
A Case of Primary Ciliary Dyskinesia Caused by a Mutation in OFD1, Which Was Diagnosed Owing to Clostridium difficile Infection


{kind=link}
{kind=link}
Abstract
1. Introduction
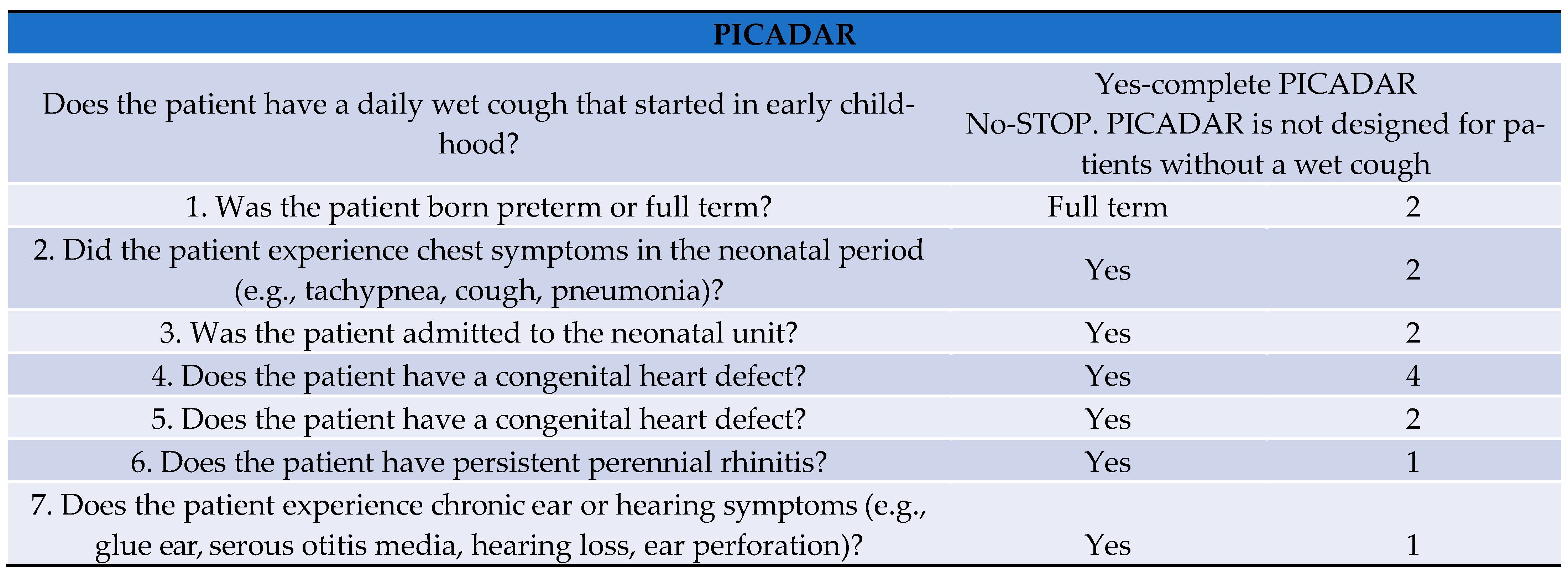
2. Case Report
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sommer, J.U.; Schäfer, K.; Omran, H.; Olbrich, H.; Wallmeier, J.; Blum, A.; Hormann, K.; Stuck, B.A. ENT manifestations in patients with primary ciliary dyskinesia: Prevalence and significance of otorhinolaryngologic co-morbidities. Eur. Arch. Oto-Rhino-Laryngol. 2010, 268, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Bukowy-Bieryllo, Z.; Rabiasz, A.; Dabrowski, M.; Pogorzelski, A.; Wojda, A.; Dmenska, H.; Grzela, K.; Sroczynski, J.; Witt, M.; Zietkiewicz, E. Truncating mutations in exons 20 and 21 of OFD1 can cause primary ciliary dyskinesia without associated syndromic symptoms. J. Med. Genet. 2019, 56, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Kitano, M.; Ishinaga, H.; Kobayashi, M.; Ogawa, S.; Nakatani, K.; Masuda, S.; Nagao, M.; Fujisawa, T. Recent advances in primary ciliary dyskinesia. Auris Nasus Larynx 2016, 43, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Behan, L.; Dimitrov, B.D.; Kuehni, C.E.; Hogg, C.; Carroll, M.; Evans, H.J.; Goutaki, M.; Harris, A.; Packham, S.; Walker, W.T.; et al. PICADAR: A diagnostic predictive tool for primary ciliary dyskinesia. Eur. Respir. J. 2016, 47, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Zariwala, M.A.; Knowles, M.R.; Leigh, M.W. Primary Ciliary Dyskinesia. In GeneReviews; Adam, P.A., Arrdinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 24 January 1993. [Google Scholar]
- Werner, C.; Lablans, M.; Ataian, M.; Raidt, J.; Wallmeier, J.; Große-Onnebrink, J.; Kuehni, C.E.; Haarman, E.G.; Leigh, M.W.; Quittner, A.L.; et al. An international registry for primary ciliary dyskinesia. Eur. Respir. J. 2015, 47, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.J.; Zariwala, M.A.; Ferkol, T.W.; Davis, S.D.; Sagel, S.D.; Dell, S.D.; Rosenfeld, M.; Olivier, K.N.; Milla, C.E.; Daniel, S.J.; et al. Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatr. Pulmonol. 2016, 51, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Hannah, W.B.; Debrosse, S.; Kinghorn, B.; Strausbaugh, S.; Aitken, M.L.; Rosenfeld, M.; Wolf, W.E.; Knowles, M.R.; Zariwala, M.A. The expanding phenotype of OFD1 -related disorders: Hemizygous loss-of-function variants in three patients with primary ciliary dyskinesia. Mol. Genet. Genom. Med. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Lamont, J.T. Assymptomatic colonization by Clostridium difficile in infants: Implications for disease in later life. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Borali, E.; De Giacomo, C. Clostridium Difficile Infection in Children: A Review. J. Pediatr. Gastroenterol. Nutr. 2016, 63, e130–e140. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasegawa, R.; Suzuki, S.; Nishimata, S.; Kashiwagi, Y.; Inagaki, N.; Kawashima, H. A Case of Primary Ciliary Dyskinesia Caused by a Mutation in OFD1, Which Was Diagnosed Owing to Clostridium difficile Infection. Pediatr. Rep. 2021, 13, 241-244. https://doi.org/10.3390/pediatric13020033
Hasegawa R, Suzuki S, Nishimata S, Kashiwagi Y, Inagaki N, Kawashima H. A Case of Primary Ciliary Dyskinesia Caused by a Mutation in OFD1, Which Was Diagnosed Owing to Clostridium difficile Infection. Pediatric Reports. 2021; 13(2):241-244. https://doi.org/10.3390/pediatric13020033
Chicago/Turabian StyleHasegawa, Rina, Shinji Suzuki, Shigeo Nishimata, Yasuyo Kashiwagi, Natsuko Inagaki, and Hisashi Kawashima. 2021. "A Case of Primary Ciliary Dyskinesia Caused by a Mutation in OFD1, Which Was Diagnosed Owing to Clostridium difficile Infection" Pediatric Reports 13, no. 2: 241-244. https://doi.org/10.3390/pediatric13020033
APA StyleHasegawa, R., Suzuki, S., Nishimata, S., Kashiwagi, Y., Inagaki, N., & Kawashima, H. (2021). A Case of Primary Ciliary Dyskinesia Caused by a Mutation in OFD1, Which Was Diagnosed Owing to Clostridium difficile Infection. Pediatric Reports, 13(2), 241-244. https://doi.org/10.3390/pediatric13020033