Abstract
In patients with sickle cell disease (SCD), transfusions pose risks like delayed hemolytic transfusion reaction (DHTR) and hyperhemolytic syndrome (HHS). We present the case of a 61-year-old Nigerian male patient with SCD, developing hyperhemolytic syndrome (HHS) post-orthopedic surgery due to alloimmunization from blood transfusions. Surgery induced massive hemorrhage, requiring RBC transfusions. Postoperatively, he developed HHS with jaundice, hemoglobinuria, and fever. Despite additional transfusions, his condition worsened, leading to hematological consultation on postoperative day +9. Laboratory findings showed positive DAT and multiple alloantibodies. The diagnosis of HHS was established and treatment involved high-dose methylprednisolone, intravenous immunoglobulin (IVIG), and erythropoietin. The patient was discharged on postoperative day +24 with stable hemoglobin levels, tapering doses of methylprednisolone, and continuous administration of hydroxyurea prescribed. HHS pathogenesis involves extensive intravascular hemolysis, exacerbated by alloimmunization. Diagnostic challenges and therapy selection complexity underscore the need for cautious transfusion strategies in HHS, reserving them for hemodynamic instability or hypoxia. This case highlights promptly recognizing and managing HHS in SCD for improved outcomes and avoiding unnecessary transfusions.
1. Introduction
Sickle cell disease (SCD) is an autosomal-recessive clinical syndrome affecting millions of people worldwide. It is caused by the homozygous form of hemoglobin S (HbS) due to a point mutation in the beta-globin chain causing a gain of function mutation in the β globin gene. HbS forms long polymers in the deoxygenated form; these semisolid HbS aggregates are formed in the blood vessels, causing red blood cells (RBC) to become less elastic, leading to the accumulation of rigid cells. This leadsto thrombosis and consequent tissue ischemia. The rate of polymerization is proportional to the HbS concentration.
In parallel, the altered morphology of RBCs makes them less resistant to shear stress, causing constant hemolysis and consequently, chronic anemia. The chronic hemolysis causes increased plasma viscosity; this, together with the rigidity of the red blood cells due to HbS polymerization and dehydration, leads to reduced blood flow through the capillaries [1]. Vaso-occlusion is also enhanced by the interactions between the highly expressed adhesion molecules on immature erythrocytes and endothelial cells [1]. It is established that vaso-occlusive crisis (VOC) is initiated by hypoxia, stress, infection, dehydration, and acidosis; thus, inflammatory cells are also activated in SCD, leading to the activation of the endothelium. Moreover, it was shown previously that the activation of coagulation pathways also contributes to vaso-occlusion [2]. The rate of hemolysis is usually stable between crises, but patients with a higher rate of hemolysis are more likely to develop organ dysfunction and vascular stiffness, and the hemolysis itself causes endothelial dysfunction. Hb is degraded to heme and heme iron which activate the innate immune system. The products of hemolysis are also considered damage-associated molecular patterns (eDAMPs), which lead to sterile inflammation in SCD patients [1].
SCD patients have various clinical manifestations that also differ in severity. The most common symptoms are recurrent episodes of pain due to VOC, fatigue, anemia, and increased sensitivity to infection (encapsulated bacteria) because of functional asplenia. Moreover, patients could have acute chest syndrome with fever, cough, angina-like pain, dyspnea usually caused by infection, fat embolism, and pulmonary infarction. Patients, especially children have an increased risk of stroke. Besides these symptoms, long-term complications are renal dysfunction, retinopathy, avascular necrosis, cholecystitis, skin ulcers, and dactylitis. Aplastic crisis can occur due to Parvovirus B19 infection causing splenic sequestration crisis, an acute painful enlargement of the spleen causing a sudden drop in hemoglobin level [3,4].
The treatment of the disease intends to minimize severe pain, hydration, and transfusions [3]. Transfusions remain a mainstay of supportive therapy, despite the increased risk in patients with SCD [5,6]. Acute transfusion is recommended in symptomatic anemia. Based on a previous pilot randomized study [7], there were no advantages of the use of transfusion on opiate use or total stay in hospital. In patients with acute ischemic stroke, acute chest syndrome exchange transfusions improve healing [5].
Chronic transfusion significantly reduces the risk of stroke by 90% in children with transcranial Doppler (TCD) velocities [8]. The goal of treatment is to reserve a HbS level below 30%; in younger patients, transfusions are recommended every 3–4 weeks, and in adults, exchange transfusions can be used [5]. Hydroxycarbamide treatment was shown to be as effective as transfusion for primary stroke prevention in children with abnormal TCD (TWiTCH trial) [9]. Alloimmunization is a significant challenge in SCD patients [5].
Delayed hemolytic transfusion reaction (DHTR) occurs in about 4% of adult patients receiving occasional transfusions [10]. DHTR is an umbrella term, primarily caused by RBC alloimmunization due to differences in blood groups between donors of Caucasian origin and recipients of African ancestry [11]. However, many cases have been described in the literature with no detectable antibodies, which raises uncertainty regarding the underlying pathogenesis [12].
In DHTR, hemolysis occurs between 24 h and 21 days after the last transfusion. However, it is hard to differentiate between other acute complications of SCD, but it has to be considered if the worsening of hemolysis is occurring immediately after the transfusion. Acute hemoglobinuria or the detection of new alloantibodies confirms the diagnosis. The direct or indirect antiglobulin (DAT) test could be positive or negative [5]. Based on previous reports, the occurrence of severe DHTR is 11.5–16% with ABO and Rh matching, while it is 4–7% in Rh and K matching [6]. The mortality of severe DHTR is high, accounting for 4.2% of all causes of death in patients with SCD [6].
The most severe form of DHTR is hyperhemolysis or hyperhemolytic syndrome (HHS), which clinically presents with fever, acute pain, and jaundice. In HHS, there is an acute increase in total bilirubin and/or lactate dehydrogenase (LDH) levels, a decrease in hemoglobin levels following RBC transfusion, and acute reticulocytopenia followed by marked reticulocytosis [11]. HHS can be classified as acute and delayed forms; the acute form occurs within 7 days of transfusion without alloantibody formation and the DAT test is negative. The delayed form occurs later than a week, alloantibodies can be detected, and the DAT test would be negative or positive [13].
The role of RBC transfusions in HHS is controversial [11,12,13,14,15]. Treatment of HHS is avoiding the use of transfusions; if transfusions are still necessary, then premedication with corticosteroids or intravenous immunoglobulin (IVIG) could be used. Based on the previous findings, in some case reports, HHS was treated with IVIG (0.5 g/kg/day for 5 days) and methylprednisolone 0.5 g/day for 2 days [16,17].
Herein, we present the case of a patient with SCD who developed HHS in the setting of alloimmunization via RBC transfusion, after orthopedic surgery.
2. Case Report
We present the case of a 61-year-old male patient from Nigeria with a history of sickle cell disease (c.20A > T; rs334 point mutation in the hemoglobin beta gene). He was hospitalized because of several vaso-occlusive crises, and his history included several uncomplicated blood transfusions. He was on hydroxyurea therapy for only a short, 6-month period. As a complication of his SCD, he developed bilateral avascular necrosis of the femoral condyles, requiring bilateral total endoprosthesis (TEP) in 1996, and thereafter, he had multiple revision procedures (in 2017 and 2019).
The patient was admitted to the Department of Traumatology, University of Debrecen, for an elective left-hip TEP revision surgery. The surgery was performed on 20 December 2022, at which time, the hemoglobin (Hgb) level was 97 g/L (normal value: >130 g/L) (Figure 1). During the surgery, a massive venous hemorrhage developed in the pelvic region. The Hgb level dropped to 65 g/L and TEP implantation could not be performed. Hemorrhagic shock emerged and four units of red blood cell (RBC) transfusions and two units of fresh frozen plasma concentrates were administered due to a vital indication. Blood-group serology testing was carried out two days before surgery and the test verified six alloantibodies (anti-D, anti-C, anti-E, anti-Fya, anti-S, and anti-Jkb) at baseline, and the direct antiglobulin test (DAT) was negative.
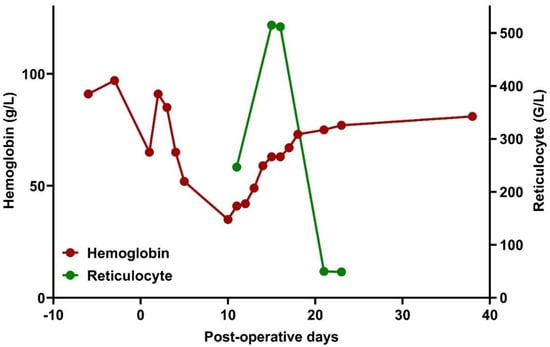
Figure 1.
Changes in hemoglobin levels and reticulocyte count in the peri-operative phase.
Considering the hemodynamic instability and the resulting need for combined high-dose vasopressor therapy, his postoperative care commenced at the Intensive Care Unit (ICU).
Under supportive therapy, his condition gradually improved; the Hgb level increased to the preoperative level of 91 g/L, the need for vasopressor therapy was resolved, and extubation could be performed. However, on the third day of ICU treatment (postoperative day +3), the patient developed jaundice, hemoglobinuria, fever, and an elevation of inflammatory markers. Hgb level decreased to 52 g/L, equivalent to a 43% reduction from baseline. The possibility of recurrent hemorrhage was ruled out, and hepatic or post-hepatic causes of jaundice were also excluded. A marked elevation of LDH and indirect bilirubin was explained by hemolysis (Figure 2).
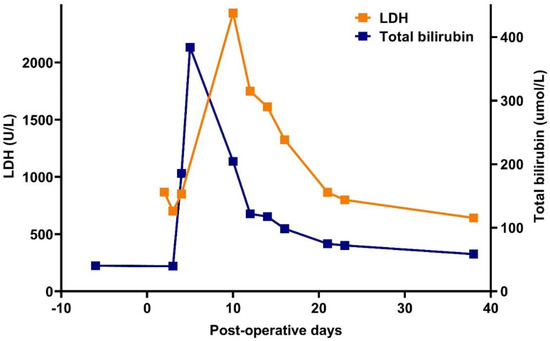
Figure 2.
Changes in lactate dehydrogenase (LDH) enzyme and total bilirubin levels in the peri-operative phase.
Based on laboratory and clinical parameters, the patient met the criteria for HHS; however, no consultation for hematology was considered at that time. The patient received an additional five units of RBC transfusion, but his Hgb decreased further (35 g/L). Peripheral smear revealed anisopoikilocytosis with numerous nucleated red blood cells, blister cells, schistocytes, and only occasional sickle cells (due to hyperhemolysis, the occurrence was reduced) (Figure 3).
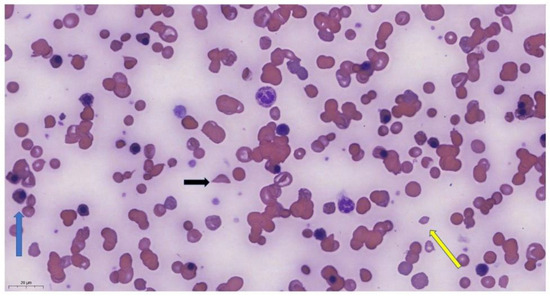
Figure 3.
Peripheral smear of the patient showing anisopoikilocytosis with schistocytes (yellow arrow), nucleated red blood cells (blue arrow), sickle cells (black arrow) (May–Grünwald–Giemsa staining, 100× oil).
On postoperative day +9, a consultation for hematology was arranged, in which a strong suspicion of HHS was raised and retrospectively established based on available laboratory data and blood group serology. He was admitted to the Division of Hematology, Department of Internal Medicine, in a frail but hemodynamically stable condition. The new serological blood testing carried out on day +9 confirmed the presence of the already known six alloantibodies (anti-D, anti-C, anti-E, anti-Fya, anti-JKB, anti-S) and DAT positivity.
For the treatment of HHS, high-dose methylprednisolone (500 mg/day for 5 days, equal to 8 mg/kg/day) and intravenous immunoglobulin (IVIG, 30 g/day for 5 days, equal to 0.4 g/kg/day) were initiated. In addition, the patient received three doses of erythropoietin (40,000 IU epoetin alpha, weekly). Methylprednisolone was gradually tapered down to a dose of 0.5 mg/kg. Although the general condition improved moderately, hemodynamic instability did not develop. A gradual increase in his hemoglobin level stabilized on postoperative day +21, with a concomitant normalization of the reticulocyte count (Figure 1). Blood products were not administered despite extreme anemia (39 g/L).
As a supportive treatment, the patient received thromboprophylaxis (enoxaparin 4000 IU/day) for immobilization. Although there were no signs of manifesting respiratory tract infection, targeted meropenem treatment was administered considering the previous mechanical ventilation, positive tracheal lavage cultures, and increased susceptibility to infection.
The patient was discharged on postoperative day +24. To prevent recurrent HHS, prolonged tapering of methylprednisolone from the dose of 0.5 mg/kg/day for 8 weeks, and permanent hydroxyurea treatment (500 mg/day) were prescribed for continuous use. Hematological control on day +38 revealed further improvement. No recurrent episodes of HHS developed, and no transfusions were required within one year of diagnosis. The patient is planning to undergo a repeated TEP replacement. The laboratory parameters of the patient are summarized in Table 1.

Table 1.
Laboratory results (Hgb—hemoglobin, LDH—lactate dehydrogenase enzyme, tBi—total bilirubin, diBi—direct bilirubin, CRP—C-reactive protein, P+—postoperative days, NA—not performed.
3. Discussion
Hyperhemolysis or HHS, defined as transfusion-associated hemolysis with a rapid decline in Hgb levels to below the pretransfusion level and an elevation in hemolysis biomarkers (indirect bilirubin and LDH), is a potentially life-threatening reaction that requires prompt intervention. The extensive intravascular hemolysis explains the severity of the condition. The sudden release of large amounts of free heme (hemopexin, hemoxygenase) generates oxidative stress, which induces symptoms of vaso-occlusive crisis and increases vascular endothelial cell damage. Simultaneously, the physiological protective mechanisms against free heme become fulminant and the complement system, activated through the classical or alternative pathways, generates extreme hemolysis via the membrane attack complex. As a result, the red blood cells are destroyed as they are not only sensitive to oxidative stress but also to the non-specific binding of activated complement factors [11,18,19,20,21].
In our case, alloimmunization due to RBC transfusion necessitated by a hemorrhagic condition led to the development of HHS. The acute hemolysis following the administration of blood products supports the diagnosis. Alloimmunization is a huge problem in patients who need long-term transfusions, especially in those SCD patients who are treated in foreign countries. The red cell antigen profile of a Nigerian SCD patient is significantly different from the donor population’s antigen profile in East-Central Europe [5]. This difference leads to new alloantibody and autoantibody formation. Our patient developed fever, jaundice, elevation of inflammatory markers, and hemoglobinuria on postoperative day 3+, developing acute hyperhemolytic crisis. Blood serology showed the same alloantibodies seen previously before the surgery, but the DAT test became positive, highlighting the autoimmune mechanism. During HHS, massive reticulocytosis was observed, which normalized as the crisis resolved. Unfortunately, the baseline reticulocyte count is unavailable, although its diagnostic value is debated [14], but on presentation, there were no signs of hemolysis, so investigation of the reticulocyte level was not justified.
HHS is suspected when both transfused and autologous red blood cells are destroyed. In HHS, the ratios of HbS and HbA both decrease concurrently after a transfusion, indicating the lysis of autologous and transfused red blood cells [20]. In this case, the HbS ratio at the peak of hemolysis (postoperative day +11) was 51.9% and the HbA ratio was 43.1%. The change in HbS ratio is unknown due to the absence of baseline data, but in homozygous SCD, the HbS ratio is usually above 90%.
The exact mechanism of HHS is not established but there are different theories to explain severe hemolysis. One theory is the “bystander complement activation”; after transfusion, both the donor and recipient RBCs are destroyed by a non-specific antibody activating the complement system [17,19], which leads to a lower post-transfusion Hgb level. This mechanism could be the background of our case. The second mechanism is “suppressed erythropoiesis”, which explains acute reticulocytopenia [17,19], just like the immune-mediated hemolysis of the RBC precursors. Moreover, sickled RBC highly expresses phosphatidylserine, drawing the activated macrophages’ attention to these cells to be removed from circulation [17].
While HHS requires prompt intervention, evidence to guide optimal therapy is limited. Currently, no guidelines exist for using steroids or IVIG as a first-line treatment, and no evidence-based studies support the use of one over the other [11,14,22]. Based on the evidence presented by previous case-control studies and according to institutional, consensus-based guidelines, we simultaneously administered IVIG and steroid therapy. EPO treatment was also applied after a cost/benefit assessment. In addition to the above, the literature data demonstrate that rituximab, a monoclonal antibody against CD20, is a good choice to treat the underlying autoimmune condition and could also rapidly re-establish the reticulocyte count [17]. Menakuru et al. administered tocilizumab, a monoclonal antibody against IL-6 (interleukin-6) and a targeted therapy used in autoimmune conditions, to treat patients with severe acute HHS, supporting the macrophage activation mechanism [22]. Eculizumab, which is a C5-convertase inhibitor, could interfere with the “bystander complement activation” by inhibiting the complement cascade [23]. The newer disease-modifying treatment approved for the treatment of SCD is voxelotor, which inhibits HbS polymerization and prevents sickle cell formation, while crizanlizumab is an antibody against P-selectin, blocking the interaction between sickled RBCs and P-selectin, thus preventing vaso-occlusive crisis. These may also be effective treatment options [24,25]. Conventional treatment, such as hydroxyurea, increases the formation of HbF in SCD patients and can prevent complications and acute crises [25].
There is no consensus in the scientific community regarding liberal versus restrictive transfusion strategies for HHS [14,26,27]. Several authors continue to recommend transfusion in HHS. Jacobs et al. demonstrated no significant difference in the median hemoglobin nadir between those who received an RBC transfusion during HHS compared to those who did not. Patients receiving a supportive RBC transfusion experienced a longer hospital stay [26]. As the risk of transfusion in HHS is high and unpredictable, we did not give any further transfusions to our patient after confirmation of HHS. A rescue transfusion in life-threatening anemia would have been indicated only in the case of hemodynamic instability or in the presence of hypoxia. No such deterioration occurred with our transfusion strategy. There were no adverse events until the beneficial effect of the immunosuppressive therapy developed.
4. Conclusions
In SCD, the transfusion of blood products increases the risk of DHTR and HHS. The risk increases with each transfusion given, especially in those cases when the recipient’s antigen profile is significantly different from the donors’ antigen profile. In such cases, prophylactic matching for the known antigens could reduce the risk of DHTR and HHS. In definitive HHS, further transfusions may lead to increased hemolysis, thereby worsening anemia. In such cases, is important to avoid using transfusions and start high-dose corticosteroid and IVIG treatment; in severe, life-threatening cases, plasmapheresis also has to be considered [28]. Our case illustrates that successful immunosuppressive treatment of HHS in SCD is possible with a restrictive transfusion approach. In HHS, the transfusion of blood products is only recommended in the presence of hemodynamic instability or tissue hypoxia.
Author Contributions
Case management: B.B., F.B. and O.O.A.-A. Manuscript review and editing for important intellectual content: O.O.A.-A., B.B., Á.I., L.I.P., F.M., F.B. and G.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Ethical review and approval were waived for this study due to the nature of data collected. This case report is published for educational purposes, with the consent of the patient. All data were collected as part of routine care.
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper.
Data Availability Statement
Data can be made available upon request to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of sickle cell disease. Annu. Rev. Pathol. 2019, 14, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Sparkenbaugh, E.; Pawlinski, R. Interplay between coagulation and vascular inflammation in sickle cell disease. Br. J. Hematol. 2013, 162, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Elendu, C.; Amaechi, D.C.; Alakwe-Ojimba, C.E.; Elendu, T.C.; Elendu, R.C.; Ayabazu, C.P.; Aina, T.O.; Aborisade, O.; Adenikinju, J.S. Understanding Sickle cell disease: Causes, symptoms, and treatment options. Medicine 2023, 102, e35237. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Brousse, V.A.M.; Brewin, J.N. Determinants of severity in sickle cell disease. Blood Rev. 2022, 56, 100983. [Google Scholar] [CrossRef]
- Rees, D.C.; Robinson, S.; Howard, J. How I manage red cell transfusions in patients with sickle cell disease. Br. J. Haematol. 2018, 180, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Pirenne, F.; Yazdanbakhsh, K. How I safely transfuse patients with sickle-cell disease and manage delayed hemolytic transfusion reactions. Blood 2018, 131, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.; Deng, X.; Hopper, C.; Style, L. A pilot randomized trial of red blood cell transfusion for acute treatment of vaso-occlusive pain episodes in sickle cell disease. Br. J. Haematol. 2015, 171, 288–290. [Google Scholar] [CrossRef]
- Adams Rj McKie, V.C.; Hsu, L.; Files, B.; Vichinsky, E.; Pegelow, C.; Abboud, M.; Gallagher, D.; Kutlar, A.; Nichols, F.T.; Bonds, D.R.; et al. Prevention of first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N. Engl. J. Med. 1998, 339, 5–11. [Google Scholar] [CrossRef]
- Ware, R.E.; Davis, B.R.; Schultz, W.H.; Brown, R.C.; Aygun, B.; Sarnaik, S.; Odame, I.; Fuh, B.; George, A.; Owen, W.; et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anemia (TWiTCH): A multicentric, open-label, phase 3, non-inferiority trial. Lancet 2016, 387, 661–670. [Google Scholar] [CrossRef]
- Narbey, D.; Habibi, A.; Chadebech, P.; Mekontso-Dessap, A.; Khellaf, M.; Lelièvre, J.D.; Godeau, B.; Michel, M.; Galactéros, F.; Djoudi, R.; et al. Incidence and predictive score for delayed hemolytic transfusion reaction in adult patients with sickle cell disease. Am. J. Hematol. 2017, 92, 1340–1348. [Google Scholar] [CrossRef]
- Pirenne, F.; Pondarré, C. Alloimmunization and hyperhemolysis in sickle cell disease. Hematology 2023, 2023, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Fasano, R.M.; Miller, M.J.; Chonat, S.; Stowell, S.R. Clinical presentation of delayed hemolytic transfusion reactions and hyperhemolysis in sickle cell disease. Transfus. Clin. Biol. 2019, 26, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Banks, M.; Shikle, J. Hyperhemolysis syndrome in patients with sickle cell disease. Arch. Pathol. Lab. Med. 2017, 142, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.W.; Stephens, L.D.; Allen, E.S.; Binns, T.C.; Booth, G.S.; Hendrickson, J.E.; Karafin, M.S.; Tormey, C.A.; Woo, J.S.; Adkins, B.D. Epidemiological and clinical features, therapeutic strategies and outcomes in patients with hyperhaemolysis: A systematic review. Br. J. Haematol. 2023, 201, 1025–1032. [Google Scholar] [CrossRef]
- Shankar, K.; Shah, D.; Huffman, D.L.; Peterson, C.; Bhagavatula, R. Hyperhemolysis Syndrome in a Patient With Sickle Cell Disease and Acute Chest Syndrome. Cureus 2021, 13, e13017. [Google Scholar] [CrossRef]
- Win, N.; New, H.; Lee, E.; de la Fuente, J. Hyperhemolysis syndrome in sickle cell disease: A case report (recurrent episode) and literature review. Transfusion 2008, 48, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, K.; Abbas, A.; Kazmi, R.; Shaaban, H.; Miller, R. Hyperhemolytic crisis following transfusion in sickle cell disease with acute hepatic crisis: A case report. Cureus 2022, 14, e27844. [Google Scholar] [CrossRef] [PubMed]
- Kalter, J.A.; Gupta, R.; Greenberg, M.R.; Miller, A.J.; Allen, J. Hyperhemolysis Syndrome in a Patient with Sickle Cell Disease: A Case Report. Clin. Pract. Cases Emerg. Med. 2021, 5, 101–104. [Google Scholar] [CrossRef]
- Win, N. Hyperhemolysis syndrome in sickle cell disease. Expert Rev. Hematol. 2009, 2, 111–115. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Bartolucci, P.; Pirenne, F. The role of Complement in Post-Transfusion Hemolysis and Hyperhemolysis Reaction. Transfus. Med. Rev. 2019, 33, 225–230. [Google Scholar] [CrossRef]
- Madu, A.J.; Ugwu, A.O.; Efobi, C. Hyperhemolytic Syndrome in Sickle Cell Disease: Clearing the Cobwebs. Med. Princ. Pract. 2021, 30, 236–243. [Google Scholar] [CrossRef]
- Menakuru, S.R.; Priscu, A.; Dhillon, V.; Salih, A. Acute Hyperhemolysis Syndrome in a Patient with Known Sickle Cell Anemia Refractory to Steroids and IVIG Treated with Tocilizumab and Erythropoietin: A Case Report and Review of Literature. Hematol. Rep. 2022, 14, 235–239. [Google Scholar] [CrossRef]
- Dumas, G.; Habibi, A.; Onimus, T.; Merle, J.C.; Razazi, K.; Mekontso Dessap, A.; Galactéros, F.; Michel, M.; Frémeaux Bacchi, V.; Noizat Pirenne, F.; et al. Eculizumab salvage therapy for delayed hemolysis transfusion reaction in sickle cell disease patients. Blood 2016, 127, 1062–1064. [Google Scholar] [CrossRef]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Kuriri, F.A. Hope on the Horizon: New and Future Therapies for sickle cell disease. J. Clin. Med. 2023, 12, 5692. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Alsawas, M.; Fasano, R.M.; Field, J.J.; Hendrickson, J.E.; Howard, J.; Kameka, M.; Kwiatkowski, J.L.; Pirenne, F.; Shi, P.A.; et al. American Society of Hematology 2020 guidelines for sickle cell disease: Transfusion support. Blood Adv. 2020, 4, 327–355. [Google Scholar] [CrossRef]
- Ballas, S.K.; Kuypers, F.A.; Gordeuk, V.R.; Hankins, J.S.; Thompson, A.A.; Vichinsky, E. Time to rethink haemoglobin threshold guidelines in sickle cell disease. Br. J. Haematol. 2021, 195, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Shaulov, A.; Rund, D.; Filon, D.; Nachmias, B.; Khalili, A.; Manny, N.; Zelig, O. Successful treatment with plasma exchange in life-threatening hyperhemolytic syndrome unrelated to sickle cell disease. Transfusion 2023, 63, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).