
From Natural History to Contemporary Management of Aortic Diseases: A State-of-the-Art Review of Thoracic Aortic Aneurysm

 , , and
, , and 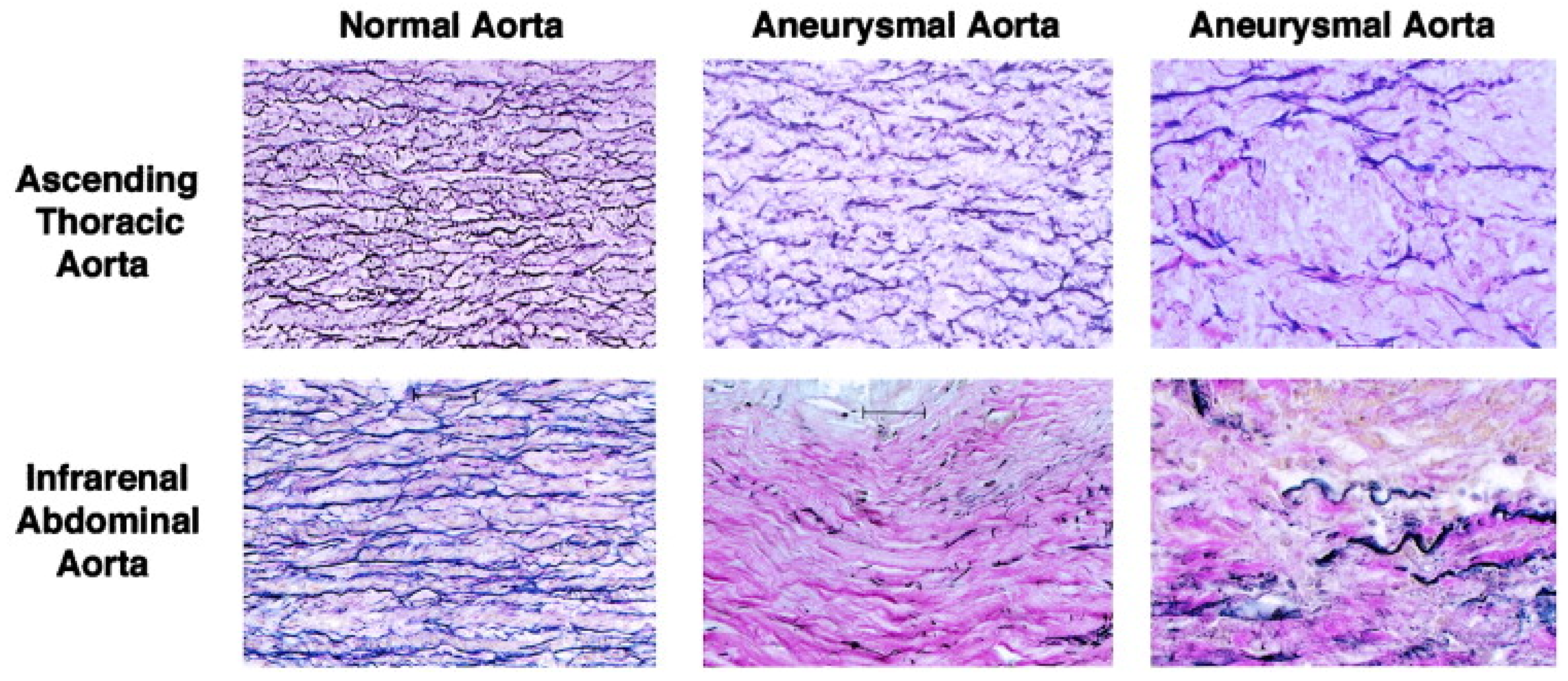{kind=link}
{kind=link}
Abstract
1. Introduction
2. Overview of Thoracic Aortic Aneurysms
2.1. Epidemiology
2.2. Pathophysiology of Thoracic Aortic Aneurysms
2.3. Gender Differences in the Pathophysiology of Thoracic Aortic Aneurysms
2.4. Risk Factors for the Formation of Thoracic Aortic Aneurysms
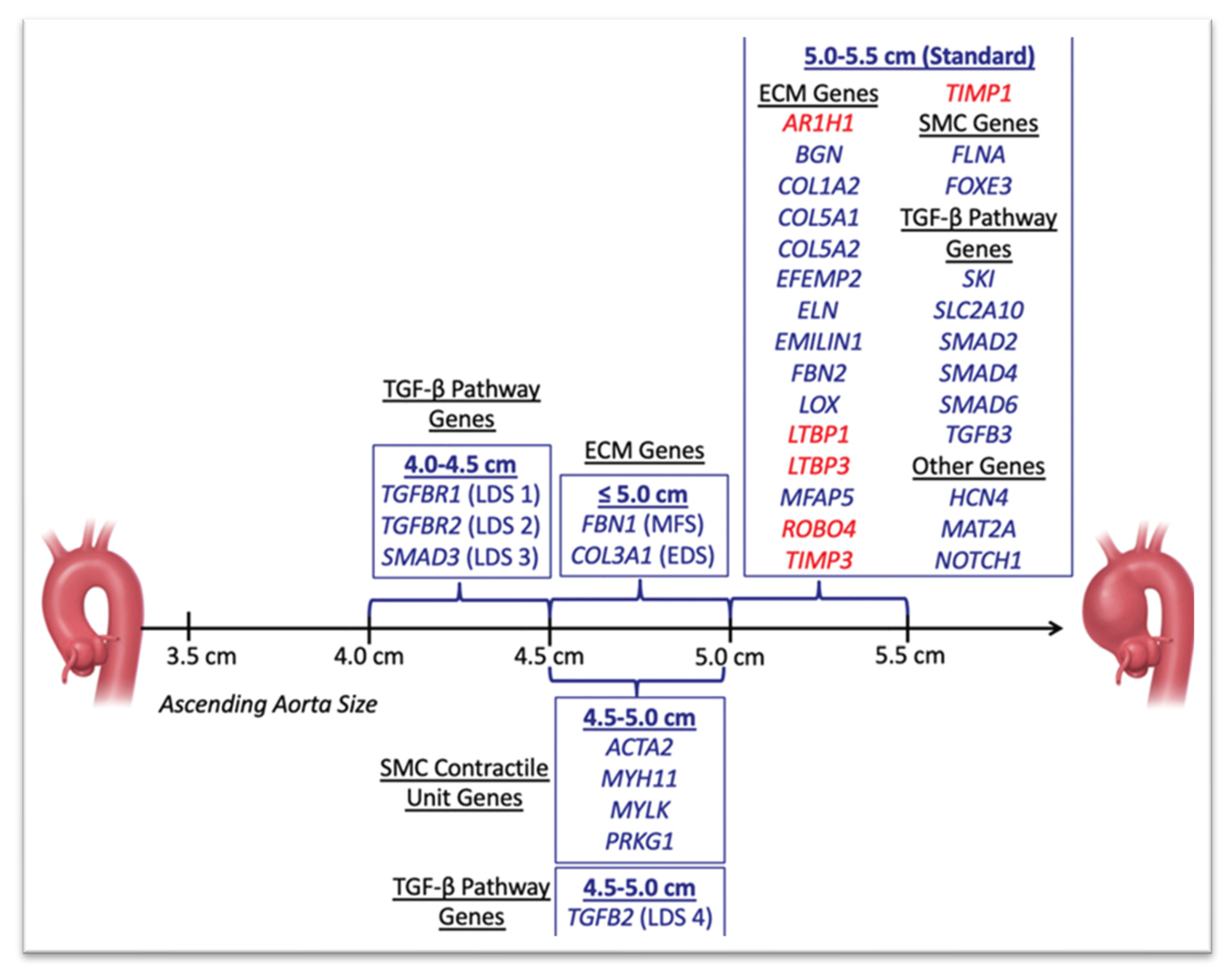
3. Genetics of Thoracic Aortic Aneurysms
3.1. Marfan Syndrome
3.2. Loeys–Dietz Syndrome
3.3. Ehlers–Danlos Syndrome
3.4. Familial Thoracic Aortic Aneurysms
3.5. Bicuspid Aortic Valve
3.6. Sporadic Thoracic Aortic Aneurysms
4. Diagnosis of Thoracic Aortic Aneurysms
4.1. Diagnostic Modalities of Thoracic Aortic Disease
4.2. Role of Mock Loop Systems in Thoracic Aortic Disease
5. Approach to Intervention
6. Prophylactic Surgical Intervention
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAA | Abdominal aortic aneurysm |
| AAS | Acute aortic syndrome |
| ACC | American College of Cardiology |
| AHA | American Heart Association |
| ARB | Angiotensin receptor antagonist |
| AVR | Aortic valve replacement |
| BAV | Bicuspid aortic valve |
| CVG | Composite valve graft |
| CT | Computed tomography |
| CTA | Computed tomography angiography |
| ECG | Electrocardiogram |
| ECM | Extracellular matrix |
| EDS | Ehlers–Danlos syndrome |
| IMH | Intramural hematoma |
| IRAD | International Registry of Aortic Dissection |
| LDS | Loeys–Dietz syndrome |
| MCL | Mock Loop Systems |
| MLCK | Myosin light chain kinase |
| MMP | Matrix metalloproteinase |
| MRA | Magnetic resonance angiography |
| MRI | Magnetic resonance imaging |
| PAU | penetrating aortic ulcer |
| PET | Positron emission tomography |
| RLC | Regulatory light chain kinase |
| SMAD | Subsequent mothers against decapentaplegic homolog |
| TAAA | Thoraco-abdominal aortic aneurysm |
| TAA | Thoracic aortic aneurysm |
| TEVAR | Thoracic endovascular aortic repair |
| TGF-β | Transforming growth factor beta |
| TGFβR | Transforming growth factor beta receptor |
| TIMP | Tissue inhibitor of metalloproteinase |
| TTE | Transthoracic echocardiography |
References
- Yiu, R.S.; Cheng, S.W.K. Natural history and risk factors for rupture of thoracic aortic arch aneurysms. J. Vasc. Surg. 2016, 63, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.; Raymond, C.E. Therapeutic Goals in Patients with Acute Aortic Dissection. J. Am. Coll. Cardiol. 2015, 65, 1599–1600. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goldfinger, J.Z.; Halperin, J.L.; Marin, M.L.; Stewart, A.S.; Eagle, K.A.; Fuster, V. Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol. 2014, 64, 1725–1739. [Google Scholar] [CrossRef] [PubMed]
- Olsson, C.; Thelin, S.; Ståhle, E.; Ekbom, A.; Granath, F. Thoracic Aortic Aneurysm and Dissection: Increasing Prevalence and Improved Outcomes Reported in a Nationwide Population-Based Study of More than 14 000 Cases from 1987 to 2002. Circulation 2006, 114, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Cecchi, A.C.; Prakash, S.K.; Milewicz, D.M. Risk Factors for Thoracic Aortic Dissection. Genes 2022, 13, 1814. [Google Scholar] [CrossRef] [PubMed]
- Frederick, J.R.; Woo, Y.J. Thoracoabdominal aortic aneurysm. Ann. Cardiothorac. Surg. 2012, 1, 277–285. [Google Scholar] [PubMed]
- Clouse, W.D.; Hallett, J.W.J.; Schaff, H.V.; Gayari, M.M.; Ilstrup, D.M.; Melton, J.I. Improved prognosis of thoracic aortic aneurysms: A population-based study. JAMA 1998, 280, 1926–1929. [Google Scholar] [CrossRef]
- Kuzmik, G.A.; Sang, A.X.; Elefteriades, J.A. Natural history of thoracic aortic aneurysms. J. Vasc. Surg. 2012, 56, 565–571. [Google Scholar] [CrossRef]
- Gouveia e Melo, R.; Silva Duarte, G.; Lopes, A.; Alves, M.; Caldeira, D.; Fernandes e Fernandes, R.; Mendes Pedro, L. Synchronous and Metachronous Thoracic Aortic Aneurysms in Patients with Abdominal Aortic Aneurysms: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2020, 9, e017468. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Guo, D.-C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic Basis of Thoracic Aortic Aneurysms and Dissections: Focus on Smooth Muscle Cell Contractile Dysfunction. Annu. Rev. Genom. Hum. Genet. 2008, 9, 283–302. [Google Scholar] [CrossRef]
- Borges, L.F.; Gomez, D.; Quintana, M.; Touat, Z.; Jondeau, G.; Leclercq, A.; Meilhac, O.; Jandrot-Perrus, M.; Gutierrez, P.S.; Freymuller, E.; et al. Fibrinolytic activity is associated with presence of cystic medial degeneration in aneurysms of the ascending aorta. Histopathology 2010, 57, 917–932. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-J.; Lin, C.-Y.; Stitziel, N.O. Genetics of the extracellular matrix in aortic aneurysmal diseases. Matrix Biol. 2018, 71–72, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Du, W.; Ren, L.; Hamblin, M.H.; Becker, R.C.; Chen, Y.E.; Fan, Y. Vascular Smooth Muscle Cells in Aortic Aneurysm: From Genetics to Mechanisms. J. Am. Heart Assoc. 2021, 10, e023601. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Guo, D.-C.; Estrera, A.L.; Safi, H.J.; Huynh, T.T.; Yin, Z.; Cao, S.-N.; Lin, J.; Kurian, T.; Buja, L.M.; et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J. Thorac. Cardiovasc. Surg. 2006, 131, 671–678.e2. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Zhang, W.; Zhang, H.; Li, T.; Wang, Y.; Qin, Y.; Gu, H.; Du, J. Mechanical stretch-induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J. Pathol. 2015, 236, 373–383. [Google Scholar] [CrossRef]
- Gillis, E.; Van Laer, L.; Loeys, B.L. Genetics of Thoracic Aortic Aneurysm: At the Crossroad of Transforming Growth Factor-β Signaling and Vascular Smooth Muscle Cell Contractility. Circ. Res. 2013, 113, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, K.B.; van Merrienboer, T.A.R.; Ket, J.C.F.; Bogunovic, N.; van der Velden, J.; Yeung, K.K. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur. J. Clin. Investig. 2022, 52, e13697. [Google Scholar] [CrossRef]
- Boczar, K.; Boodwhani, M.; Beauchesne, L.; Chan, K.; Dennie, C.; Coutinho, T. Sex Differences in Thoracic Aortic Aneurysm Growth: Role of Aortic Stiffness. Hypertension 2019, 73, 190–196. [Google Scholar] [CrossRef]
- Huckaby, L.V.; Sultan, I.; Trimarchi, S.; Leshnower, B.; Chen, E.P.; Brinster, D.R.; Myrmel, T.; Estrera, A.L.; Montgomery, D.G.; Korach, A.; et al. Sex-Based Aortic Dissection Outcomes From the International Registry of Acute Aortic Dissection. Ann. Thorac. Surg. 2022, 113, 498–505. [Google Scholar] [CrossRef]
- Davies, R.R.; Goldstein, L.J.; Coady, A.M.; Tittle, S.L.; Rizzo, A.J.; Kopf, G.S.; Elefteriades, J.A. Yearly rupture or dissection rates for thoracic aortic aneurysms: Simple prediction based on size. Ann. Thorac. Surg. 2002, 73, 17–28. [Google Scholar] [CrossRef]
- Nienaber, C.A.; Fattori, R.; Mehta, R.H.; Richartz, B.M.; Evangelista, A.; Petzsch, M.; Cooper, J.V.; Januzzi, J.L.; Ince, H.; Sechtem, U.; et al. Gender-Related Differences in Acute Aortic Dissection. Circulation 2004, 109, 3014–3021. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.; Boodhwani, M.; Chan, K.L.; Beauchesne, L.; Dick, A.; Coutinho, T. Thoracic Aortic Aneurysm Growth: Role of Sex and Aneurysm Etiology. J. Am. Heart Assoc. 2017, 6, e003792. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Khatri, J.J. Matrix Metalloproteinases in Vascular Remodeling and Atherogenesis: The Good, the Bad, and the Ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [CrossRef]
- Sokolis, D.P.; Iliopoulos, D.C. Impaired mechanics and matrix metalloproteinases/inhibitors expression in female ascending thoracic aortic aneurysms. J. Mech. Behav. Biomed. Mater. 2014, 34, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Chew, D.K.W.; Conte, M.S.; Khalil, R.A. Matrix metalloproteinase-specific inhibition of Ca2+ entry mechanisms of vascular contraction. J. Vasc. Surg. 2004, 40, 1001–1010. [Google Scholar] [CrossRef]
- Waddell, T.K.; Dart, A.M.; Gatzka, C.D.; Cameron, J.D.; Kingwell, B.A. Women exhibit a greater age-related increase in proximal aortic stiffness than men. J. Hypertens. 2001, 19, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Koullias, G.J.; Ravichandran, P.; Korkolis, D.P.; Rimm, D.L.; Elefteriades, J.A. Increased Tissue Microarray Matrix Metalloproteinase Expression Favors Proteolysis in Thoracic Aortic Aneurysms and Dissections. Ann. Thorac. Surg. 2004, 78, 2106–2110. [Google Scholar] [CrossRef]
- Landenhed, M.; Engström, G.; Gottsäter, A.; Caulfield, M.P.; Hedblad, B.; Newton-Cheh, C.; Melander, O.; Smith, J.G. Risk Profiles for Aortic Dissection and Ruptured or Surgically Treated Aneurysms: A Prospective Cohort Study. J. Am. Heart Assoc. 2015, 4, e001513. [Google Scholar] [CrossRef]
- Saliba, E.; Sia, Y.; Dore, A.; El Hamamsy, I. The ascending aortic aneurysm: When to intervene? IJC Heart Vasc. 2015, 6, 91–100. [Google Scholar] [CrossRef]
- Groenink, M. The influence of aging and aortic stiffness on permanent dilation and breaking stress of the thoracic descending aorta. Cardiovasc. Res. 1999, 43, 471–480. [Google Scholar] [CrossRef]
- Mathur, A.; Mohan, V.; Ameta, D.; Gaurav, B.; Haranahalli, P. Aortic aneurysm. J. Transl. Intern. Med. 2016, 4, 35–41. [Google Scholar] [CrossRef]
- Caglayan, A.O.; Dundar, M. Inherited diseases and syndromes leading to aortic aneurysms and dissections. Eur. J. Cardiothorac. Surg. 2009, 35, 931–940. [Google Scholar] [CrossRef]
- Meester, J.A.N.; Verstraeten, A.; Schepers, D.; Alaerts, M.; Van Laer, L.; Loeys, B.L. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann. Cardiothorac. Surg. 2017, 6, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Bradley, T.J.; Bowdin, S.C.; Morel CF, J.; Pyeritz, R.E. The Expanding Clinical Spectrum of Extracardiovascular and Cardiovascular Manifestations of Heritable Thoracic Aortic Aneurysm and Dissection. Can. J. Cardiol. 2016, 32, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.W.Y.; Yang, H.H.C.; Radomski, M.W.; Van Breemen, C. Long-Term Doxycycline Is More Effective Than Atenolol to Prevent Thoracic Aortic Aneurysm in Marfan Syndrome Through the Inhibition of Matrix Metalloproteinase-2 and -9. Circ. Res. 2008, 102, 73–85. [Google Scholar] [CrossRef]
- Robinson, P.N.; Arteaga-Solis, E.; Baldock, C.; Collod-Beroud, G.; Booms, P.; De Paepe, A.; Dietz, H.C.; Guo, G.; Handford, A.P.; Judge, D.P.; et al. The molecular genetics of Marfan syndrome and related disorders. J. Med. Genet. 2006, 43, 769–787. [Google Scholar] [CrossRef]
- Maeda, J.; Kosaki, K.; Shiono, J.; Kouno, K.; Aeba, R.; Yamagishi, H. Variable severity of cardiovascular phenotypes in patients with an early-onset form of Marfan syndrome harboring FBN1 mutations in exons 24–32. Heart Vessels 2016, 31, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Carande, E.J.; Bilton, S.J.; Adwani, S. A Case of Neonatal Marfan Syndrome: A Management Conundrum and the Role of a Multidisciplinary Team. Case Rep. Pediatr. 2017, 2017, 8952428. [Google Scholar] [CrossRef] [PubMed]
- Porciani, M.C.; Attanasio, M.; Lepri, V.; Lapini, I.; Demarchi, G.; Padeletti, L.; Pepe, G.; Abbate, R.; Gensini, G.F. Prevalence of cardiovascular manifestations in Marfan syndrome. Ital. Heart J. Suppl. Off. J. Ital. Fed. Cardiol. 2004, 5, 647–652. [Google Scholar]
- Krause, K.J. Marfan syndrome: Literature review of mortality studies. J. Insur. Med. N. Y. 2000, 32, 79–88. [Google Scholar]
- Chan, Y.; Ting, C.; Ho, P.; Poon, J.; Cheung, G.; Cheng, S. Ten-Year Epidemiological Review of In-Hospital Patients with Marfan Syndrome. Ann. Vasc. Surg. 2008, 22, 608–612. [Google Scholar] [CrossRef]
- Shores, J.; Berger, K.R.; Murphy, E.A.; Pyeritz, R.E. Progression of Aortic Dilatation and the Benefit of Long-Term β-Adrenergic Blockade in Marfan’s Syndrome. N. Engl. J. Med. 1994, 330, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Rossi-Foulkes, R.; Roman, M.J.; Rosen, E.S.; Kramer-Fox, R.; Ehlers, K.H.; O’loughlin, J.E.; Davis, J.G.; Devereux, R.B. Phenotypic features and impact of Beta blocker or calcium antagonist therapy on Aortic lumen size in the Marfan syndrome. Am. J. Cardiol. 1999, 83, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, A.; Spata, E.; Emberson, J.; Davies, K.; Halls, H.; Holland, L.; Wilson, K.; Reith, C.; Child, A.H.; Clayton, T.; et al. Angiotensin receptor blockers and β blockers in Marfan syndrome: An individual patient data meta-analysis of randomised trials. Lancet 2022, 400, 822–831. [Google Scholar] [CrossRef]
- Groenink, M.; Hartog, A.W.D.; Franken, R.; Radonic, T.; de Waard, V.; Timmermans, J.; Scholte, A.J.; Berg, M.P.v.D.; Spijkerboer, A.M.; Marquering, H.A.; et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: A randomized controlled trial. Eur. Heart J. 2013, 34, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Forteza, A.; Evangelista, A.; Sánchez, V.; Teixidó-Turà, G.; Sanz, P.; Gutiérrez, L.; Gracia, T.; Centeno, J.; Rodríguez-Palomares, J.; Rufilanchas, J.J.; et al. Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: A randomized clinical trial. Eur. Heart J. 2016, 37, 978–985. [Google Scholar] [CrossRef]
- Chiu, H.H.; Wu, M.H.; Wang, J.K.; Lu, C.W.; Chiu, S.N.; Chen, C.A.; Lin, M.T.; Hu, F.C. Losartan Added to β-Blockade Therapy for Aortic Root Dilation in Marfan Syndrome: A Randomized, Open-Label Pilot Study. Mayo Clin. Proc. 2013, 88, 271–276. [Google Scholar] [CrossRef]
- Isselbacher, E.M.; Preventza, O.; Iii, J.H.B.; Augoustides, J.G.; Beck, A.W.; Bolen, M.A.; Braverman, A.C.; Bray, B.E.; Brown-Zimmerman, M.M.; Chen, E.P.; et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease. J. Am. Coll. Cardiol. 2022, 80, e223–e393. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Dietz, H.C.; Miller, D.C. Treatment of Aortic Disease in Patients with Marfan Syndrome. Circulation 2005, 111, e150–e157. [Google Scholar] [CrossRef]
- Zeigler, S.M.; Sloan, B.; Jones, J.A. Pathophysiology and Pathogenesis of Marfan Syndrome. In Progress in Heritable Soft Connective Tissue Diseases; Halper, J., Ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2021; Volume 1348, pp. 185–206. [Google Scholar]
- Vinholo, T.F.; Brownstein, A.J.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: 2019 Update and Clinical Implications. AORTA 2019, 7, 99–107. [Google Scholar]
- MacCarrick, G.; Black, J.H.; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C. Loeys–Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef]
- Nickol, J.L.; Malik, S.A.; Yetman, A.T. Case report of Loeys-Dietz syndrome presenting with coronary artery aneurysm. Eur. Heart J. Case Rep. 2022, 6, ytac383. [Google Scholar] [CrossRef] [PubMed]
- Fattori, R.; Sangiorgio, P.; Mariucci, E.; Ritelli, M.; Wischmeijer, A.; Greco, C.; Colombi, M. Spontaneous coronary artery dissection in a young woman with Loeys-Dietz syndrome. Am. J. Med. Genet. A 2012, 158A, 1216–1218. [Google Scholar] [CrossRef]
- Cury, M.; Zeidan, F.; Lobato, A.C. Aortic Disease in the Young: Genetic Aneurysm Syndromes, Connective Tissue Disorders, and Familial Aortic Aneurysms and Dissections. Int. J. Vasc. Med. 2013, 2013, 267215. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm Syndromes Caused by Mutations in the TGF-β Receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Gouda, P.; Kay, R.; Habib, M.; Aziz, A.; Aziza, E.; Welsh, R. Clinical features and complications of Loeys-Dietz syndrome: A systematic review. Int. J. Cardiol. 2022, 362, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Erbel, R.; Germany, C.; Aboyans, V.; France, C.; France, C.B. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2873–2926. [Google Scholar]
- Writing Group Members; Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E., Jr.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients with Thoracic Aortic Disease: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010, 121, e266–e3369. [Google Scholar]
- Appoo, J.J.; Bozinovski, J.; Chu, M.W.; El-Hamamsy, I.; Forbes, T.L.; Moon, M.; Ouzounian, M.; Peterson, M.D.; Tittley, J.; Boodhwani, M. Canadian Cardiovascular Society/Canadian Society of Cardiac Surgeons/Canadian Society for Vascular Surgery Joint Position Statement on Open and Endovascular Surgery for Thoracic Aortic Disease. Can. J. Cardiol. 2016, 32, 703–713. [Google Scholar] [CrossRef][Green Version]
- Boodhwani, M.; Andelfinger, G.; Leipsic, J.; Lindsay, T.; McMurtry, M.S.; Therrien, J.; Siu, S.C. Canadian Cardiovascular Society Position Statement on the Management of Thoracic Aortic Disease. Can. J. Cardiol. 2014, 30, 577–589. [Google Scholar] [CrossRef]
- Eagleton, M.J. Arterial complications of vascular Ehlers-Danlos syndrome. J. Vasc. Surg. 2016, 64, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Kuivaniemi, H.; Tromp, G. Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases. Gene 2019, 707, 151–171. [Google Scholar] [CrossRef]
- Germain, D.P. Ehlers-Danlos syndrome type IV. Orphanet J. Rare Dis. 2007, 2, 32. [Google Scholar] [CrossRef]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and Genetic Features of Ehlers–Danlos Syndrome Type IV, the Vascular Type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef]
- Ong, K.-T.; Perdu, J.; De Backer, J.; Bozec, E.; Collignon, P.; Emmerich, J.; Fauret, A.-L.; Fiessinger, J.-N.; Germain, D.P.; Georgesco, G.; et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: A prospective randomised, open, blinded-endpoints trial. Lancet 2010, 376, 1476–1484. [Google Scholar] [CrossRef]
- Bowen, J.M.; Hernandez, M.; Johnson, D.S.; Green, C.; Kammin, T.; Baker, D.; Keigwin, S.; Makino, S.; Taylor, N.; Watson, O.; et al. Diagnosis and management of vascular Ehlers-Danlos syndrome: Experience of the UK national diagnostic service, Sheffield. Eur. J. Hum. Genet. 2023, 31, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Byers, P.H.; Belmont, J.; Black, J.; De Backer, J.; Frank, M.; Jeunemaitre, X.; Johnson, D.; Pepin, M.; Robert, L.; Sanders, L.; et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 40–47. [Google Scholar] [CrossRef]
- Lee, C.; Tully, A.; Fang, J.C.; Sugeng, L.; Elmariah, S.; Grubb, K.J.; Young, M.N. Building and Optimizing the Interdisciplinary Heart Team. J. Soc. Cardiovasc. Angiogr. Interv. 2023, 2, 101067. [Google Scholar] [CrossRef]
- Batchelor, W.B.; Anwaruddin, S.; Wang, D.D.; Perpetua, E.M.; Krishnaswami, A.; Velagapudi, P.; Wyman, J.F.; Fullerton, D.; Keegan, P.; Phillips, A.; et al. The Multidisciplinary Heart Team in Cardiovascular Medicine. JACC Adv. 2023, 2, 100160. [Google Scholar] [CrossRef]
- Takeda, N.; Komuro, I. Genetic basis of hereditary thoracic aortic aneurysms and dissections. J. Cardiol. 2019, 74, 136–143. [Google Scholar] [CrossRef]
- Wang, L.; Guo, D.-C.; Cao, J.; Gong, L.; Kamm, K.E.; Regalado, E.; Li, L.; Shete, S.; He, W.-Q.; Zhu, M.-S.; et al. Mutations in Myosin Light Chain Kinase Cause Familial Aortic Dissections. Am. J. Hum. Genet. 2010, 87, 701–707. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Peng, Y.; Zhang, W.; Lv, N.; Tang, J.; Chen, C.; Zhang, C.; Gao, S.; Chen, H.; Zhi, G.; et al. Myosin Light Chain Kinase Is Central to Smooth Muscle Contraction and Required for Gastrointestinal Motility in Mice. Gastroenterology 2008, 135, 610–620.e2. [Google Scholar] [CrossRef] [PubMed]
- Vinholo, T.F.; Brownstein, A.J.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: An Update and Clinical Implications. AORTA 2017, 5, 11–20. [Google Scholar]
- Guo, D.-C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in Smooth Muscle Alpha-Actin (ACTA2) Cause Coronary Artery Disease, Stroke, and Moyamoya Disease, Along with Thoracic Aortic Disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493. [Google Scholar] [CrossRef]
- Regalado, E.S.; Guo, D.-C.; Prakash, S.; Bensend, T.A.; Flynn, K.; Estrera, A.; Safi, H.; Liang, D.; Hyland, J.; Child, A.; et al. Aortic Disease Presentation and Outcome Associated with ACTA2 Mutations. Circ. Cardiovasc. Genet. 2015, 8, 457–464. [Google Scholar] [CrossRef]
- Zhu, L.; Vranckx, R.; Van Kien, P.K.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.-M.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef]
- Allaire, E.; Schneider, F.; Saucy, F.; Dai, J.; Cochennec, F.; Michineau, S.; Zidi, M.; Becquemin, J.-P.; Kirsch, M.; Gervais, M. New Insight in Aetiopathogenesis of Aortic Diseases. Eur. J. Vasc. Endovasc. Surg. 2009, 37, 531–537. [Google Scholar] [CrossRef]
- Guo, D.-C.; Regalado, E.; Casteel, D.E.; Santos-Cortez, R.L.; Gong, L.; Kim, J.J.; Dyack, S.; Horne, S.G.; Chang, G.; Jondeau, G.; et al. Recurrent Gain-of-Function Mutation in PRKG1 Causes Thoracic Aortic Aneurysms and Acute Aortic Dissections. Am. J. Hum. Genet. 2013, 93, 398–404. [Google Scholar] [CrossRef]
- Albornoz, G.; Coady, M.A.; Roberts, M.; Davies, R.R.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Familial Thoracic Aortic Aneurysms and Dissections—Incidence, Modes of Inheritance, and Phenotypic Patterns. Ann. Thorac. Surg. 2006, 82, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.-G.; Chou, A.S.; Mok, S.C.; Ziganshin, B.A.; Charilaou, P.; Zafar, M.A.; Sieller, R.S.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Positive family history of aortic dissection dramatically increases dissection risk in family members. Int. J. Cardiol. 2017, 240, 132–137. [Google Scholar] [CrossRef]
- Biddinger, A.; Rocklin, M.; Coselli, J.; Milewicz, D.M. Familial thoracic aortic dilatations and dissections: A case control study. J. Vasc. Surg. 1997, 25, 506–511. [Google Scholar] [CrossRef]
- Robertson, E.N.; van der Linde, D.; Sherrah, A.G.; Vallely, M.P.; Wilson, M.; Bannon, P.G.; Jeremy, R.W. Familial non-syndromal thoracic aortic aneurysms and dissections—Incidence and family screening outcomes. Int. J. Cardiol. 2016, 220, 43–51. [Google Scholar] [CrossRef]
- Verhagen, J.M.; Kempers, M.; Cozijnsen, L.; Bouma, B.J.; Duijnhouwer, A.L.; Post, J.G.; Hilhorst-Hofstee, Y.; Bekkers, S.C.; Kerstjens-Frederikse, W.S.; van Brakel, T.J.; et al. Expert consensus recommendations on the cardiogenetic care for patients with thoracic aortic disease and their first-degree relatives. Int. J. Cardiol. 2018, 258, 243–248. [Google Scholar] [CrossRef]
- Weinsaft, J.W.; Devereux, R.B.; Preiss, L.R.; Feher, A.; Roman, M.J.; Basson, C.T.; Geevarghese, A.; Ravekes, W.; Dietz, H.C.; Holmes, K.; et al. Aortic Dissection in Patients with Genetically Mediated Aneurysms. J. Am. Coll. Cardiol. 2016, 67, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- Saeyeldin, A.; Zafar, M.A.; Li, Y.; Tanweer, M.; Abdelbaky, M.; Gryaznov, A.; Brownstein, A.J.; Velasquez, C.A.; Buntin, J.; Thombre, K.; et al. Decision-making algorithm for ascending aortic aneurysm: Effectiveness in clinical application? J. Thorac. Cardiovasc. Surg. 2019, 157, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B. Bicuspid Aortic Valve and Thoracic Aortic Aneurysm: Three Patient Populations, Two Disease Phenotypes, and One Shared Genotype. Cardiol. Res. Pract. 2012, 2012, 926975. [Google Scholar] [CrossRef] [PubMed]
- Borger, M.A.; Fedak, P.W.; Stephens, E.H.; Gleason, T.G.; Girdauskas, E.; Ikonomidis, J.S.; Khoynezhad, A.; Siu, S.C.; Verma, S.; Hope, M.D.; et al. The American Association for Thoracic Surgery consensus guidelines on bicuspid aortic valve–related aortopathy: Full online-only version. J. Thorac. Cardiovasc. Surg. 2018, 156, e41–e74. [Google Scholar] [CrossRef] [PubMed]
- Michelena, H.I.; Prakash, S.K.; Della Corte, A.; Bissell, M.M.; Anavekar, N.; Mathieu, P.; Bossé, Y.; Limongelli, G.; Bossone, E.; Benson, D.W.; et al. Bicuspid Aortic Valve: Identifying Knowledge Gaps and Rising to the Challenge From the International Bicuspid Aortic Valve Consortium (BAVCon). Circulation 2014, 129, 2691–2704. [Google Scholar] [CrossRef] [PubMed]
- Yassine, N.M.; Shahram, J.T.; Body, S.C. Pathogenic Mechanisms of Bicuspid Aortic Valve Aortopathy. Front. Physiol. 2017, 8, 687. [Google Scholar] [CrossRef]
- Cotrufo, M.; Della Corte, A.; De Santo, L.S.; Quarto, C.; De Feo, M.; Romano, G.; Amarelli, C.; Scardone, M.; Di Meglio, F.; Guerra, G.; et al. Different patterns of extracellular matrix protein expression in the convexity and the concavity of the dilated aorta with bicuspid aortic valve: Preliminary results. J. Thorac. Cardiovasc. Surg. 2005, 130, 504.e1–504.e9. [Google Scholar] [CrossRef]
- Michelena, H.I.; Khanna, A.D.; Mahoney, D.; Margaryan, E.; Topilsky, Y.; Suri, R.M.; Eidem, B.; Edwards, W.D.; Sundt, T.M.; Enriquez-Sarano, M. Incidence of Aortic Complications in Patients with Bicuspid Aortic Valves. JAMA 2011, 306, 1104. [Google Scholar] [CrossRef]
- Michelena, H.I.; Desjardins, V.A.; Avierinos, J.F.; Russo, A.; Nkomo, V.T.; Sundt, T.M.; Pellikka, P.A.; Tajik, A.J.; Enriquez-Sarano, M. Natural History of Asymptomatic Patients with Normally Functioning or Minimally Dysfunctional Bicuspid Aortic Valve in the Community. Circulation 2008, 117, 2776–2784. [Google Scholar] [CrossRef] [PubMed]
- Tzemos, N. Outcomes in Adults with Bicuspid Aortic Valves. JAMA 2008, 300, 1317. [Google Scholar] [CrossRef] [PubMed]
- Siu, S.C.; Silversides, C.K. Bicuspid Aortic Valve Disease. J. Am. Coll. Cardiol. 2010, 55, 2789–2800. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, S.W. Accentuating and Opposing Factors Leading to Development of Thoracic Aortic Aneurysms Not Due to Genetic or Inherited Conditions. Front. Cardiovasc. Med. 2015, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Danyi, P.; Elefteriades, J.A.; Jovin, I.S. Medical therapy of thoracic aortic aneurysms. Trends Cardiovasc. Med. 2012, 22, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Oguri, M.; Kato, N.; Hibino, T.; Yajima, K.; Yoshida, T.; Metoki, N.; Yoshida, H.; Satoh, K.; Watanabe, S.; et al. Assessment of Genetic Risk Factors for Thoracic Aortic Aneurysm in Hypertensive Patients. Am. J. Hypertens. 2008, 21, 1023–1027. [Google Scholar] [CrossRef]
- Daugherty, A.; Rateri, D.L.; Charo, I.F.; Owens, A.P.; Howatt, D.A.; Cassis, L.A. Angiotensin II infusion promotes ascending aortic aneurysms: Attenuation by CCR2 deficiency in apoE−/− mice. Clin. Sci. 2010, 118, 681–689. [Google Scholar] [CrossRef]
- Kanematsu, Y.; Kanematsu, M.; Kurihara, C.; Tsou, T.L.; Nuki, Y.; Liang, E.I.; Makino, H.; Hashimoto, T. Pharmacologically Induced Thoracic and Abdominal Aortic Aneurysms in Mice. Hypertension 2010, 55, 1267–1274. [Google Scholar] [CrossRef]
- Kallianos, K.G.; Burris, N.S. Imaging Thoracic Aortic Aneurysm. Radiol. Clin. N.Am. 2020, 58, 721–731. [Google Scholar] [CrossRef]
- Ho, D.; Squelch, A.; Sun, Z. Modelling of aortic aneurysm and aortic dissection through 3D printing. J. Med. Radiat. Sci. 2017, 64, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Bhave, N.M.; Nienaber, C.A.; Clough, R.E.; Eagle, K.A. Multimodality Imaging of Thoracic Aortic Diseases in Adults. JACC Cardiovasc. Imaging 2018, 11, 902–919. [Google Scholar] [CrossRef]
- Kato, K.; Nishio, A.; Kato, N.; Usami, H.; Fujimaki, T.; Murohara, T. Uptake of 18 F-FDG in Acute Aortic Dissection: A Determinant of Unfavorable Outcome. J. Nucl. Med. 2010, 51, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Almarzooq, Z.; Salata, B.; Devereux, R.B. Role of molecular imaging with positron emission tomographic in aortic aneurysms. J. Thorac. Dis. 2017, 9, S333–S342. [Google Scholar] [CrossRef] [PubMed]
- Hyafil, F.; Cornily, J.-C.; Rudd, J.H.; Machac, J.; Feldman, L.J.; Fayad, Z.A. Quantification of Inflammation within Rabbit Atherosclerotic Plaques Using the Macrophage-Specific CT Contrast Agent N1177: A Comparison with 18 F-FDG PET/CT and Histology. J. Nucl. Med. 2009, 50, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; De Santis, G.; Negri, F.; Tresoldi, D.; Rizzo, G.; Ponzini, R.; Massai, D.; Deriu, M.; Cadioli, M.; Verhegghe, B.; et al. On the Use of In Vivo Measured Flow Rates as Boundary Conditions for Image-Based Hemodynamic Models of the Human Aorta. In Proceedings of the ASME 2011 Summer Bioengineering Conference, Parts A and B, Farmington, PA, USA, 22–25 June 2011; American Society of Mechanical Engineers: New York, NY, USA, 2011; pp. 15–16. [Google Scholar] [CrossRef]
- Morbiducci, U.; Ponzini, R.; Gallo, D.; Bignardi, C.; Rizzo, G. Inflow boundary conditions for image-based computational hemodynamics: Impact of idealized versus measured velocity profiles in the human aorta. J. Biomech. 2013, 46, 102–109. [Google Scholar] [CrossRef]
- Xuan, Y.; D’souza, S.N.; Wang, Z.; Pierre, A.S.; Lawton, J.S.; Ge, L.; Tseng, E.E. Patient-Specific Biomechanics in Marfan Ascending Thoracic Aortic Aneurysms. Ann. Thorac. Surg. 2022, 114, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Vignali, E.; Gasparotti, E.; Mariotti, A.; Haxhiademi, D.; Ait-Ali, L.; Celi, S. High-Versatility Left Ventricle Pump and Aortic Mock Circulatory Loop Development for Patient-Specific Hemodynamic In Vitro Analysis. ASAIO J. 2022, 68, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, Y.; Spears, M.; Boustred, C.; May, M.; Anderson, S.; Benjamin, E.; Boutouyrie, P.; Cameron, J.; Chen, C.-H.; Cruickshank, J.K.; et al. Aortic Pulse Wave Velocity Improves Cardiovascular Event Prediction. J. Am. Coll. Cardiol. 2014, 63, 636–646. [Google Scholar] [CrossRef]
- Grabenwöger, M.; Alfonso, F.; Bachet, J.; Bonser, R.; Czerny, M.; Eggebrecht, H.; Evangelista, A.; Fattori, R.; Jakob, H.; Lönn, L.; et al. Thoracic Endovascular Aortic Repair (TEVAR) for the treatment of aortic diseases: A position statement from the European Association for Cardio-Thoracic Surgery (EACTS) and the European Society of Cardiology (ESC), in collaboration with the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur. Heart J. 2012, 33, 1558–1563. [Google Scholar]
- McCarthy, A.; Gray, J.; Sastry, P.; Sharples, L.; Vale, L.; Cook, A.; Mcmeekin, P.; Freeman, C.; Catarino, P.; Large, S. Systematic review of endovascular stent grafting versus open surgical repair for the elective treatment of arch/descending thoracic aortic aneurysms. BMJ Open 2021, 11, e043323. [Google Scholar] [CrossRef] [PubMed]
- Bavaria, J.E.; Appoo, J.J.; Makaroun, M.S.; Verter, J.; Yu, Z.-F.; Mitchell, R.S. Endovascular stent grafting versus open surgical repair of descending thoracic aortic aneurysms in low-risk patients: A multicenter comparative trial. J. Thorac. Cardiovasc. Surg. 2007, 133, 369–377.e4. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-S.; Haider, S.; Makaroun, M.S. Endovascular Therapy of Thoracic Aneurysms: Gore TAG Trial Results. Semin. Vasc. Surg. 2006, 19, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-S.; Haider, S.; Makaroun, M.S. US multicenter trials of endoprostheses for the endovascular treatment of descending thoracic aneurysms. J. Vasc. Surg. 2006, 43, A12–A19. [Google Scholar] [CrossRef]
- Go, M.R.; Cho, J.-S.; Makaroun, M.S. Mid-term Results of a Multicenter Study of Thoracic Endovascular Aneurysm Repair Versus Open Repair. Perspect. Vasc. Surg. Endovasc. Ther. 2007, 19, 124–130. [Google Scholar] [CrossRef]
- Von Allmen, R.S.; Anjum, A.; Powell, J.T. Outcomes after endovascular or open repair for degenerative descending thoracic aortic aneurysm using linked hospital data. Br. J. Surg. 2014, 101, 1244–1251. [Google Scholar] [CrossRef]
- Goodney, P.P.; Travis, L.; Lucas, F.L.; Fillinger, M.F.; Goodman, D.C.; Cronenwett, J.L.; Stone, D.H. Survival After Open Versus Endovascular Thoracic Aortic Aneurysm Repair in an Observational Study of the Medicare Population. Circulation 2011, 124, 2661–2669. [Google Scholar] [CrossRef]
- Mori, M.; Shioda, K.; Wang, X.; Mangi, A.A.; Yun, J.J.; Darr, U.; Elefteriades, J.A.; Geirsson, A. Perioperative Risk Profiles and Volume-Outcome Relationships in Proximal Thoracic Aortic Surgery. Ann. Thorac. Surg. 2018, 106, 1095–1104. [Google Scholar] [CrossRef]
- Writing Group Members; Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E., Jr.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients with Thoracic Aortic Disease. J. Am. Coll. Cardiol. 2010, 55, e27–e129. [Google Scholar] [CrossRef]
- Guo, M.H.; Appoo, J.J.; Saczkowski, R.; Smith, H.N.; Ouzounian, M.; Gregory, A.J.; Herget, E.J.; Boodhwani, M. Association of Mortality and Acute Aortic Events with Ascending Aortic Aneurysm: A Systematic Review and Meta-analysis. JAMA Netw. Open 2018, 1, e181281. [Google Scholar] [CrossRef] [PubMed]
- Ashton, H.A.; Buxton, M.J.; Day, N.E.; Kim, L.G.; Marteau, E.M.; Scott, R.A.P.; Thompson, S.G.; Walker, N.M. Multicentre Aneurysm Screening Study Group. The Multicentre Aneurysm Screening Study (MASS) into the effect of abdominal aortic aneurysm screening on mortality in men: A randomised controlled trial. Lancet 2002, 360, 1531–1539. [Google Scholar] [PubMed]
- Lederle, F.A.; Wilson, S.E.; Johnson, G.R.; Reinke, D.B.; Littooy, F.N.; Acher, C.W.; Ballard, D.J.; Messina, L.M.; Gordon, I.L.; Chute, E.P.; et al. Immediate repair compared with surveillance of small abdominal aortic aneurysms. N. Engl. J. Med. 2002, 346, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.H.; Appoo, J.J.; Wells, G.A.; Chu, M.; Ouzounian, M.; Fortier, J.; Boodhwani, M. Protocol for a randomised controlled trial for Treatment in Thoracic Aortic Aneurysm: Surgery versus Surveillance (TITAN: SvS). BMJ Open 2021, 11, e052070. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulenka, Y.; Lee, C.; Tawayha, M.; Dow, S.; Shah, K.; Henkin, S.; Mosleh, W. From Natural History to Contemporary Management of Aortic Diseases: A State-of-the-Art Review of Thoracic Aortic Aneurysm. Cardiogenetics 2023, 13, 154-172. https://doi.org/10.3390/cardiogenetics13040015
Paulenka Y, Lee C, Tawayha M, Dow S, Shah K, Henkin S, Mosleh W. From Natural History to Contemporary Management of Aortic Diseases: A State-of-the-Art Review of Thoracic Aortic Aneurysm. Cardiogenetics. 2023; 13(4):154-172. https://doi.org/10.3390/cardiogenetics13040015
Chicago/Turabian StylePaulenka, Yuliya, Christopher Lee, Mays Tawayha, Sam Dow, Kajal Shah, Stanislav Henkin, and Wassim Mosleh. 2023. "From Natural History to Contemporary Management of Aortic Diseases: A State-of-the-Art Review of Thoracic Aortic Aneurysm" Cardiogenetics 13, no. 4: 154-172. https://doi.org/10.3390/cardiogenetics13040015
APA StylePaulenka, Y., Lee, C., Tawayha, M., Dow, S., Shah, K., Henkin, S., & Mosleh, W. (2023). From Natural History to Contemporary Management of Aortic Diseases: A State-of-the-Art Review of Thoracic Aortic Aneurysm. Cardiogenetics, 13(4), 154-172. https://doi.org/10.3390/cardiogenetics13040015