
DNA Repair Inhibitors: Potential Targets and Partners for Targeted Radionuclide Therapy



Abstract
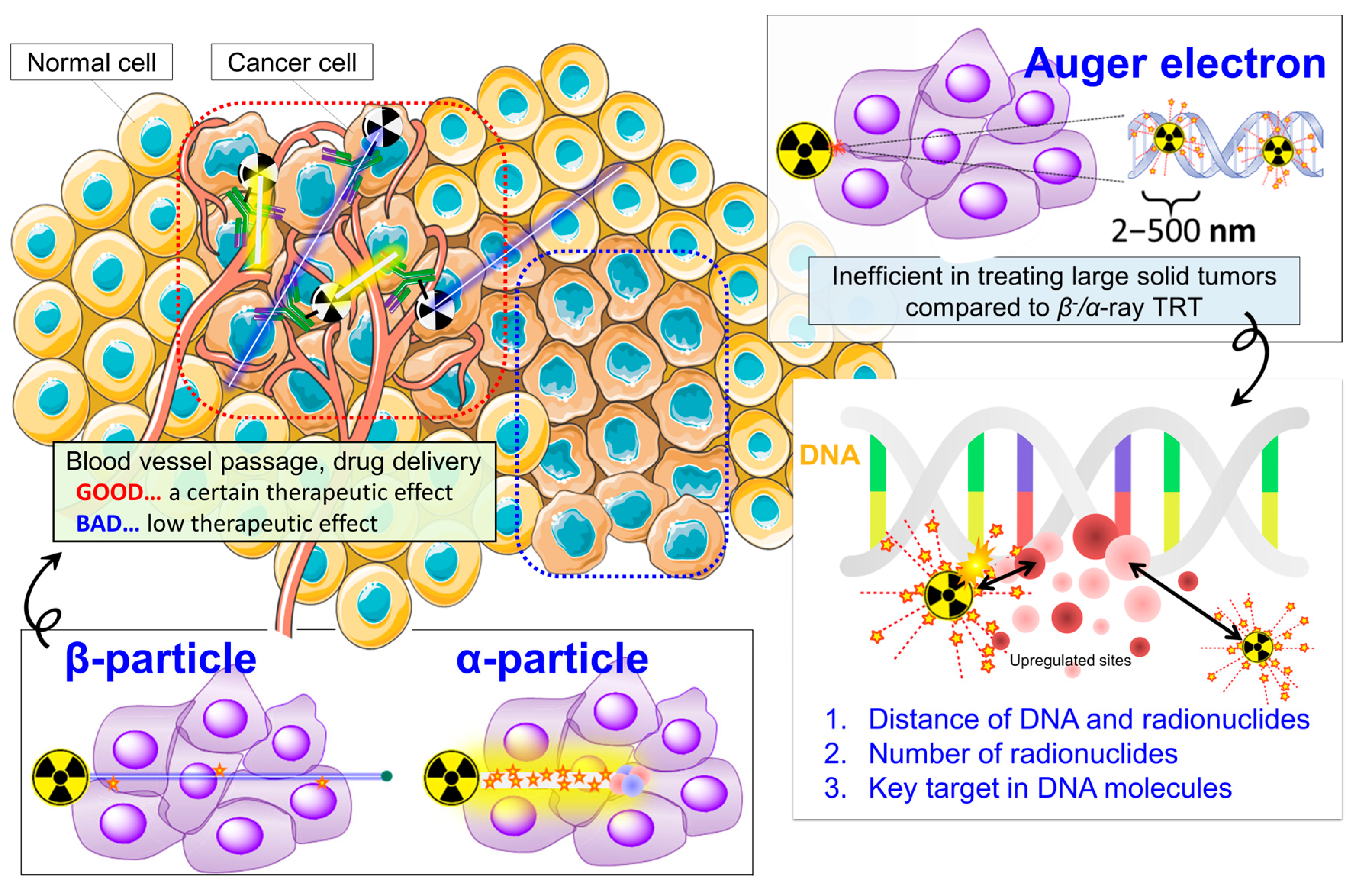
1. Overview of the Current Status of Targeted Radionuclide Therapy (TRT)
1.1. β−-Particle or α-Particle-Based TRT Exhibits Therapeutic Efficacy: On the Way to General Utility
1.2. Auger-Electron TRT Remains Attractive but Success Is Elusive
2. A New Therapeutic Strategy to Break through Current TRT Limitations: Making TRT More Effective
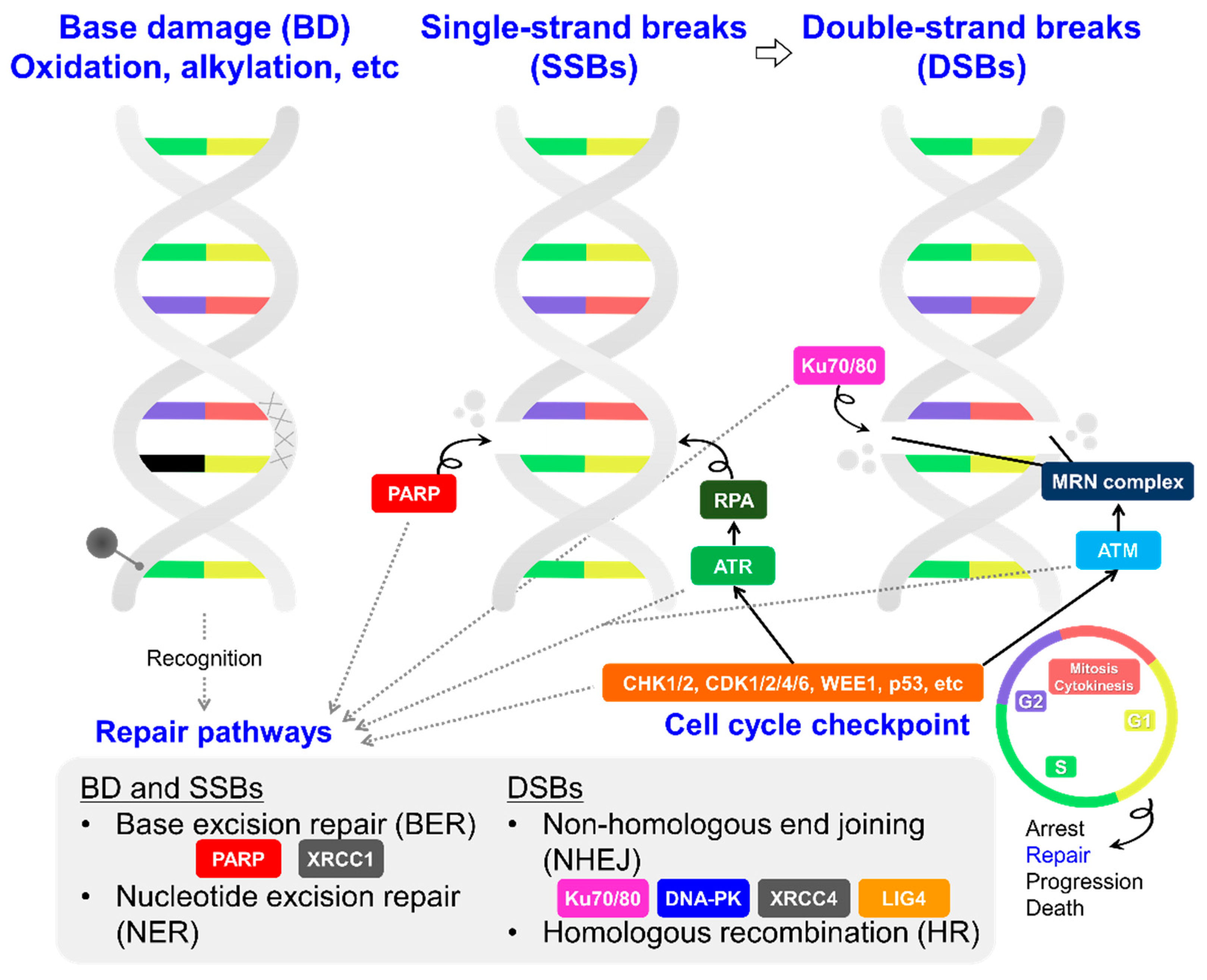
3. Potential Molecular Targets and Their Inhibitors as TRT Partners: Focusing on DNA Damage Response/Repair and Cell Cycle Maintenance Systems in Cancer Cells
3.1. DNA Damage Response/Repair Factors Controlling the Cell Cycle
3.1.1. PARP
3.1.2. ATM and ATR
3.1.3. DNA-PK, LIG4, and XRCC4
3.1.4. Others
3.2. Amplified and Overexpressed Factors Regulating the Cancer Genome
3.2.1. Transcription-Related Factor
3.2.2. DNA Methyltransferase and Epigenetic Regulators
4. Perspectives: Possible Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug. Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef] [PubMed]
- Kassis, A.I.; Adelstein, S.J. Radiobiologic principles in radionuclide therapy. J. Nucl. Med. 2005, 46, 4S–12S. [Google Scholar] [PubMed]
- Pons, F.; Herranz, R.; Garcia, A.; Vidal-Sicart, S.; Conill, C.; Grau, J.J.; Alcover, J.; Fuster, D.; Setoain, J. Strontium-89 for palliation of pain from bone metastases in patients with prostate and breast cancer. Eur. J. Nucl. Med. 1997, 24, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Witzig, T.E.; Gordon, L.I.; Cabanillas, F.; Czuczman, M.S.; Emmanouilides, C.; Joyce, R.; Pohlman, B.L.; Bartlett, N.L.; Wiseman, G.A.; Padre, N.; et al. Randomized controlled trial of yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin’s lymphoma. J. Clin. Oncol. 2002, 20, 2453–2463. [Google Scholar] [CrossRef]
- Wilson, J.S.; Gains, J.E.; Moroz, V.; Wheatley, K.; Gaze, M.N. A systematic review of 131I-meta iodobenzylguanidine molecular radiotherapy for neuroblastoma. Eur. J. Cancer 2014, 50, 801–815. [Google Scholar] [CrossRef]
- NuDat 3.0. Available online: https://www.nndc.bnl.gov/nudat3/ (accessed on 21 April 2023).
- U.S. Food and Drug, Administration. FDA Approves Lutetium Lu 177 Dotatate for Treatment of GEP-NETS. 2018. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lutetium-lu-177-dotatate-treatment-gep-nets (accessed on 27 April 2023).
- U.S. Food and Drug, Administration. FDA Approves Pluvicto for Metastatic Castration-Resistant Prostate Cancer. 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pluvicto-metastatic-castration-resistant-prostate-cancer (accessed on 27 April 2023).
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225Ac-PSMA-617 for PSMA-targeted α-radiation therapy of metastatic castration-resistant prostate cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef]
- Eychenne, R.; Chérel, M.; Haddad, F.; Guérard, F.; Gestin, J.F. Overview of the Most, Promising Radionuclides for Targeted, Alpha Therapy: The “Hopeful, Eight”. Pharmaceutics 2021, 13, 906. [Google Scholar] [CrossRef]
- Kluetz, P.G.; Pierce, W.; Maher, V.E.; Zhang HTang, S.; Song, P.; Liu, Q.; Haber, M.T.; Leutzinger, E.E.; Al-Hakim, A.; Chen, W.; et al. Radium, Ra 223 dichloride injection: U.S. Food and Drug, Administration drug approval summary. Clin. Cancer Res. 2014, 20, 9–14. [Google Scholar] [CrossRef]
- Albertsson, P.; Bäck, T.; Bergmark, K.; Hallqvist, A.; Johansson, M.; Aneheim, E.; Lindegren, S.; Timperanza, C.; Smerud, K.; Palm, S. Astatine-211 based radionuclide therapy: Current clinical trial landscape. Front. Med. 2022, 9, 1076210. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; Pavel, M.E.; et al. 177Lu-Dotatate plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef]
- Ruiz, M.E.R.; Box, C.V.; Melero, I.; Formenti, S.C.; Demaria, S. Immunological mechanisms responsible for radiation-induced abscopal effect. Trends Immunol. 2018, 39, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Chen, D.; Yu, J. Radiotherapy combined with immunotherapy: The dawn of cancer treatment. Signal. Transduct. Target. Ther. 2022, 7, 258. [Google Scholar] [CrossRef] [PubMed]
- Rebischung, C.; Hoffmann, D.; Stéfani, L.; Desruet, M.D.; Wang, K.; Adelstein, S.J.; Artignan, X.; Vincent, F.; Gauchez, A.S.; Zhang, H.; et al. First human treatment of resistant neoplastic meningitis by intrathecal administration of MTX Plus 125IUdR. Int. J. Radiat. Biol. 2008, 84, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Welt, S.; Scott, A.M.; Divgi, C.R.; Kemeny, N.E.; Finn, R.D.; Daghighian, F.; Germain, J.S.; Richards, E.C.; Larson, S.M.; Old, L.J. Phase, I/II study of iodine 125-labeled monoclonal antibody A33 in patients with advanced colon cancer. J. Clin. Oncol. 1996, 14, 1787–1797. [Google Scholar] [CrossRef]
- Quang, T.S.; Brady, L.W. Radioimmunotherapy as a novel treatment regimen: 125I-labeled monoclonal antibody 425 in the treatment of high-grade brain gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 972–975. [Google Scholar] [CrossRef]
- Jong, M.; Valkema, R.; Jamar, F.; Kvols, L.K.; Kwekkeboom, D.J.; Breeman, W.A.P.; Bakker, W.H.; Smith, C.; Pauwels, S.; Krenning, E.P. Somatostatin receptor-targeted radionuclide therapy of tumors: Preclinical and clinical findings. Semin. Nucl. Med. 2002, 32, 133–140. [Google Scholar] [CrossRef]
- Delpassand, E.S.; Samarghandi, A.; Mourtada, J.S.; Zamanian, S.; Espenan, G.D.; Sharif, R.; MacKenzie, S.; Kosari, K.; Barakat, O.; Naqvi, S.; et al. Long-term survival, toxicity profile, and role of F-18 FDG PET/CT scan in patients with progressive neuroendocrine tumors following peptide receptor radionuclide therapy with high activity In-111 pentetreotide. Theranostics 2012, 2, 472–480. [Google Scholar] [CrossRef]
- Vallis, K.A.; Reilly, R.M.; Scollard, D.; Merante, P.; Brade, A.; Velauthapillai, S.; Caldwell, C.; Chan, I.; Freeman, M.; Lockwood, G.; et al. Phase, I trial to evaluate the tumor and normal tissue uptake, radiation dosimetry and safety of 111In-DTPA-human epidermal growth factor in patients with metastatic EGFR-positive breast cancer. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 181–192. [Google Scholar]
- Rosenkranz, A.A.; Slastnikova, T.A.; Karmakova, T.A.; Vorontsova, M.S.; Morozova, N.B.; Petriev, V.M.; Abrosimov, A.S.; Khramtsov, Y.V.; Lupanova, T.N.; Ulasov, A.V.; et al. Antitumor activity of auger electron emitter 111In delivered by modular nanotransporter for treatment of bladder cancer with EGFR overexpression. Front. Pharmacol. 2018, 9, 1331. [Google Scholar] [CrossRef]
- Santoro, L.; Boutaleb, S.; Garambois, V.; Mollevi, C.B.; Boudousq, V.; Kotzki, P.O.; Pèlegrin, M.; Teulon, I.N.; Pèlegrin, A.; Pouget, J.P. Noninternalizing monoclonal antibodies are suitable candidates for 125I radioimmunotherapy of small-volume peritoneal carcinomatosis. J. Nucl. Med. 2009, 50, 2033–2041. [Google Scholar] [CrossRef]
- Sankaranarayanan, R.A.; Florea, A.; Allekotte, S.; Vogg, A.T.J.; Maurer, J.; Schäfer, L.; Bolm, C.; Terhorst, S.; Classen, A.; Bauwens, M.; et al. PARP targeted Auger emitter therapy with [125I]PARPi-01 for triple-negative breast cancer. EJNMMI Res. 2022, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Riad, A.; Martorano, P.; Mansfield, A.; Samanta, M.; Batra, V.; Mach, R.H.; Maris, J.M.; Pryma, D.A.; Makvandi, M. PARP-1-targeted Auger emitters display high-LET cytotoxic properties in vitro but show limited therapeutic utility in solid tumor models of human neuroblastoma. J. Nucl. Med. 2020, 61, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, A.A.; Slastnikova, T.A.; Georgiev, G.P.; Zalutsky, M.R.; Sobolev, A.S. Delivery systems exploiting natural cell transport processes of macromolecules for intracellular targeting of Auger electron emitters. Nucl. Med. Biol. 2020, 80, 45–56. [Google Scholar] [CrossRef]
- Buchegger, F.; Adamer, F.P.; Dupertuis, Y.M.; Delaloye, A.B. Auger radiation targeted into DNA: A therapy perspective. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 1352–1363. [Google Scholar] [CrossRef] [PubMed]
- UNSCEAR. Annex E: Occupational radiation exposures. In Sources and Effects of Ionizing Radiation: United Nations Scientific Committee on the Effects of Atomic Radiation UNSCEAR 2000 Report to the General Assembly, with Scientific Annexes, Volume II: Effects of ionizing radiation; United Nations: San Francisco, CA, USA, 2000. [Google Scholar]
- Balagurumoorthy, P.; Xu, X.; Wang, K.; Adelstein, S.J.; Kassis, A.I. Effect of distance between decaying 125I and DNA on Auger-electron induced double-strand break yield. Int. J. Radiat. Biol. 2012, 88, 998–1008. [Google Scholar] [CrossRef]
- Pereira, E.; Quental, L.; Palma, E.; Oliveira, M.C.; Mendes, F.; Raposinho, P.; Correia, I.; Lavrado, J.; Maria, S.D.; Belchior, A.; et al. Evaluation of acridine orange derivatives as DNA-targeted radiopharmaceuticals for auger therapy: Influence of the radionuclide and distance to DNA. Sci. Rep. 2017, 7, 42544. [Google Scholar] [CrossRef]
- Reissig, F.; Mamat, C.; Steinbach, J.; Pietzsch, H.J.; Freudenberg, R.; Retamal, C.N.; Caballero, J.; Kotzerke, J.; Wunderlich, G. Direct and auger electron-induced, single and double-strand breaks on plasmid DNA caused by 99mTc-labeled pyrene derivatives and the effect of bonding distance. PLoS ONE 2016, 11, e0161973. [Google Scholar] [CrossRef]
- Karagiannis, T.C. Comparison of different classes of radionuclides for potential use in radioimmunotherapy. Hell. J. Nucl. Med. 2007, 10, 82–88. [Google Scholar]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar] [CrossRef]
- Aloj, L.; Attili, B.; Lau, D.; Caraco, C.; Lechermann, L.M.; Mendichovszky, I.A.; Harper, I.; Cheow, H.; Casey, R.T.; Sala, E.; et al. The emerging role of cell surface receptor and protein binding radiopharmaceuticals in cancer diagnostics and therapy. Nucl. Med. Biol. 2021, 92, 53–64. [Google Scholar] [CrossRef]
- Marnett, L.J.; Plastaras, J.P. Endogenous, DNA damage and mutation. Trends Genet. 2001, 17, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous, DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Schär, P. Spontaneous, DNA damage, genome instability, and cancer–when DNA replication escapes control. Cell 2001, 104, 329–332. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Kastan, M.B. Cell cycle control and cancer. Science 1994, 266, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Zheng, C.C.; Huang, Y.N.; He, M.L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo-and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2021, 2, 315–340. [Google Scholar] [CrossRef]
- Smirnov, D.A.; Morley, M.; Shin, E.; Spielman, R.S.; Cheung, V.G. Genetic analysis of radiation-induced changes in human gene expression. Nature 2009, 459, 587–591. [Google Scholar] [CrossRef]
- Valerie, K.; Yacoub, A.; Hagan, M.P.; Curiel, D.T.; Fisher, P.B.; Grant, S.; Dent, P. Radiation-induced cell signaling: Inside-out and outside-in. Mol. Cancer Ther. 2007, 6, 789–801. [Google Scholar] [CrossRef]
- Tang, L.; Wei, F.; Wu, Y.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Deng, H.; et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res. 2018, 37, 87. [Google Scholar] [CrossRef]
- Daguenet, E.; Louati, S.; Wozny, A.S.; Vial, N.; Gras, M.; Guy, J.B.; Vallard, A.; Lafrasse, C.R.; Magné, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer 2020, 123, 339–348. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Victor, C.T.S.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Khan, M.K.; Wu, X.; Simone, C.B.; Fan, J.; Gressen, E.; Zhang, X.; Limoli, C.L.; Bahig, H.; Tubin, S.; et al. Spatially fractionated radiation therapy: History, present and the future. Clin. Transl. Radiat. Oncol. 2019, 20, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Prise, K.M.; Sullivan, J.M.O. Radiation-induced bystander signalling in cancer therapy. Nat. Rev. Cancer 2009, 9, 351–360. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal. Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes. Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef]
- Amours, D.D.; Desnoyers, S.; Silva, I.D.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef]
- Swindall, A.F.; Stanley, J.A.; Yang, E.S. PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis? Cancers 2013, 5, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Andronikou, C.; Rottenberg, S. Studying, PAR-dependent chromatin remodeling to tackle PARPi resistance. Trends, Mol. Med. 2021, 27, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog. Nucleic Acid. Res. Mol. Biol. 1988, 35, 95–125. [Google Scholar]
- Le, X.C.; Xing, J.Z.; Lee, J.; Leadon, S.A.; Weinfeld, M. Inducible repair of thymine glycol detected by an ultrasensitive assay for DNA damage. Science 1998, 280, 1066–1069. [Google Scholar] [CrossRef]
- Pouget, J.P.; Ravanat, J.L.; Douki, T.; Richard, M.J.; Cadet, J. Measurement of DNA base damage in cells exposed to low doses of gamma-radiation: Comparison between the HPLC-EC and comet assays. Int. J. Radiat. Biol. 1999, 75, 51–58. [Google Scholar] [CrossRef]
- Lesueur, P.; Chevalier, F.; Austry, J.B.; Waissi, W.; Burckel, H.; Noël, G.; Habrand, J.L.; Saintigny, Y.; Joly, F. Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: A systematic review of pre-clinical and clinical human studies. Oncotarget. 2017, 8, 69105–69124. [Google Scholar] [CrossRef]
- Michmerhuizen, A.R.; Pesch, A.M.; Moubadder, L.; Chandler, B.C.; Wilder-Romans, K.; Cameron, M.; Olsen, E.; Thomas, D.G.; Zhang, A.; Hirsh, N. PARP1 inhibition radiosensitizes models of inflammatory breast cancer to ionizing radiation. Mol. Cancer Ther. 2019, 18, 2063–2073. [Google Scholar] [CrossRef]
- Senra, J.M.; Telfer, B.A.; Cherry, K.E.; McCrudden, C.M.; Hirst, D.G.; O’Connor, M.J.; Wedge, S.R.; Stratford, I.J. Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol. Cancer Ther. 2011, 10, 1949–1958. [Google Scholar] [CrossRef]
- Dungey, F.A.; Löser, D.A.; Chalmers, A.J. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-Ribose) polymerase: Mechanisms and therapeutic potential. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 1188–1197. [Google Scholar] [CrossRef]
- Noël, G.; Godon, C.; Fernet, M.; Giocanti, N.; Mégnin-Chanet, F.; Favaudon, V. Radiosensitization by the poly(ADP-ribose) polymerase inhibitor 4-amino-1,8-naphthalimide is specific of the S phase of the cell cycle and involves arrest of DNA synthesis. Mol. Cancer Ther. 2006, 5, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Löser, D.A.; Shibata, A.; Shibata, A.K.; Woodbine, L.J.; Jeggo, P.A.; Chalmers, A.J. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol. Cancer Ther. 2010, 9, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, C.; Waldeck, K.; Kirby, L.; Rogers, B.E.; Eu, P.; Tothill, R.W.; Hicks, R.J. Enhancing the anti-tumour activity of 177Lu-DOTA-octreotate radionuclide therapy in somatostatin receptor-2 expressing tumour models by targeting PARP. Sci. Rep. 2020, 10, 10196. [Google Scholar] [CrossRef]
- O’Neill, E.; Mosley, M.; Cornelissen, B. A screen of 350 combination therapies with 177Lu-DOTATATE reveal distinct differences to EBRT and novel action of CDK4/6 inhibitors. J. Nucl. Med. 2020, 61 (Suppl. S1), 32. [Google Scholar]
- Dabagian, H.; Taghvaee, T.; Martorano, P.; Martinez, D.; Samanta, M.; Watkins, C.M.; Chai, R.; Mansfield, A.; Graham, T.J.; Maris, J.M.; et al. PARP targeted alpha-particle therapy enhances response to PD-1 immune-checkpoint blockade in a syngeneic mouse model of glioblastoma. ACS Pharmacol. Transl. Sci. 2021, 4, 344–351. [Google Scholar] [CrossRef]
- Makvandi, M.; Lee, H.; Puentes, L.N.; Reilly, S.W.; Rathi, K.S.; Weng, C.C.; Chan, H.S.; Hou, C.; Raman, P.; Martinez, D.; et al. Targeting, PARP-1 with alpha-particles is potently cytotoxic to human neuroblastoma in preclinical models. Mol. Cancer Ther. 2019, 18, 1195–1204. [Google Scholar] [CrossRef]
- Wilson, T.; Pirovano, G.; Xiao, G.; Samuels, Z.; Roberts, S.; Viray, T.; Guru, N.; Zanzonico, P.; Gollub, M.; Pillarsetty, N.V.K.; et al. PARP-targeted Auger therapy in p53 mutant colon cancer xenograft mouse models. Mol. Pharm. 2021, 18, 3418–3428. [Google Scholar] [CrossRef]
- Pirovano, G.; Jannetti, S.A.; Carter, L.M.; Sadique, A.; Kossatz, S.; Guru, N.; França, P.D.S.; Maeda, M.; Zeglis, B.M.; Lewis, J.S.; et al. Targeted brain tumor radiotherapy using an Auger emitter. Clin. Cancer Res. 2020, 26, 2871–2881. [Google Scholar] [CrossRef]
- Destro, G.; Chen, Z.; Chan, C.Y.; Fraser, C.; Dias, G.; Mosley, M.; Guibbal, F.; Gouverneur, V.; Cornelissen, B. A radioiodinated rucaparib analogue as an Auger electron emitter for cancer therapy. Nucl. Med. Biol. 2023, 116–117, 108312. [Google Scholar] [CrossRef]
- Makvandi, M.; Xu, K.; Lieberman, B.P.; Anderson, R.C.; Effron, S.S.; Winters, H.D.; Zeng, C.; McDonald, E.S.; Pryma, D.A.; Greenberg, R.A.; et al. A radiotracer strategy to quantify PARP-1 expression in vivo provides a biomarker that can enable patient selection for PARP inhibitor therapy. Cancer Res. 2016, 76, 4516–4524. [Google Scholar] [CrossRef]
- Sankaranarayanan, R.A.; Peil, J.; Vogg, A.T.J.; Bolm, C.; Terhorst, S.; Classen, A.; Bauwens, M.; Maurer, J.; Mottaghy, F.; Morgenroth, A. Auger emitter conjugated PARP inhibitor for therapy in triple negative breast cancers: A comparative in-vitro study. Cancers 2022, 4, 230. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. Mechanisms of ATM activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Kurz, E.U.; Lees-Miller, S.P. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair. 2004, 3, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Vo, Q.N.; Zuo, C.; Li, L.; Ling, W.; Suen, J.Y.; Hanna, E.; Brown, K.D.; Fan, C.Y. Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: Promoter hypermethylation with clinical correlation in 100 cases. Cancer Epidemiol. Biomark. Prev. 2004, 13, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Kubota, E.; Williamson, C.T.; Ye, R.; Elegbede, A.; Peterson, L.; Lees-Miller, S.P.; Bebb, D.G. Low, ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell. Cycle 2014, 13, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.M.; Munoz-Alegre, M.; Henderson, S.; Tang, T.; Sun, P.; Johnson, N.; Fletcher, O.; Silva, I.D.S.; Peto, J.; Boshoff, C.; et al. Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Hum. Mol. Genet. 2009, 18, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Garcia-Closas, M.; Orr, N.; Fletcher, O.; Jones, M.; Ashworth, A.; Swerdlow, A.; Thorne, H.; Riboli, E.; Vineis, P.; et al. Intragenic, ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer Res. 2012, 72, 2304–2313. [Google Scholar] [CrossRef]
- Eich, M.; Roos, W.P.; Nikolova, T.; Kaina, B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol. Cancer Ther. 2013, 12, 2529–2540. [Google Scholar] [CrossRef]
- Kühne, M.; Riballo, E.; Rief, N.; Rothkamm, K.; Jeggo, P.A.; Löbrich, M. A double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Res. 2004, 64, 500–508. [Google Scholar] [CrossRef]
- Xu, Y.; Nowsheen, S.; Deng, M. DNA repair deficiency regulates immunity response in cancers: Molecular mechanism and approaches for combining immunotherapy. Cancers 2023, 15, 1619. [Google Scholar] [CrossRef]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM mutations in cancer: Therapeutic implications. Mol. Cancer, Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Duedal, S.; Kirner, J.; McGuffog, L.; Last, J.; Reiman, A.; Byrd, P.; Taylor, M.; Easton, D.F. Cancer risks and mortality in heterozygous ATM mutation carriers. J. Natl. Cancer Inst. 2005, 97, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Lecona, E.; Fernandez-Capetillo, O. Targeting, ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, S.; Fitzpatrick, K.; Liu, D.; Cai, M.Y.; Elmarakeby, H.A.; Park, J.; Ricker, C.; Kochupurakkal, B.S.; Choudhury, A.D.; Hahn, W.C.; et al. ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Cancer Res. 2020, 80, 2094–2100. [Google Scholar] [CrossRef] [PubMed]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, A.; Casals, T.; Carmichael, J.; Rodrigues, D.N. Advanced prostate cancer with ATM loss: PARP and ATR inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-human trial of the oral ataxia telangiectasia and RAD3-related (ATR) inhibitor BAY 1895344 in patients with advanced solid tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef]
- Smith, G.C.M.; Cary, R.B.; Lakin, N.D.; Hann, B.C.; Teo, S.H.; Chen, D.J.; Jackson, S.P. Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. Proc. Natl. Acad. Sci. USA 1999, 96, 11134–11139. [Google Scholar] [CrossRef]
- Unsal-Kaçmaz, K.; Makhov, A.M.; Griffith, J.D.; Sancar, A. Preferential binding of ATR protein to UV-damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 6673–6678. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, B.P.C.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair. 2014, 17, 21–29. [Google Scholar] [CrossRef]
- Goodwin, J.F.; Knudsen, K.E. Beyond, DNA repair: DNA-PK function in cancer. Cancer Discov. 2014, 4, 1126–1139. [Google Scholar] [CrossRef]
- Lee, H.S.; Choe, G.; Park, K.U.; Park, D.J.; Yang, H.K.; Lee, B.; Kim, W.H. Altered expression of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) during gastric carcinogenesis and its clinical implications on gastric cancer. Int. J. Oncol. 2007, 31, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Wu, X.; Vaporciyan, A.A.; Spit, M.R.; Gu, J. Prognostic significance ofataxia-telangiectasia mutated, DNA-dependent protein kinase catalytic subunit, and Ku heterodimeric regulatory complex 86-kD subunit expression in patients with nonsmall cell lung cancer. Cancer 2008, 112, 2756–2764. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous, DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wang, A.; Lin, J.; Wu, L.; Zhang, H.; Yang, X.; Wan, X.; Miao, R.; Sang, X.; Zhao, H. Mps1/TTK: A novel target and biomarker for cancer. J. Drug. Target. 2017, 25, 112–118. [Google Scholar] [CrossRef]
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting mitosis in cancer: Emerging strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef]
- Maia, A.R.R.; Man, J.; Boon, U.; Janssen, A.; Song, J.Y.; Omerzu, M.; Sterrenburg, J.G.; Prinsen, M.B.W.; Willemsen-Seegers, N.; Roos, J.A.D.M.; et al. Inhibition of the spindle assembly checkpoint kinase TTK enhances the efficacy of docetaxel in a triple-negative breast cancer model. Ann. Oncol. 2015, 26, 2180–2192. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional addiction in cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Michalak, E.M.; Burr, M.L.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell. Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Mazzone, R.; Zwergel, C.; Mai, A.; Valente, S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin. Ep. 2017, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Baretti, M.; Yarchoan, M. Epigenetic modifiers synergize with immune-checkpoint blockade to enhance long-lasting antitumor efficacy. J. Clin. Investig. 2021, 131, e151002. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ren, Y.; Weng, S.; Xu, H.; Li, L.; Han, X. A new trend in cancer treatment: The combination of epigenetics and immunotherapy. Front. Immunol. 2022, 13, 809761. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Bae, S.C.; Chuang, L.S.H. The RUNX family: Developmental regulators in cancer. Nat. Rev. Cancer 2015, 15, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Mitsuda, Y.; Morita, K.; Kashiwazaki, G.; Taniguchi, J.; Bando, T.; Obara, M.; Hirata, M.; Kataoka, T.R.; Muto, M.; Kaneda, Y.; et al. RUNX1 positively regulates the ErbB2/HER2 signaling pathway through modulating SOS1 expression in gastric cancer cells. Sci. Rep. 2018, 8, 6423. [Google Scholar] [CrossRef]
- Liu, C.; Xu, D.; Xue, B.; Liu, B.; Li, J.; Huang, J. Upregulation of RUNX1 Suppresses proliferation and migration through repressing VEGFA expression in hepatocellular carcinoma. Pathol. Oncol. Res. 2020, 26, 1301–1311. [Google Scholar] [CrossRef]
- Ferrari, N.; Mohammed, Z.M.A.; Nixon, C.; Mason, S.M.; Mallon, E.; McMillan, D.C.; Morris, J.S.; Cameron, E.R.; Edwards, J.; Blyth, K. Expression of RUNX1 correlates with poor patient prognosis in triple negative breast cancer. PLoS ONE 2014, 9, e100759. [Google Scholar] [CrossRef]
- Mendler, J.H.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Becker, H.; Metzeler, K.H.; Schwind, S.; Whitman, S.P.; Khalife, J.; Kohlschmidt, J. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J. Clin. Oncol. 2012, 30, 3109–3118. [Google Scholar] [CrossRef]
- Martin, J.W.; Zielenska, M.; Stein, G.S.; Wijnen, A.J.; Squire, J.A. The role of RUNX2 in osteosarcoma oncogenesis. Sarcoma 2011, 2011, 282745. [Google Scholar] [CrossRef]
- Pratap, J.; Lian, J.B.; Javed, A.; Barnes, G.L.; Wijnen, A.J.; Stein, J.L.; Stein, G.S. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev. 2006, 25, 589–600. [Google Scholar] [CrossRef]
- Vishal, M.; Swetha, R.; Thejaswini, G.; Arumugam, B.; Selvamurugan, N. Role of Runx2 in breast cancer-mediated bone metastasis. Int. J. Biol. Macromol. 2017, 99, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Date, Y.; Ito, K. Oncogenic, RUNX3: A link between p53 deficiency and MYC dysregulation. Mol. Cells 2020, 43, 176–181. [Google Scholar] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Koivisto, P.; Kononen, J.; Palmberg, C.; Tammela, T.; Hyytinen, E.; Isola, J.; Trapman, J.; Cleutjens, K.; Noordzij, A.; Visakorpi, T.; et al. Androgen receptor gene amplification: A possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997, 57, 314–319. [Google Scholar]
- Harenza, J.L.; Diamond, M.A.; Adams, R.N.; Song, M.M.; Davidson, H.; Hart, L.S.; Dent, M.H.; Fortina, P.; Reynolds, C.P.; Maris, J.M. Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines. Sci. Data 2017, 4, 170033. [Google Scholar] [CrossRef]
- Kang, J.H.; Rychahou, P.G.; Ishola, T.A.; Qiao, J.; Evers, B.M.; Chung, D.H. MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochem. Biophys. Res. Commun. 2006, 351, 192–197. [Google Scholar] [CrossRef]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, B.; Wu, W.; Li, L.; Broom, B.M.; Basourakos, S.P.; Korentzelos, D.; Luan, Y.; Wang, J.; Yang, G.; et al. Targeting the MYCN-PARP-DNA Damage response pathway in neuroendocrine prostate cancer. Clin. Cancer Res. 2018, 24, 696–707. [Google Scholar] [CrossRef]
- Yin, Y.; Xu, L.; Chang, Y.; Zeng, T.; Chen, X.; Wang, A.; Groth, J.; Foo, W.C.; Liang, C.; Hu, H.; et al. N-Myc promotes therapeutic resistance development of neuroendocrine prostate cancer by differentially regulating miR-421/ATM pathway. Mol. Cancer 2019, 18, 11. [Google Scholar] [CrossRef]
- Thompson, T.C.; Liu, B.; Li, L. N-MYC regulation of DNA damage response in neuroendocrine prostate cancer: Mechanistic insight and novel combination therapy approaches. Oncoscience 2018, 5, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Sawai, H.; Ogihara-Umeda, I.; Tanada, S.; Kim, E.E.; Yonekura, Y.; Sasaki, Y. Molecular therapy of human neuroblastoma cells using Auger electrons of 111In-labeled N-myc antisense oligonucleotides. J. Nucl. Med. 2006, 47, 1670–1677. [Google Scholar] [PubMed]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target specificity of the CRISPR-Cas9 system. Quant. Biol. 2014, 2, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pandian, G.N.; Hidaka, T.; Sugiyama, H. Therapeutic gene regulation using pyrrole–imidazole polyamides. Adv. Drug. Deliv. Rev. 2019, 147, 66–85. [Google Scholar] [CrossRef]
- Igarashi, J.; Fukuda, N.; Inoue, T.; Nakai, S.; Saito, K.; Fujiwara, K.; Matsuda, H.; Ueno, T.; Matsumoto, Y.; Watanabe, T.; et al. Preclinical study of novel gene silencer pyrrole-imidazole polyamide targeting human TGF-β1 promoter for hypertrophic scars in a common marmoset primate model. PLoS ONE 2015, 10, e0125295. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Baylin, S.B.; Ohm, J.E. Epigenetic gene silencing in cancer—A mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 2006, 6, 107–116. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Nepali, K.; Liou, J.P. Recent developments in epigenetic cancer therapeutics: Clinical advancement and emerging trends. J. Biomed. Sci. 2021, 28, 27. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, S. Cancer’s epigenetic drugs: Where are they in the cancer medicines? Pharm. J. 2020, 20, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Licht, J.D.; Bennett, R.L. Leveraging epigenetics to enhance the efficacy of immunotherapy. Clin. Epi. 2021, 13, 115. [Google Scholar] [CrossRef]
- Shen, J.Z.; Qiu, Z.; Wu, Q.; Finlay, D.; Garcia, G.; Sun, D.; Rantala, J.; Barshop, W.; Hope, J.L.; Gimple, R.C.; et al. FBXO44 promotes DNA replication-coupled repetitive element silencing in cancer cells. Cell 2021, 184, 352–369. [Google Scholar] [CrossRef] [PubMed]
- Griffin, G.K.; Wu, J.; Vellve, A.I.; Patti, J.C.; Hsu, J.; Davis, T.; Oni, D.D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef]
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Esteller, M.; Lee, S.H.; Choi, J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019, 10, 4278. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.C.; Pasero, P.; Vinay, S.P. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef]
- Eckerman, K.F.; Endo, A. MIRD: Radionuclide, Data and Decay, Schemes, 2nd ed.; Society of Nuclear, Medicine: Reston, VA, USA, 2008. [Google Scholar]
- Ku, A.; Facca, V.J.; Cai, Z.; Reilly, R.M. Auger electrons for cancer therapy—A review. EJNMMI Radiopharm. Chem. 2019, 4, 27. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Stage of Investigation | Category | Drug | Cancer Type | Year | Country | Notes | Reference |
|---|---|---|---|---|---|---|---|
| Clinical (adjuvant use) | antimetabolite | 5-[125I]iodo-2’-deoxyuridine | Recurrent and advanced pancreatic cancer relapsed with cytologically proven neoplastic meningitis | 2008 | USA | Intrathecal administration as adjuvant treatment (1850 MBq). Feasible without acute neurological toxicity. Seemed to have produced a biological response. | [16] |
| Clinical (dose escalation study) | antibody | 125I-labeled mAb A33 | Advanced chemotherapy-resistant colon cancer | 1996 | USA | Phase 1/2 study to determine the maximum tolerated dose (21 patients with doses up to 350 mCi/m2). No bowel or bone marrow toxicity, modest antitumor activity. | [17] |
| Clinical (adjuvant use) | 125I-labeled MAb 425 (targeting EGFR) | High-grade glioma | 1990~2004 | USA | Significant increase in median survival as an adjuvant treatment following initial surgery and postoperative external beam radiation therapy | [18] | |
| Clinical (dose escalation study) | somatostatin receptor targeted peptide | [111In-DTPA0]octreotide | Somatostatin receptor-positive neuroendocrine tumors | 2002 | The Netherlands | 50 patients (cumulative doses of 20 to 160 GBq). Therapeutic effects were seen in 21 patients: partial remission (1), minor remissions (6), and stabilization of previously progressive tumors (14). | [19] |
| Clinical (dose escalation study) | 111In-labeled pentetreotide | Progressive, disseminated, and unresectable neuroendocrine tumor (stage III and IV) | 2012 | USA | 112 patients, 500 mCi (18.5 GBq) per cycle and ~4 cycles. Majority (85%) of patients had stable disease. No significant acute toxicity, but grade II/III toxicity was observed. | [20] | |
| Clinical (dose escalation study) | epidermal growth factor | [111In]In-DTPA-hEGF | EGFR-positive metastatic breast cancer | 2014 | Canada | 16 patients with doses of up to 2290 MBq. No objective antitumor responses; most common adverse events were flushing, chills, nausea, and vomiting occurring during or immediately post-injection. | [21] |
| Preclinical (animal) | modular nanotransporter (MNT) targeting EGFR | [111In]In-NOTA-MNT | EGFR-overexpressing tumor xenograft (human bladder cancer) | 2018 | Russia | Significant dose-dependent tumor growth delay (up to 90% growth inhibition) after local infusion of 111In-NOTA-MNT in EJ xenograft-bearing mice. | [22] |
| Preclinical (animal) | antibodies (35A7 mAbs: anti-CEA; m225 mAbs: anti-HER2) | 125I-labeled 35A7 mAb (non-internalizing), 125I-labeled m225 mAb (internalizing) | Vulvar squamous carcinoma cell line A-431 tumor xenograft | 2009 | France | Compared with unlabeled mAb, non-internalizing 125I-labeled 35A7mAb had a significant increase in survival. | [23] |
| Preclinical (animal) | PARP inhibitor | [125I]PARPi-01 | Triple-negative breast cancer cell line MDA-MB-231 tumor xenograft | 2022 | Germany, The Netherlands | Endogenous therapy induced a significant delay in tumor growth. No significant survival advantage, but significantly higher apoptosis ratio, no radiotoxicity in liver or thyroid. | [24] |
| Preclinical (cell) | PARP inhibitor | 125I-labeled KX1 | High-risk neuroblastoma cell line | 2020 | USA | 125I-labeled KX1 was approximately twice as effective as 131I-labeled KX1 | [25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obata, H.; Ogawa, M.; Zalutsky, M.R. DNA Repair Inhibitors: Potential Targets and Partners for Targeted Radionuclide Therapy. Pharmaceutics 2023, 15, 1926. https://doi.org/10.3390/pharmaceutics15071926
Obata H, Ogawa M, Zalutsky MR. DNA Repair Inhibitors: Potential Targets and Partners for Targeted Radionuclide Therapy. Pharmaceutics. 2023; 15(7):1926. https://doi.org/10.3390/pharmaceutics15071926
Chicago/Turabian StyleObata, Honoka, Mikako Ogawa, and Michael R. Zalutsky. 2023. "DNA Repair Inhibitors: Potential Targets and Partners for Targeted Radionuclide Therapy" Pharmaceutics 15, no. 7: 1926. https://doi.org/10.3390/pharmaceutics15071926
APA StyleObata, H., Ogawa, M., & Zalutsky, M. R. (2023). DNA Repair Inhibitors: Potential Targets and Partners for Targeted Radionuclide Therapy. Pharmaceutics, 15(7), 1926. https://doi.org/10.3390/pharmaceutics15071926