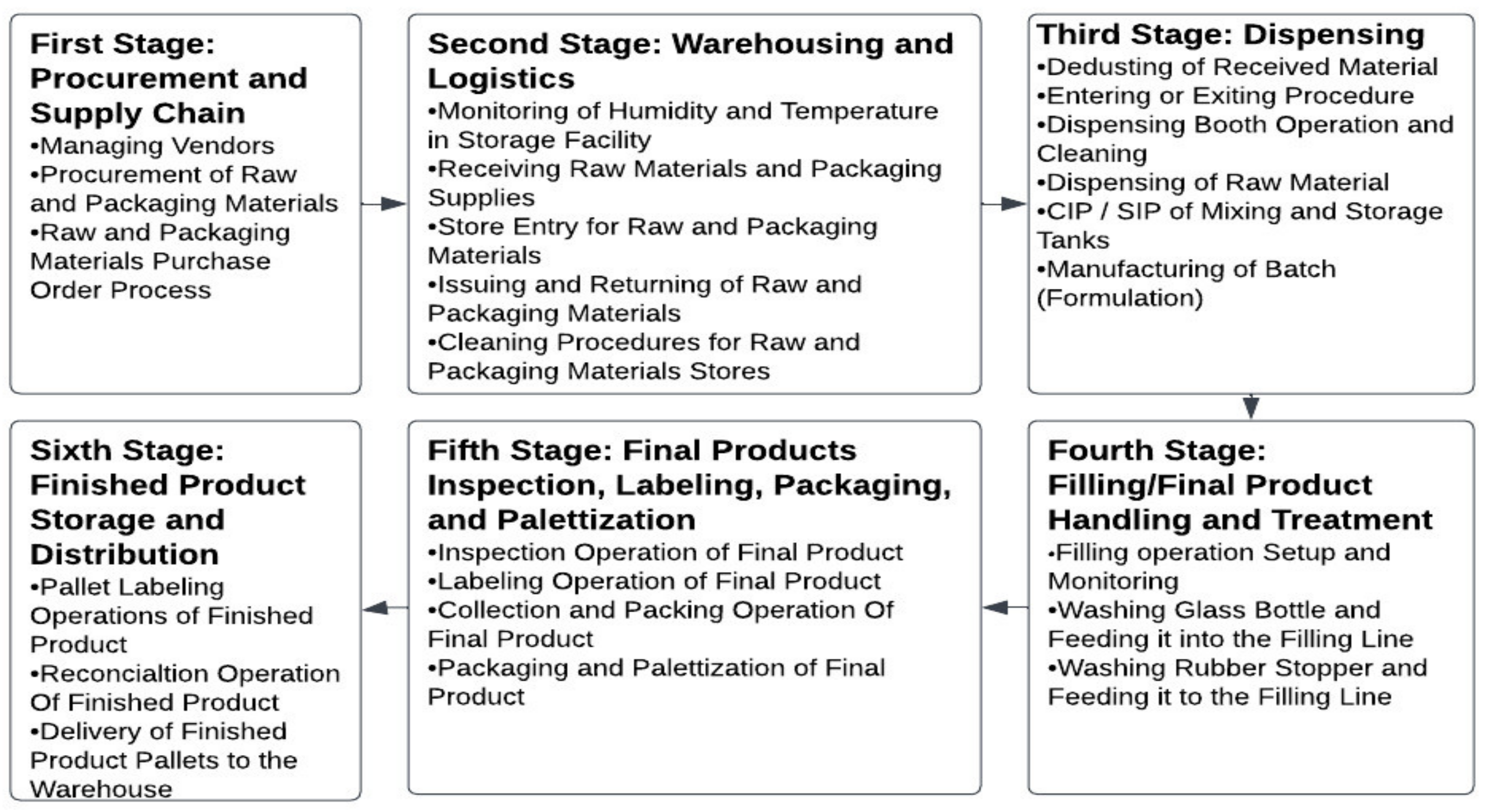
1. Introduction
2. Materials and Methods
- A
- Reading and comprehending the applicable standard operating procedure of the selected procedure.
- B
- Meeting with both the process owners and supervisors to simplify the procedure into specific, well-defined steps.
- C
- Using a brainstorming technique and in cooperation with a risk management specialist, all possible risks connected with every step are identified.
- D
- The risk table for risk analysis is filled out by addressing well-known risk specific questions such as, “What could go wrong?” What is the possibility (likelihood) that something could go wrong? What are the effects (severity)? What is detection capability (detectability)? As presented in Figure 2, this is a straightforward implementation of the FMEA risk assessment tool.
- E
- FMEA risk evaluation can identify severity, probability of occurrence, and likelihood of detection ratings on a scale from 1 to 10. 1 is attributed to the lowest risk and 10 to the worst risk to the safety of the product. Risk priority number (RPN) is determined by multiplying the three specified scores: [Severity of effect] × [Likelihood of occurrence] × [Unlikelihood of detection]. Table 1.
- F
- Risk control can be carried out by putting in place new policies or standards, making physical or design changes, or making changes to how work is performed that can completely remove (when possible) or lessen the risk.



{kind=link}
{kind=link}
| Risk Priority Number (Severity × Occurrence × Detection) | Level * | Action |
|---|---|---|
| 1–34 | Low | Risk is acceptable |
| 35–104 | Medium | Risk can be acceptable. Reduce risk as much as it is practically possible |
| 105–1000 | High | Risk cannot be accepted. Risk reduction and mitigation are required. |
- Changes are made to the existing process or design.
- A change is made to the operation conditions.
- An improvement goal is created for the existing process.
- New regulations are introduced.
- Customer feedback.
3. Results and Discussion
3.1. Risk Assessment Associated with Entry and Exit Procedure to Cleanrooms
3.2. Risk Assessment Associated with Glass Bottle Washing Machine and Tunnel Operation and Its Related Activity
3.3. Risk Assessment Associated with Glass Filling Operation and Process Checks
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, N.; Jha, A. Quality risk management during pharmaceutical ‘good distribution practices’—A plausible solution. Bull. Fac. Pharm. Cairo Univ. 2018, 56, 18–25. [Google Scholar] [CrossRef]
- Chavda, V.P.; Maru, S.; Patel, B. How Quality Risk Management is Useful for Pharmaceuticals. Glob. Res. J. Sci. Nat. 2015, 1, 10–13. [Google Scholar]
- ICH. Committee Report, International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, Quality Risk Management. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-3.pdf (accessed on 20 April 2022).
- Nauman, M.; Bano, R. Implementation of Quality Risk (QRM) in Pharmaceutical Manufacturing. J. Pharm. Biol. Sci. 2014, 9, 95–101. [Google Scholar]
- Sivadasu, S.; Gangadharappa, H.; Kiran, H.; Jose, A. Quality Risk Management: A Review. Int. J. Pharm. Sci. Rev. Res. 2017, 44, 142–148. [Google Scholar]
- Das, A.; Kadwey, P.; Mishra, J.K.; Moorkoth, S. Quality Risk Management (QRM) in Pharmaceutical Industry: Tools and Methodology. Int. J. Pharm. Qual. Assur. 2014, 5, 13–21. [Google Scholar]
- Krishna, V.; Srivastava, P. Risk management in pharmaceuticals. Mintage J. Pharm. Med. Sci. 2014, 3, 32–39. [Google Scholar]
- Snee, R.; Rodebaugh, W.F. Failure Modes and Effects Analysis. In Encycelopedia of Statistics in Quality and Reliability, 1st ed.; Ruggeri, F., Kenett, R., Faltin, F., Eds.; Wiley: Hoboken, NJ, USA, 2007; Volume 3, pp. 154–196. [Google Scholar]
- Vartak, R.; Bhagure, G. Quality Risk Management in Pharmaceutical Industry—A Overview. Asian J. Chem. 2012, 24, 5576–5578. [Google Scholar]
- Perez, M.; Foinar, A.; Barthélémy, C.; Décaudin, B.; Odou, P. Particulate Matter in Injectable Drugs: Evaluation of Risks to Patients. Pharm. Technol. Hosp. Pharm. 2016, 1, 91–103. [Google Scholar] [CrossRef]
- Sandle, T. Sterile Ophthalmic Preparations and Contamination Control. J. GXP Compliance 2014, 18, 1–5. [Google Scholar]
- Alsaidalani, R.; Elmadhoun, B. Quality Risk Management in Pharmaceutical Supply Chain Warehousing and Dispensing. Int. J. Pharm. Sci. Rev. Res. 2021, 68, 155–164. [Google Scholar]
- Ismael, O.A.; Ahmed, M.I. Using Quality Risk Management in Pharmaceutical Industries. Qual.-Access Success 2020, 21, 106–113. [Google Scholar]
- Chitmetha, M.; Prombanpong, S.; Somboonwiwat, T. Quality Risk Management in Pharmaceutical Dispensing. Int. J. Chem. Eng. Appl. 2013, 4, 241–248. [Google Scholar] [CrossRef]
- Carlin, B. Quality Risk Management of Compliant Excipients. J. Excip. Food Chem. 2012, 3, 143–153. [Google Scholar]
- Sandle, T. Aseptic Transfer Risk Assessment: A Case Study. J. Valid. Technol. 2015, 21, 1–10. [Google Scholar]
| Risk Assessment | Risk Control | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Risk Identification | Risk Analysis | Risk Evaluation * | Risk Reduction and Acceptance | Compliance of Action | Risk Re-Evaluation * | |||||||||||
| Step No. | Process Step/Input | Potential Failure Mode | Potential Failure Effects | SEVERITY (S) (1–10) | Potential Occurrence | OCCURRENCE (O) (1–10) | Current Controls | DETECTION (D) (1–10) | RPN (S × O × D) | Risk Acceptance | Action Recommended | Resp. | Actions Taken | OCCURRENCE (O) | DETECTION (D) | RPN (S × O × D) |
| What Controls Exist That Will Either Prevent or Detect Failure? | What Are the Recommended Actions to Reduce the Occurrence of the Causes or Enhance Detection? | Who is Responsible for Assuring That the Actions Are Carried Out? | What Actions have Been Done Regarding the Rpn? | |||||||||||||
| 1 | Photographs are not allowed unless permitted by an authorized person. | Photographs may be intentionally or unintentionally taken in the restricted area. | Company private property is jeopardized. Company confidential information is exposed. Photos may be misused. | 8 | Absence of site supervision. No or inadequate control on visitors entering cleanrooms. No posters stating photographs are prohibited. Lack of staff awareness | 6 | Area supervision is available for every shift. Visitors are not allowed in the area without a supervisor. Posters are available stating no photographs. | 4 | 192 | No. Risk mitigation is required | Revise SOP and add new instructions and control stating that no camera, mobile, or any device has a camera to accompany staff or visitors in the restricted area. | Production | Action completed | 4 | 2 | 64 |
| Risk Assessment | Risk Control | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Risk Identification | Risk Analysis | Risk Evaluation * | Risk Reduction and Acceptance | Compliance of Action | Risk Re-Evaluation * | |||||||||||
| Step No. | Process Step/Input | Potential Failure Mode | Potential Failure Effects | SEVERITY (S) (1-10) | Potential Occurrence | OCCURRENCE (O) (1 –10) | Current Controls | DETECTION (D) (1 –10) | RPN (S × O × D) | Risk Acceptance | Action Recommended | Resp. | Actions Taken | OCCURRENCE (O) | DETECTION (D) | RPN (S × O × D) |
| What Controls Exist That Will Either Prevent or Detect Failure? | What Are the Recommended Actions to Reduce the Occurrence of the Causes or Enhance Detection? | Who Is Responsible for Assuring That the Actions Are Carried Out? | What Actions Have Been Done Regarding the Rpn? | |||||||||||||
| 1 | Assure that password level protection is in place and complies with the principles of data integrity. | Loss of protection and possible manipulation and change in setting | Questionable data integrity. GMP and GDocP guideline noncompliance. Regulatory Auditor concern. Negative impact on product quality | 8 | Improper password level protection. Sharing or delegating password to unauthorized person(s). lack of awareness of data integrity. Inadequate staff training on GDocP and data integrity | 4 | All authorized machine operators received GDocP and data Integrity training. | 6 | 192 | No. Risk mitigation is required | Regular checks of audit trails and report any violation of password level protection. Regular personnel training on the importance of data integrity. Self-inspection should cover the implementation of data integrity principles. | Production/QA and IT | Completed | 3 | 4 | 96 |
| 2 | Assure that line clearance activity is conducted, documented, and approved before startup. | No line clearance. Improper line clearance. Line clearance Not documented. Absence of QA check and approval. | Possibility of a mix-up. Product quality is questionable. GMP violation and non-compliance. Regulatory concern, negative impact on patient safety. | 9 | GMP guidelines not implemented. Absence of proper GxP training. Insufficient quality assurance monitoring and management | 6 | Clearance procedure is available. Staff training records are documented. Production supervisor review and approve clearance document. Records are available. | 5 | 270 | No. Risk mitigation is required | Clearance SOP and associated clearance format should be revised to include involvement of QA inspector. Clearance document should be finally approved by QA before commencing production process. | Production and QA | completed | 3 | 3 | 81 |
| 3 | All primary packaging materials, e.g., rubber stopper, shall be transferred through dynamic pass box by stacking on stainless steel trolleys to filling room. | Primary packaging materials transferred through personal entry. Dynamic pass box is not in function. | Violation of company procedure and GMP guidelines. Disturbances in the cleanroom classification may cause product contamination. | 8 | Lack of proper production supervision. Ineffective QA inspection. Failure in dynamic pass box due to improper routine maintenance. | 4 | SOP for handling PPM in glass filling unit is available. Staff is trained, and production supervision exists. QA inspectors are available. The current procedure lacks the provision of checking dynamic pass box operation during checklist before startup of machine or during line clearance | 4 | 128 | No. Risk mitigation is required | Process-related SOP should be revised, and provision for checking dynamic pass box should be part of area checking before start and/or during line clearance. Line clearance checklist needs to be changed to cover dynamic pass box status. | Production and QA | completed | 3 | 3 | 72 |
| 4 | No empty washed bottles shall be left inside washing machine during break time or end of shift. | Some empty washed bottles are leftover inside washing machine. | Contaminated bottles may be used in subsequent filling. Lot reconciliation is not accurate. Chance of mix-up. Negative impact on product quality. GMP violation and regulatory concern | 7 | Unqualified staff handling the process. Lack of monitoring and supervision. No checklist to document the absence of any empty bottles inside glass bottles washing machine. No counter-check. | 4 | Staff training records on related SOP are available. | 4 | 112 | No. Risk mitigation is required | Related SOP should be revised to cover the use of a checklist to assure the absence of any empty glass bottles inside bottle washing machine during breaks and at the end of shift. The checklist should be counter-signed by unit supervisor. | Production department | Completed | 2 | 2 | 28 |
| Risk Assessment | Risk Control | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Risk Identification | Risk Analysis | Risk Evaluation * | Risk Reduction and Acceptance | Compliance of Action | Risk Re-Evaluation * | |||||||||||
| Step No. | Process Step/Input | Potential Failure Mode | Potential Failure Effects | SEVERITY (S) (1–10) | Potential Occurrence | OCCURRENCE (O) (1–10) | Current Controls | DETECTION (D) (1–10) | RPN (S × O × D) | Risk Acceptance | Action Recommended | Resp. | Actions Taken | OCCURRENCE (O) | DETECTION (D) | RPN (S × O × D) |
| What Controls Exist That Will Either Prevent or Detect Failure? | What Are the Recommended Actions to Reduce the Occurrence of the Causes or Enhance Detection? | Who Is Responsible for Assuring That the Actions Are Carried Out? | What Actions Have Been Done Regarding the Rpn? | |||||||||||||
| 1 | Assure that line clearance activity is conducted, demented, and checked before startup | No line clearance. Line clearance is not double-checked. | Product mix-up. Poor lot reconciliation. Violation of GMP standards. Regulatory authority concern. | 8 | Line clearance SOP is not available. Line clearance is not propyl conducted. Filling line operators are not trained. No intervention or approval from QA | 5 | No records are available for filling line clearance. No formal, detailed, and specific SOP for filling line clearance. Line clearance of filling room is conducted by filling line operators without formal documents or double-checking. | 5 | 200 | No. Risk mitigation is required | SOP for filling line clearance should be produced, reviewed, and approved. Filling line clearance should be conducted by quailed production personnel, checked, and approved by QA personnel. The filling process should not be started before approved line clearance. Filling line clearance report should be available in BMR | Production and QA | completed | 2 | 3 | 48 |
| 2 | Solution filter shall be wetted with product solution, its integrity is tested, and activity is recorded in BMR. | Filter integrity test is not done or done incorrectly, or failed results are manipulated. | Unqualified solution filter is used. Microbial contamination of product solution. Overall product quality is negatively impacted. GMP non-compliance. | 8 | Unqualified personnel doing the test. Unavailability of filter integrity test machine. Unqualified testing machine. Manual filter integrity testing | 5 | Production staff is trained on filter integrity testing procedures. Solution filter integrity test is conducted manually. Testing results are recorded manually in the form. Second operator is double-checking the recorded result. | 5 | 200 | No. Risk mitigation is required | Manual filter integrity test shall not be used. Filter test shall be carried out using machine, and testing results should be automatically saved and printed. No manual recording of the result. | Production, engineering, and QA | completed | 4 | 3 | 96 |
| 3 | Ensure that the scales in the filling machine are calibrated through IPC station, and such activity shall be done under production condition | Scales of filling machine during adjusting weighing modules are not checked and may be out of calibration. | Inaccurate product volume may be produced. Product volume is out of specification. Non-compliance to GMP guidelines and regulatory concerns. | 7 | Related procedure is not clear and not understood by line operators. No filling machine checklist. Lack of QA monitoring. Lack of proper training. | 3 | Activity-related SOP is available and followed. Line operators are well trained on the related SOP. Record is available in BMR and checked by the production supervisor. | 4 | 84 | No. Risk mitigation is required | Activity-related SOP shall be revised to implement using a checklist covering scales calibration status and be available in BMR. | Production and QA | Completed | 2 | 3 | 42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsaidalani, R.; Elmadhoun, B. Quality Risk Management in Pharmaceutical Manufacturing Operations: Case Study for Sterile Product Filling and Final Product Handling Stage. Sustainability 2022, 14, 9618. https://doi.org/10.3390/su14159618
Alsaidalani R, Elmadhoun B. Quality Risk Management in Pharmaceutical Manufacturing Operations: Case Study for Sterile Product Filling and Final Product Handling Stage. Sustainability. 2022; 14(15):9618. https://doi.org/10.3390/su14159618
Chicago/Turabian StyleAlsaidalani, Rawidh, and Bassam Elmadhoun. 2022. "Quality Risk Management in Pharmaceutical Manufacturing Operations: Case Study for Sterile Product Filling and Final Product Handling Stage" Sustainability 14, no. 15: 9618. https://doi.org/10.3390/su14159618
APA StyleAlsaidalani, R., & Elmadhoun, B. (2022). Quality Risk Management in Pharmaceutical Manufacturing Operations: Case Study for Sterile Product Filling and Final Product Handling Stage. Sustainability, 14(15), 9618. https://doi.org/10.3390/su14159618