Abstract
The management of inherited neuropathies relies mostly on the treatment of symptoms. In recent years, a better understanding of the pathogenic mechanisms that underlie neuropathies has allowed for the development of disease-modifying therapies. Here, we systematically review the therapies that have emerged in this field over the last five years. An updated list of diseases with peripheral neuropathy as a clinical feature was created based on panels of genes used clinically to diagnose inherited neuropathy. This list was extended by an analysis of published data by the authors and verified by two experts. A comprehensive search for studies of human patients suffering from one of the diseases in our list yielded 28 studies that assessed neuropathy as a primary or secondary outcome. Although the use of various scales and scoring systems made comparisons difficult, this analysis identified diseases associated with neuropathy for which approved therapies exist. An important finding is that the symptoms and/or biomarkers of neuropathies were assessed only in a minority of cases. Therefore, further investigation of treatment efficacy on neuropathies in future trials must employ objective, consistent methods such as wearable technologies, motor unit indexes, MRI or sonography imaging, or the use of blood biomarkers associated with consistent nerve conduction studies.
1. Introduction
Hereditary neuropathies are a broad range of diseases that differ in terms of inheritance (dominant, recessive, X-linked, or mitochondrial), electrophysiological features (demyelinating, axonal, or intermediate), and clinical properties. Indeed, neuropathy may be the primary feature of a disease, such as in Charcot–Marie–Tooth disease (CMT), hereditary sensory and autonomic neuropathies (HSAN), or hereditary motor neuropathies (HMN), in which patients may present different levels of muscle atrophy and weakness, sensory and autonomic disturbances, and/or skeletal deformities. On the other hand, neuropathy may be part of a more complex systemic disease, as in peroxisomal, lysosomal, or mitochondrial diseases, for which neuropathy can be the first symptom but can also remain subclinical, making diagnosis more challenging [1,2,3]. The global prevalence of inherited neuropathies is unknown, but CMT has an estimated prevalence of 1/2500, making it the most common inherited disorder of the peripheral nervous system. The management of inherited neuropathies relies mostly on symptomatic treatments such as physiotherapy, analgesics, or surgery [4]. In recent years, a better understanding of the pathological mechanisms underlying some of these diseases has allowed the development of disease-modifying therapies or diets, although these treatments have been validated for a very small subset of diseases [2,5,6]. Well-conducted reviews have been published regarding the management of inherited neuropathies [7,8]; however, the ever-growing landscape of these diseases means that these reviews are outdated. We conducted a review of the recent literature to identify approaches for the treatment of primary hereditary neuropathies and also more complex systemic diseases with peripheral neuropathy as a clinical feature. Our systematic review demonstrates that patients with certain inherited neuropathies benefit from specific treatment and that objective, consistent methods for the evaluation of responses to treatment are needed.
2. Methods
2.1. Compilation of List of Inherited Neuropathies
Two authors (MH and A-MS) identified genes used clinically to determine inherited neuropathies by merging genes from panels used in the UK (PanelApp) [9], France (Paris-Saclay) [10], and the USA (Mayo Clinic) [11]. These genes were merged into one list and linked to specific diseases using Orphanet [12] and Genecards [13] databases. The list was then extended based on the expertise of the authors and two experts (Dr Tania Stojkovic, Nord/Est/Ile-de-France Neuromuscular Reference Center, Institut of Myology, Pitié-Salpêtrière Hospital, APHP, Sorbonne University, Paris, France, and Dr Isabelle Lievens, Neuromuscular Reference Center, Department of Neurology, University Hospital Liège and University of Liège, Liège, Belgium). Finally, our results were crossed with existing published data from Fernandez-Eulate et al. [2], Rossor et al. [1], Masingue et al. [3], and Finsterer et al. [14]. Appendix A provides the list of genes associated with inherited neuropathies.
2.2. Literature and Clinical Trial Database Searches
The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) checklist and flow diagram were followed for the design and reporting of this work, as detailed below. Two independent authors (MH and A-MS) conducted a systematic search of Ovid Medline [15] on 24 February 2023 for original, full-text articles published in English after 1 January 2018. The studies that were included described the assessment of treatments in any of the inherited neuropathies identified (Appendix A) with a clinical, biological, or electrophysiological outcome. Non-pharmacological interventions such as physiotherapy, genetic counselling, or surgery were excluded. Retrospective observational studies were not included. A search was also carried out using the Clinicaltrial.gov [16] database on 24 February 2023. Database-specific filters were applied to ensure that only interventional studies completed with results published after 1 January 2018 were included and that studies describing diseases such as cancer, carpal tunnel, amyloid cardiomyopathy, X-linked cerebral adrenoleukodystrophy, and bone assessment in Gaucher disease were not included. Finally, relevant articles were extracted from the reference sections of systematic or expert reviews and meta-analyses. The search strategy is shown schematically in Figure 1. A table displaying information about the diseases for which clinical trials were identified is presented in Appendix B.
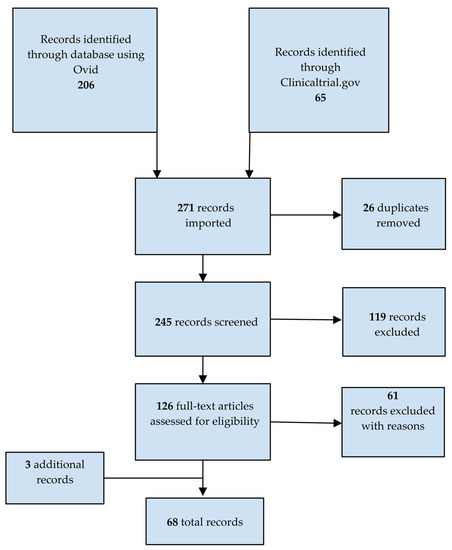
Figure 1.
PRISMA flowchart of studies identified as relevant to the treatment of inherited neuropathies.
2.3. Selection of Studies
Two researchers (MH and A-MS) independently screened titles and abstracts and then full texts for eligibility. To be included, studies had to meet the following criteria: (1) the study was performed on human patients, (2) patients had been diagnosed with one of the diseases included in our list (Appendix A), (3) a pharmacological treatment or diet was evaluated, and (4) the results were presented. No restriction was applied regarding gender, age at onset, severity, ethnicity, or phenotypic features. The two reviewers compared their findings and potential disagreements were resolved by consensus.
2.4. Data Extraction and Analysis
For each study that fulfilled our criteria, information was collected on the study (authors, year of publication, type, duration), the population (sample, specific characteristics where applicable), the treatment (drug/dietary supplement name, dosing and administration, mechanism), and the outcome (clinical, biological, or electrophysiological). Clinical and biological outcomes that were related to symptoms other than neuropathies were not extracted. All biomarkers were included as they were considered a way of attesting to the disease-modifying aspect of treatments. Only the most recent updates on pilot projects were considered. Eligible papers were assessed for risk of bias using the Jadad score (score ranging from 0 to 5 according to whether the study was described as randomized and double-blinded and including a thorough description of withdrawals) [17] (Appendix C). Data extraction was carried out by MH, reviewed by A-MS, and then reviewed by LS. Due to differences in clinical and biological outcomes, meta-analysis was precluded.
3. Results
3.1. Identification of Studies That Assessed Treatments for Inherited Neuropathies
We first compiled a list of genes associated with neuropathies. Based on a literature and clinical trials database search for studies of pharmacological treatments or dietary interventions in patients diagnosed with one of the diseases identified in our list (Appendix A), we identified 271 studies, from which 26 duplicates were removed. Of the remaining studies, 119 were excluded based on titles and abstracts. The remaining 126 studies were read in full by two assessors, resulting in the exclusion of 61 studies. Three additional relevant studies were captured from the reference sections of systematic or expert reviews and meta-analyses. In the end, 68 studies met our criteria (Figure 1). Among these, only 28 assessed neuropathy as a primary or secondary outcome. These studies assessed 14 different drugs in nine inherited neuropathies (Table 1).

Table 1.
Summary of clinical trial results in which neuropathy was assessed as a primary or secondary outcome.
Moreover, 40 articles that assessed biomarkers or neurological clinical aspects potentially related to neuropathy were identified (Table 2). Changes in disease-related biomarkers or the modification of neurological features such as balance, gait, strength, or dexterity can be indicative of therapeutic efficacy, even if neuropathy was not assessed as a primary or secondary outcome. The inclusion of biomarkers or neurological clinical aspects potentially related to neuropathy allowed us to capture early-phase, proof-of-concept studies.
Table 2.
Summary of clinical trial results in which neuropathy was not assessed as a primary or secondary outcome.
Appendix B lists modes of inheritance, clinical presentation, and standards of care for each disease for which a relevant clinical trial was identified.
3.2. Treatments for Conditions Where Neuropathy Is the Sole or Predominant Feature of the Disease
3.2.1. Familial Dysautonomia
A randomized, placebo-controlled, crossover phase 2 study of the effect of DOPA decarboxylase inhibitor carbidopa was conducted in 22 patients with familial dysautonomia [41]. At the end of the three 4-week treatment periods, the two co-primary endpoints, reduction in systolic blood pressure variability and systolic blood pressure peaks, were met. Although this is a symptomatic treatment, this could improve quality of life.
3.2.2. Hereditary Sensory and Autonomic Neuropathy Type 1 (HSAN1)
A randomized, placebo-controlled, phase 1/2 study of L-serine with an open-label extension (OLE) was conducted on 18 patients with HSAN1 [42]. The total duration of the trial was 2 years. This study showed a non-significant improvement in the Charcot–Marie–Tooth Neuropathy Score (CMTNS) compared to placebo, a significant reduction in the neurotoxic deoxysphinganine levels, and evidence of reinnervation at 1 year from distal biopsies. No significant electrophysiological differences were detected in the tested nerves after 1 year; however, there was a paucity of recordable responses at baseline, and the statistical power of the study was limited by the small sample size. Further studies are needed to determine the efficacy of this potentially disease-modifying treatment.
3.2.3. Charcot–Marie–Tooth 1A (CMT1A)
After a favourable phase 2 trial [94], a randomized, double-blind, placebo-controlled phase 3 study was conducted to evaluate the efficacy and safety of a high- and low-dose combination of baclofen, naltrexone, and sorbitol (PTX3003) in 323 CMT1A patients over 15 months. Although crystal formation in the high-dose formulation led to early discontinuation, significant clinical improvements were observed in this group compared to the placebo. In the ongoing open-label continuation study, participants are being treated with twice the volume of the low-dose formulation instead of the high dose to limit this stability issue [43].
3.3. Treatments for Conditions Where Neuropathy Is One Symptom of the Disease
3.3.1. Hereditary Transthyretin Amyloidosis Polyneuropathy (ATTRv-PN)
Transthyretin (TTR) stabilizers: The safety and efficacy of tafamidis in delaying neurologic disease progression in patients with early-stage ATTRv-PN (stage 1) have been demonstrated in several clinical trials, extension studies [95], and around 10 years of clinical follow-up [18]. Tafamidis was approved in Europe in 2011 and subsequently in several other countries for the treatment of stage 1 ATTRv-PN. This drug has not been approved in the USA for this condition due to a lack of robust efficacy in clinical trials [21].
Tolcapone is another TTR stabilizer approved for clinical use in the treatment of Parkinson’s disease. It is currently under development to treat ATTRv-PN and other forms of ATTR, including leptomeningeal amyloidosis, due to its ability to cross the blood–brain barrier [56]. It was shown to have efficacy in a phase 2 study of subjects with ATTRv-PN [55].
Acoramidis (AG10) is an orally bioavailable, selective TTR stabilizer that is currently being assessed for effects on mortality and hospitalization for cardiovascular causes in subjects with transthyretin amyloid cardiomyopathy in an ongoing phase 3 trial, with topline data expected in mid-2023. The phase 3 study in ATTRv-PN patients was terminated by the sponsor after a review of the currently available treatments [53,96,97].
Gene silencers: Inotersen, which is an antisense oligonucleotide that targets TTR mRNA for degradation, proved its efficacy in a 15-month, randomized, placebo-controlled, double-blind phase 3 trial conducted on 172 adult patients with stage 1 or 2 ATTRv-PN [31,32,33,34]. Glomerulonephritis occurred in three patients and thrombocytopenia occurred in three patients treated with inotersen, with one death associated with grade 4 thrombocytopenia [31]. In the OLE, an update at 3 years showed a sustained efficacy compared with natural history, regardless of the mutation, prior treatment, or stage of the disease (except in the group treated first with the placebo and then inotersen in stage 2). In the OLE, no cases of acute glomerulonephritis were reported, but approximately 50% of patients in both the group treated first with the placebo and then with inotersen and the group continuously treated with inotersen experienced thrombocytopenia (<100 × 109/L), although none had a grade 4 platelet count decrease [36]. Narayanan et al. [98] reported that patients with grade 4 thrombocytopenia in the phase 3 trial had higher levels of proinflammatory cytokines at baseline, which suggests a predisposition to immune-mediated thrombocytopenia via antiplatelet IgG antibodies. A post hoc analysis demonstrated that the higher health-related quality of life of patients treated with inotersen compared to placebo-treated subjects during the pivotal study was sustained through week 104 of the OLE study, highlighting the importance of early treatment [37].
Eplontersen is an antisense oligonucleotide targeting TTR mRNA that is conjugated to N-acetylgalactosamine, a ligand for a receptor expressed on hepatocytes, which improves distribution to these liver cells compared to unconjugated oligonucleotides. Two randomized, double-blind, placebo-controlled phase 1 studies showed greater TTR reduction compared to inotersen (88% at 45 mg q4w relative to inotersen 74% at 300 mg weekly) [51]. The efficacy and safety of eplontersen are currently being investigated in phase 3 trials.
Patisiran is a small interfering RNA (siRNA) that targets TTR mRNA for degradation through the endogenous RNA interference pathway to reduce the expression of TTR [22]. The efficacy and safety of patisiran were assessed in an 18-month randomized, placebo-controlled, double-blind phase 3 trial in 225 adult patients with a polyneuropathy disability score of IIIb or lower. Positive effects were seen across all subgroups, irrespective of age, sex, race, body weight, mutation, prior use of tafamidis, and mild/moderate renal or hepatic involvement [24]. Most of the adverse events were mild or moderate and the rate of serious adverse events was similar between the patisiran-treated and placebo groups [22]. Another 24-month phase 2 OLE study confirmed this favourable safety and efficacy profile, with concomitant tafamidis or diflunisal use [25]. A phase 3b open-label trial proved efficacy and tolerability in subjects with ATTRv-PN after liver transplantation, and no apparent drug interactions were observed between patisiran and immunosuppressive treatments [27].
Vutrisiran is another siRNA that reduces the synthesis of variant and wild-type transthyretin. Unlike patisiran, it is given subcutaneously and hence does not require premedication. In a phase 3 open-label study, vutrisiran reduced TTR levels to an extent similar to patisiran and was more efficacious than patisiran in multiple assessments. Vutrisiran was well tolerated and most adverse events were consistent with an expected ATTRv natural history progression [29]. A randomized treatment extension period is currently ongoing in which patients receive vutrisiran injections once every 6 months or once every 3 months.
Gene editing: NTLA-2001 is a gene-editing therapy based on CRISPR-Cas9. The two active components of this therapeutic are the mRNA encoding the Cas9 endonuclease and a single guide RNA complementary to the gene encoding TTR. These components are encapsulated in a lipid nanoparticle that enables targeted delivery to hepatocytes. The interim results of an ongoing phase 1 study (NCT04601051) in six patients with ATTRv-PN showed a reduction in TTR concentration in a dose-dependent manner after a single intravenous dose. In terms of safety, all the adverse events occurring during or after treatment were mild in severity. Increased D-dimer levels were noticed 4 to 24 h after infusion in five of the six patients but values returned to baseline after 7 days [54,97].
Comparison of approved ATTRv-PN treatments: No head-to-head comparisons of the therapeutics approved for the treatment of ATTRv-PN have been made. Indirect comparisons based on available data suggest that patisiran has a greater effect on neuropathy and quality of life than inotersen or tafamidis [99,100]. The choice of treatment should be based on the physician’s expertise, considering the disease’s stage and phenotype and country-specific recommendations. Further questions that need to be addressed include the optimal timing of treatment initiation or whether combinations of treatments might be more efficacious than single agents.
3.3.2. Metachromatic Leukodystrophy (MLD)
Enzyme replacement therapy (ERT) with recombinant human arylsulfatase A (rhASA) has been tested in two phase 1/2 open-label trials in subjects with MLD, one with intravenous (IV) delivery and one with intrathecal (IT) delivery [44,45]. The study assessing the IV delivery of ERT was conducted in a cohort of 13 children. The trial lasted 52 weeks, and there was an additional 24-month extension period. The study with the IT delivery of ERT, on the other hand, enrolled 24 children and lasted for 38 weeks. The ongoing extension study should be completed by December 2024 (NCT01887938). Both ERT dosing routes resulted in a reduction in cerebrospinal fluid (CSF) sulfatide levels and no serious drug-related adverse events, but a clinical improvement was not demonstrated and electrophysiological parameters did not change compared to baseline. The IV study was terminated after 24 months due to a lack of efficacy, suggesting that the recombinant protein may not cross the blood–brain barrier in therapeutic quantities. In the study evaluating the IT delivery of ERT, 10 patients developed anti-rhASA antibodies in serum, with seven having an in vitro neutralizing status, but no correlation was found with the occurrence of adverse events. Anti-rhASA antibodies were also detected in CSF in some patients but a neutralizing status has not been determined.
A gene therapy, atidarsagene autotemcel (arsa-cel), has also been tested in subjects with MLD. The therapeutic is an autologous haematopoietic stem and progenitor cell (HSPC) population transduced ex vivo with a lentiviral vector encoding ASA. This novel approach was evaluated in a phase 1/2 study conducted on 29 children with pre-symptomatic or early-symptomatic early-onset MLD compared to a natural history cohort [46]. Promising results were obtained, such as increased ARSA activity and considerable clinical improvement. Some patients were found to have gross motor development similar to healthy children. There were significant differences in nerve conduction velocities between treated patients with late-infantile MLD and the natural history cohort. In terms of safety, arsa-cel was well tolerated with busulfan conditioning. Four patients developed anti-ARSA antibodies but this did not affect clinical outcomes.
3.3.3. Spinal and Bulbar Muscular Atrophy (SBMA or Kennedy Disease)
Patients with SBMA may experience myotonia-like symptoms under cold exposure. It has been hypothesized that this is linked to sodium channel dysfunction in skeletal muscles [101]. Yamada et al. [49] conducted an observational study on 51 SBMA patients and noticed that ulnar nerve distal latency was prolonged under cold exposure compared to healthy controls (p < 0.001), and this was correlated with grip strength. They then conducted a randomized, placebo-controlled, double-blind study on 20 subjects over 4 weeks to evaluate mexiletine hydrochloride. Statistically significant improvements in some clinical parameters were observed, although there was not a significant change in nerve conduction velocity. No serious adverse events were reported. Further trials are warranted to evaluate the symptomatic effect of mexiletine hydrochloride on motor function.
Insulin-like growth factor 1 (IGF-1) stimulates Akt-mediated phosphorylation of the androgen receptor, which promotes its clearance. BVS857 is a pegylated IGF-1 mimetic with a longer half-life than endogenous IGF-1. The safety and efficacy of BVS857 were evaluated in a randomized, placebo-controlled trial in 27 adults that lasted 12 weeks. The subcutaneous delivery of BVS857 resulted in erythema at the injection site and low and variable exposure. Thus, IV delivery was employed in phase B of the study. Five participants developed neutralizing antibodies to endogenous IGF-1, but this did not have a clinical or pharmacokinetic impact. The half-life of BVS857 was less than theorized, barely 24 h. Nevertheless, a significant increase was observed in thigh muscle volume over the short period of the study, although this was not correlated with the improvement of any of the motor functional measures [90].
3.3.4. Adult Polyglucosan Body Disease (APBD)
Polyglucosan bodies were detected in the astrocytes of a patient with confirmed glycogen branching enzyme (GBE) deficiency [102]. Therefore, it is believed that the accumulation of abnormal glycogen in astrocytes leads to a lack of energy substrates for neurons and is responsible for certain symptoms of APBD. Due to its anaplerotic properties, triheptanoin was tested in a randomized, cross-over, placebo-controlled trial on 23 participants with APBD over 1 year (6 months on treatment and 6 months on placebo), followed by a 4-year OLE. Neither the 6 min walk test (6MWT) nor the secondary endpoints were met at the end of the study. However, two patients who were mildly disabled at baseline remained clinically stable after treatment discontinuation, as assessed by the 6MWT and Expanded Disability Status Scale (EDSS), and this was associated with stable nerve conduction studies under treatment. This study confirmed the safety profile of triheptanoin, but further investigations are needed to evaluate its effect on peripheral nerve deficits [50].
3.3.5. Fabry Disease (FB)
The mechanisms underlying the neuropathy experienced by FB patients are not fully understood, but globotriaosylceramide (Gb-3) has been demonstrated to accumulate in dorsal root ganglia [103,104], myelinated axons [105], and microvascular endothelial cells, which can result in ischemic axonal degeneration [106]. These data suggest that Gb3 drives peripheral neuron dysfunction. Two approved ERTs (agalsidase alfa and beta) have shown favourable effects on peripheral neuropathies and reduce Gb3 and globotriaosylsphingosine (lyso-Gb3), especially when initiated soon after the onset of symptoms [107,108,109]. Lower dose regimens of agalsidase beta were tested in a paediatric cohort by Ramaswami et al., but it did not show consistent benefit, and the approved dose of 1 mg/kg/2-weekly was supported by this trial [62]. More recently, a new agalsidase beta (ISU303) with an almost identical structure to Fabrazyme was developed by ISU Abxis. This alternative agalsidase beta is of lower cost, but close monitoring is required to ensure its quality, safety, and efficacy [63]. A PEGylated ERT, pegunigalsidase alfa, was developed with the aim of increasing plasma half-life and reducing immunogenicity. A 1-year, dose-ranging, open-label, phase 1/2 trial (NCT01981720) of the PEGylated compound was conducted on 18 adult FB patients [64]. One patient was withdrawn from the study due to bronchospasm after the first infusion, and three patients developed treatment-induced antibodies, but this did not influence the efficacy or safety. The results of the 60-month extension study were recently published on clinicaltrial.gov, but no peer-review publications were available at the time of this review.
Migalastat is an approved orally administered chaperone that enhances the activity of endogenous patients’ alpha-galactosidase-A. It is only suitable for selected patients with certain mutations, as determined by an in vitro assay [110]. Results from the ATTRACT trial, an 18-month randomized, open-label, active-controlled trial and its 12-month OLE showed a relatively safe profile among adult FB patients after a switch from an ERT to migalastat. Migalastat had comparable or superior effects on renal function, reducing cardiac mass and FB-related clinical events, compared to the ERT. The effect on neuropathy was not monitored in this trial. Interestingly, plasma lyso-Gb3 levels remained low from baseline to month 30 in migalastat-treated subjects. White blood cell alpha-galactosidase-A activity in male patients increased in the group treated with migalastat during the randomized period but remained stable in the group treated with the ERT [58,59]. The efficacy of migalastat in males with classic phenotypes was confirmed in the phase 3 FACETS trial and its extension study, but, again, the impact on neuropathy was not assessed [60].
Venglustat, a substrate reduction therapy, was assessed in a 26-week open-label phase 2 study and its 130-week extension study in 11 ERT-naïve adult male patients with classic FB phenotypes. Biomarkers all decreased upon treatment with venglustat, but the effect on pain was not significantly sustained at the end of the study and specific neuropathy endpoints were not assessed [61]. Phase 3 trials are ongoing.
3.3.6. Acute Intermittent Porphyria (AIP)
A siRNA that silences the expression of the gene encoding ALAS1 has been approved for the treatment of AIP [111]. The siRNA, givosiran, prevents the accumulation of delta-aminolevulinic (ALA) and porphobilinogen (PBG), which are intermediates in heme synthesis. In the ENVISION trial, givosiran treatment resulted in sustained reductions in hepatic ALAS1 mRNA and urinary ALA and PBG levels and annualized attack rate compared to placebo [74,76]. Givosiran had an acceptable safety profile, although two patients discontinued the study because of increased homocysteine levels and one because of alanine aminotransferase levels greater than eight times the upper limit of normal. Small decreases in eGFR observed early in therapy stabilized over months 12 to 24, and no patients discontinued givosiran due to renal events during the extension period [75]. By lowering the annualized attack rate, givosiran reduces the probability that patients will develop acute motor neuropathy during an attack or more chronic neuropathies that appear in subjects not treated promptly or with recurrent attacks [112].
3.4. Treatments for Conditions Where Neuropathy Plays a Role in Ataxia
3.4.1. Spinocerebellar Ataxia Type 38 (SCA 38)
It has been shown that mutations in ELOVL5 are responsible for a reduction in serum docosahexanoic acid (DHA) in patients with SCA 38 [113]. Oral DHA given to nine patients in a 2-year OLE study showed clear clinical benefits and nerve conduction velocities were stable during the treatment period [40]. To determine whether DHA has an effect on neuropathy, additional information, such as analyses of compound muscle action potential and sensory nerve action potential, which are relevant considering the predominantly axonal nature of the polyneuropathy experienced by SCA 38 patients [114], and a longer follow-up are needed.
3.4.2. Spinocerebellar Ataxia Type 2 (SCA2)
Two randomized, placebo-controlled, double-blind studies were recently conducted in SCA2 cohorts. The first assessed the safety and efficacy of riluzole, which had previously shown benefits in inherited ataxias [115]. The study was conducted over one year in 45 moderately affected adult patients; riluzole treatment did not result in any improvement compared to placebo [65]. The second study enrolled 34 mildly or moderately affected Cuban adult patients to test the efficacy and safety of nasally administered human recombinant erythropoietin (EPO). Supporting the utility of this treatment, endogenous EPO is abnormally low in the cerebrospinal fluid (CSF) of SCA2 patients [116], and EPO has been shown to have an additional neurotrophic role [117]. The proportion of high responders in the spinocerebellar ataxia functional index (SCAFI ≥ 0.75), which is a composite score of motor performances, was slightly higher among the EPO-treated patients than those given a placebo despite a considerable placebo effect. Moreover, the drug was relatively well tolerated including in terms of erythropoiesis activity [66]. Further studies are warranted to evaluate the effect of EPO on neuropathy.
3.4.3. Ataxia-Telangiectasia (A-T)
The efficacy of low-dose betamethasone was studied in a 2-year open-label study of patients with A-T. Although transient efficacy was observed after 6 months, sustained benefits were not observed at the end of the trial [87]. Although it was not used in this specific trial, the encapsulation of steroids within autologous erythrocytes (EryDex) allows a slow release for up to 1 month. This novel encapsulation approach was used with dexamethasone sodium phosphate in a phase 3, randomized-controlled, double-blind trial (ATTeST) in A-T patients and showed clear clinical benefits after 6 months [88].
3.4.4. X-Linked Adrenoleukodystrophy (X-ALD)
Redox imbalance plays an important role in the pathogenesis of X-ALD, and this is the reason why a combination of antioxidants (α-tocopherol, N-acetylcysteine, and α-lipoic acid) was evaluated in a pilot open-label phase 2 study in 13 subjects [48]. In patients with X-ALD, there is a negative correlation between the levels of the chemokine MCP1 and the pro-inflammatory metabolite 15-hydroxyeicosatetraenoic with 6MWT, hence these oxidative damage markers are considered predictors of disease progression. Treatment with the combination of antioxidants resulted in a considerable decrease in all oxidative damage markers and pro-inflammatory markers and significant clinical improvements. Nerve conduction velocities were stable after 1 year in treated patients, whereas the conduction time of motor-evoked potential decreased, suggesting an improvement in upper motor neuron function. Laser-evoked potentials increased in two-thirds of treated patients. Although these positive effects do not seem to be linked to an impact on neuropathy, further testing of antioxidants is warranted.
ADVANCE was a randomized, double-blind, phase 2/3 study evaluating the safety and efficacy of oral leriglitazone in 116 men with X-ALD who had adrenomyeloneuropathy for a total of 96 weeks [89]. A change from baseline in the 6MWT, the primary outcome measure, was not observed. Therefore, the hierarchical testing of all secondary endpoints was not performed. Moreover, adverse effects, including weight gain and peripheral oedema, were observed. In post hoc subgroup analyses, patients with early-stage disease who were treated with leriglitazone had less decline in the 6MWT and in EDSS scores relative to those treated with a placebo at week 96. Finally, no leriglitazone-treated patients developed cerebral adrenoleukodystrophy (CALD), demonstrating that the drug may slow the progression of CALD. An OLE is ongoing (NCT03231878).
3.4.5. Friedreich Ataxia (FRDA)
Multiple trials have been conducted in subjects with FRDA in recent years, and our search yielded seven publications since 2018, assessing six different drugs. Most trials used the Friedreich Ataxia Rating Scale (FARS) or modified FARS (mFARS), 9-hole peg test (9HPT), and 25 or 8 min walk tests to assess the efficacies of the drugs. However, none of these tests are specific to neuropathy progression. For example, the mFARS omits the peripheral nervous system subscore section of the FARS. Three drugs, luvadaxistat [81], (+)-epicatechin [80], and IFN-γ1b [78] did not demonstrate efficacy in terms of neurological outcomes. EPI-743 treatment was associated with a statistically significant improvement in neurological function and disease progression relative to a natural history cohort and also showed clinically meaningful improvement in the FARS-neuro at 6 months [79]. RT001 improved peak workload relative to baseline in a double-blind phase 1/2 study, and there was not a significant difference in FARS-neuro scores between the drug and comparator over a short period of 28 days [84]. Omaveloxolone statistically improved mFARS over placebo in a 12-week, dose-ranging study and in an international, double-blind, randomized, placebo-controlled, phase 2 trial conducted on 103 patients with a daily dose of 150 mg [82,83]. This drug has been approved by the FDA in 2023 but its efficacy on neuropathy is unknown.
3.4.6. Fragile-X Associated Tremor/Ataxia Syndrome (FXTAS)
An open-label phase 2 trial was conducted on 10 FXTAS patients to assess the safety and efficacy of citicoline on motor and cognitive functions over a 1-year treatment period. There were no significant changes in FXTAS rating scale scores in treated subjects, although worsening was expected in this population. Moreover, most of the secondary outcome measures remained stable, and citicoline was well tolerated [93]. These findings suggest that citicoline may stabilize disease progression, but a larger study will be necessary to confirm these results.
3.5. Treatments for Conditions Where Neuropathy Is Inconsistent and/or Subclinical
3.5.1. Acid Sphingomyelinase Deficiency (ASMD)
ASMD, also known as Niemann–Pick disease, is a lysosomal storage disease. Olipudase alfa is a human recombinant acid sphingomyelinase. IV administration is used in non-central nervous system manifestations as it does not cross the blood–brain barrier. Two open-label studies assessed this therapy in ASMD patients. In one study, five adults were treated for 30 months [92], and the other enrolled 20 paediatric patients with chronic ASMD [91]. Both studies showed a reduction in biomarkers and benefits in non-neurological parameters; however, neuropathy was not assessed. No adult patients developed antidrug antibodies, but antidrug antibodies were detected in eight paediatric patients. None tested positive for neutralizing antibodies that would interfere with enzyme uptake into cells, although one tested transiently positive for the inhibition of enzyme catalytic activity. One of the patients had an anaphylactic reaction but continued with treatment after desensitization. Further studies are required to evaluate the effects of olipudase alfa on neuropathy.
3.5.2. Mitochondrial Encephalomyopathy with Lactic Acidosis and Stroke-like Episodes (MELAS)
Three peer-reviewed publications were found that describe the testing of amino acids L-arginine [67], taurine [69], and glutamine [68] in subjects with MELAS. None of these studies assessed neuropathy as a primary or secondary endpoint. Neither of the two open-label phase 3 studies conducted in Japan that evaluated taurine and L-arginine demonstrated improvement in treated subjects based on the Japanese Mitochondrial Disorder Rating Scale, but the frequencies of stroke-like episodes were decreased by treatment, and both amino acids had good safety profiles. In a study of the effects of glutamine, Guerrero-Moline et al. [68] showed that high-dose oral supplementation resulted in a decrease in CSF glutamate levels and an increase in glutamine levels in subjects with MELAS. Thus, L-arginine, taurine, and glutamine have potential and should be further evaluated.
3.5.3. Gaucher Disease Type 1 (GD1)
Although GD1 is usually considered to be non-neuronopathic, there is growing evidence that peripheral nervous system manifestations are part of the clinical spectrum of this disease [14,118]. However, the aetiology of polyneuropathy associated with GD1 is unclear. It is not known if the imbalance in calcium homeostasis seen in the central nervous system of certain patients with GD1 [119] is also to blame for peripheral nerve injuries. Therefore, no biomarkers of peripheral nervous system involvement in GD1 have been validated.
Several therapies have been approved for the treatment of GD1. Three are ERTs: the recombinant β-glucocerebrosidases imiglucerase, velaglucerase alfa, and taliglucerase alfa. Another approach is substrate reduction therapy with glucosylceramide synthase inhibitors miglustat and eliglustat [120]. None of the identified trials assessed neuropathy as an outcome. In the phase 3 placebo-controlled, double-blind ENGAGE trial, eliglustat led to a reduction in several biomarkers, including median chitotriosidase, glucosylceramide (the primary sphingolipid that accumulates in GD1), glucosylsphingosine (a sphingolipid that is a highly specific validated biomarker of GD1), 4-monosialodihexosylganglioside (GM3, a precursor for more complex gangliosides), and macrophage inflammatory protein MIP-1β, which is a marker of metabolic inflammation [70]. Similar findings were reported after 8 years in previously untreated adults with GD1 who completed an open-label, phase 2 trial of eliglustat [71]. Further trials are required to test the efficacy of these drugs on neuropathy. It will be important to determine whether there is a correlation between neuropathy and any of the biomarkers, as this could help reveal the underlying mechanism of peripheral nervous system involvement in GD1.
3.5.4. Bile Acid Synthesis Disorder (BASD)
Cholic acid and chenodeoxycholic acid, depending on the subtype of BASD, downregulate bile acid synthesis and are used clinically to treat BASD. The efficacy of cholic acid was demonstrated in a phase 3 open-label continuation study on 53 paediatric patients [85]. This study confirmed that cholic acid downregulates the production of atypical bile acids and improves liver biochemistries and growth in patients with BASD. Interestingly, one patient discontinued the study because of peripheral neuropathy that was reported as a treatment-emergent adverse event. It is possible that this neuropathy was a natural history progression. Further studies are warranted to evaluate the potential benefit of cholic acid on neuropathy in BASD.
4. Discussion
4.1. Limitations of This Analysis
Our search was limited to studies with results published after 1 January 2018. For this reason, several clinically validated therapies for inherited neuropathies (e.g., vitamin E for AVED, riboflavin for Brown–Vialetto–Van–Laere, diet to limit pristanic acid for Refsum disease, etc.) were not captured in our search (but are well described by Fernandez-Eulate et al. [2]), nor were unsuccessful trials prior to 2018 (e.g., ascorbic acid, progesterone antagonists/modulators in CMT1A) [8]. Similarly, suspended studies (e.g., the gene therapy scAAV1.tMCK.NTF3 for CMT1A; NCT03520751) were not considered. Finally, we did not include preclinical studies or recruiting trials, thereby missing potential future therapeutic approaches (e.g., gene therapies for Charcot–Marie–Tooth type 4J and X, AT007 for sorbitol dehydrogenase deficiency patients).
4.2. Added Value of This Analysis
Inherited neuropathies are challenging to diagnose and treat. Here, we provided an up-to-date list of conditions that may have neuropathy as a clinical feature, highlighting those that are treatable or for which treatment could soon be available.
The list of genes causative of neuropathy continues to grow, and whilst it is established that certain conditions (e.g., Friedreich ataxia) are associated with neuropathy, the latter can be a very minor and inconstant aspect of an inherited condition, making it difficult to confirm the association of neuropathy with a particular disease, especially in small cohorts of patients. This review identified neuropathies by beginning with gene panels used for the diagnosis of inherited neuropathies in the UK, France, and the USA; this list was extended by the authors and through searches of published data [1,2,3,14] and revised by two experts. Although it may not be exhaustive, the list provided here includes genes not described in recent publications and highlights the inconsistencies among published data and the different gene panels used for this analysis. Indeed, only a few genes were consistently found in all three panels, and some genes mentioned in the literature were not found in any of these three panels (Appendix A).
In addition to providing an updated list of conditions associated with inherited neuropathies, this review summarized data from clinical trials conducted in the last 5 years, evaluating therapies for these specific conditions. This analysis allowed us to identify 28 studies evaluating the effects of drugs or dietary interventions on neuropathy. Among these, 14 different drugs were assessed in nine inherited neuropathies. Nine additional therapies were identified that were not included in the systematic review published by Jennings et al. in 2021 [7], attesting to the increased interest in this field over recent years. Our search yielded several disease-modifying drugs, including small molecules, nucleic acid-based drugs, and gene therapy, especially in patients with ATTRv-PN and MLD. Some trials looked promising (e.g., L-serine for HSAN1 and PTX3003 for CMT1A) and will require close monitoring in the future, whereas others showed relatively stable nerve conduction studies upon treatment (e.g., DHA for SCA 38, rhASA for MLD) and warrant longer follow-ups and/or comparisons to natural history cohorts to assess the effects on neuropathy. Finally, some treatments of symptoms were beneficial (e.g., carbidopa in familial dysautonomia).
Although not designed to evaluate neuropathy as an endpoint, several therapeutics were demonstrated to cause changes in biomarkers thought to be causal of neuropathies (i.e., NTLA-2001 and eplontersen for ATTRv-PN, givosiran for AIP, and ERT, EET, and SRT for Fabry disease). These therapeutics should be tested with a focus on neuropathy in further trials. Other therapeutics caused changes in biomarkers but the association with neuropathy is unknown (i.e., taurine and glutamine for MELAS, SRT and ERT for Gaucher disease, several trials in FRDA, cholic acid in BASD, leriglitazone in X-ALD, and olipudase alfa in ASMD). Finally, improvements in neurological assessments were observed for patients with FRDA treated with EPI-743 and omaveloxolone and for patients with X-ALD treated with leriglitazone, but further investigations are needed in terms of efficacy on neuropathy, especially since omaveloxolone has recently been approved by the FDA.
In addition, this research highlighted two general findings. Firstly, most clinical trials have been conducted with subjects diagnosed with complex disorders with a possible neuropathology component rather than pure hereditary neuropathies. As neuropathy is only one of a number of symptoms that can be experienced by these subjects, fewer than 50% of studies monitored nerve conduction or employed specific neuropathy scales to assess treatment efficacy. Secondly, among clinical trials with neuropathy as an outcome measure, various clinical scales or scoring systems were used, which prevented comparisons between studies. Moreover, some scoring systems, although validated for use in the evaluation of the progression of neuropathy, may not be appropriate in all circumstances. For example, heart rate to deep breathing from the modified Neuropathy Impairment Score +7 scale, which is validated for use in neuropathies involving autonomic dysfunction [121], was not performed in the NEURO-TTR trial evaluating inotersen, because most ATTRv-PN patients included in this trial had active pacing or atrial fibrillation. Therefore, other endpoints must be used to assess autonomic dysfunction in such subjects. Additional limitations are that the scoring systems are subjective and cannot reliably detect subtle motor changes, especially in small cohorts.
Therefore, it will be important to assess neuropathy as an outcome in further trials, aiming to support an association of various complex conditions with neuropathies amongst larger cohorts. In addition, it will be critical to develop objective and reliable methods for this analysis. Research is ongoing to evaluate disease progression in subjects with peripheral neuropathies, including the use of wearable technologies [122,123], nerve sonography [124], intramuscular fat accumulation demonstrated by MRI imaging of the lower limbs [125], elevated plasma neurofilaments light chain concentration [126], and changes in the motor unit index (MUNIX) (NCT03715283). Associated with systematic nerve conduction studies, this could allow a better characterization of the type of neuropathy, their prevalence, and their potential correlation with readily monitored biomarkers to lead to a better understanding of the underlying physiopathological mechanisms of neuropathy and more effective treatment.
Author Contributions
M.H. conceived of the idea, conducted the research, and wrote the protocol; M.H. and A.-M.S. contributed equally to the review of the literature; L.S. supervised the project and was the third evaluator in case of disagreement between M.H. and A.-M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This review does not describe any studies with human participants performed by any of the authors.
Acknowledgments
We acknowledge the help of two experts, Tania Stojkovic and Isabelle Lievens, in the review of our list of inherited neuropathies.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Table A1.
List of inherited neuropathies and associated genes.
Table A1.
List of inherited neuropathies and associated genes.
| Classification | Disease and Synonyms | Gene | UK Panel | USA Panel | France Panel | Literature | |
|---|---|---|---|---|---|---|---|
| Mechanism depending on the variant | Charcot–Marie–Tooth, also known as hereditary motor and sensory neuropathy, Dejerine-Sottas syndrome, and peroneal muscular atrophy | Several causative genes | X | X | X | [3,14] | |
| Hereditary sensory and autonomic neuropathy (HSAN), also known as hereditary sensory neuropathy (HSN) | Several causative genes | X | X | X | [3,14] | ||
| Hereditary motor neuropathy (HMN) | Several causative genes | X | X | X | [3,14] | ||
| Spinocerebellar ataxia (SCA) (1–38) | Several causative genes | X | X | X | [1,3,14,122] | ||
| Hereditary spastic paraplegia (HSP), also known as familial spastic paraparesis (1, 2, 3A, 4, 5A, 6, 7, 9A/B, 10, 11, 12, 14, 15, 17, 20, 25, 26, 27, 28, 30, 31, 36, 38, 39, 43, 46, 47, 49, 55, 56, 57, 61, 76) | Several causative genes | X | X | X | [1,3,14] | ||
| Porphyria | Coproporphyria | CPOX | X | X | |||
| Variegata | PPOX | X | X | ||||
| Acute intermittent | HMBS | X | X | ||||
| DOSS porphyria | ALAD1 | [14] | |||||
| Peroxisomal | Zellweger spectrum disorder | PEX10, PXMP2 | X | ||||
| Refsum disease | PHYH, PEX7 | X | X | ||||
| X-linked adrenoleukodystrophy (X-ALD) | ABCD1 | X | |||||
| Alpha-methylacyl-CoA racemase deficiency (AMACRD) | AMACR | X | |||||
| Lysosomal | GM2-gangliosidose AB, also known as Tay-Sachs disease and Sandhoff disease | GM2A, HEXA, HEXB | X | ||||
| Metachromatic leukodystrophy (MLD) | ARSA | X | X | ||||
| DEGS1 insufficiency | DEGS1 | X | |||||
| Krabbe disease, also known as globoid leukodystrophy | GALC | X | X | ||||
| Fabry disease | GLA | X | X | X | |||
| Chediak–Higashi syndrome | LYST | X | |||||
| Kanzaki, also known as Schindler type II | NAGA | X | |||||
| Acid sphingomyelinase deficiency (ASMD), also known as Niemann–Pick A, B | SMPD1 | [3,127,128] | |||||
| B-mannosidosis | MANBA | [3,14] | |||||
| Sialidosis type 1 | NEU1 | [129] | |||||
| Salla disease | SLC17A | [130] | |||||
| Acid ceramidase deficiency, also known as Farber disease | ASAH1 | [131] | |||||
| Multiple sulfatase deficiency, also known as Austin disease | SUMF1 | [132] | |||||
| Glucocerebrosidase deficiency, also known as Gaucher disease | GBA | [14,118] | |||||
| Galactosialidosis | CTSA | [1,14] | |||||
| Mitochondrial | Mitochondrial neurogastrointestinal encephalopathy (MNGIE) | RRM2B | X | ||||
| POLG | X | X | X | ||||
| TYMP | X | X | |||||
| Neuropathy, ataxia, and retinitis pigmentosa (NARP) syndrome | MT-ATP6 | X | |||||
| Mitochondrial encephalomyopathy lactic acidosis and stroke-like episodes (MELAS) | MT-TL1 | X | |||||
| Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) | POLG | X | X | X | |||
| TWNK also known as C10ORF2 | X | X | |||||
| Myoclonic epilepsy associated with ragged red fibres (MERRF) | MTTK | [1,3,14] | |||||
| Infantile-onset spinocerebellar ataxia, ophthalmoplegia, hypotonia, ataxia, hypoacusis, and athetosis (IOSCA), also known as OHAHA syndrome | TWNK also known as C10ORF2 | X | X | ||||
| Leigh Syndrome | SURF1 | X | X | ||||
| Kearns–Sayre syndrome | deletion mtDNA | [1] | |||||
| Friedreich ataxia (FRDA) | FXN | X | X | X | |||
| Pyruvate carrier deficiency | MPC1 | X | |||||
| Pyruvate dehydrogenase complex deficiency | PDHA1 | X | |||||
| Trifunctional protein deficiency with myopathy and neuropathy (MTP), also known as LCHAD deficiency | HADHA/HADHB | X | X | ||||
| Multiple Acyl-Coa Dehydrogenase Deficiency (MADD) | ETFHD | [2] | |||||
| Infantile-onset multisystem neurologic, endocrine, and pancreatic disease (IMNEPD) | PTRH2 | X | |||||
| Mitochondrial complex IV deficiency nuclear type 2 (Mc4dn2), also known as cardioencephalomyopathy, fatal infantile, due to cytochrome c oxidase deficiency (CEMCOX1) | SCO2 | X | |||||
| Combined oxidative phosphorylation deficiency 3 (CoxPD3) | TSFM | X | |||||
| Leucoencephalopathy with brain stem and spinal cord involvement and lactate elevation (LBSL) | DARS2 | X | |||||
| Coenzyme Q10 deficiency primary, 8 | COQ7 | X | |||||
| Cataract, growth hormone deficiency, sensory neuropathy and hearing loss, and skeletal dysplasia (CAGSSS) | IARS2 | X | X | ||||
| Mitochondrial DNA depletion syndrome type 6 (MTDPS6), also known as Navajo neuropathy | MPV17 | X | X | X | |||
| Mitochondrial DNA depletion syndrome type 3 (MTDPS3), also known as DGUOK deficiency | DGUOK | [1] | |||||
| Mitochondrial DNA depletion syndrome type 5 (MTDPS5), also known as Booth–Haworth–Dilling Syndrome | SUCLA2 | X | |||||
| Parkinsonism, deafness, and sensory-motor axonal neuropathy | MT-RNR1 | X | |||||
| Dominant optic atrophy plus (DOA+), also known as Behr syndrome | OPA1 | X | X | ||||
| Dominant optic atrophy (DOA), also known as Costeff syndrome | OPA3 | X | |||||
| Mitochondrial Complex 4 deficiency, Nuclear Type 11 | COX20 | X | |||||
| Mitochondrial Complex 5 deficiency | MTATP8 | [1] | |||||
| Congenital disorder of deglycosylation 1 (CDDG1) | NGLY1 | [1] | |||||
| Ornithine aminotransferase deficiency | MT-TK | [3] | |||||
| Adult-Onset Chronic Progressive External Ophthalmoplegia with Mitochondrial Myopathy | RNASEH1 | X | |||||
| DNA repair/replication | Ataxia-oculomotor apraxia 1 (OA1), also known as ataxia early-onset with oculomotor apraxia and hypoalbuminemia (EAOH) | APTX | X | X | |||
| Ataxia-oculomotor apraxia 2 (AOA2), also known as spinocerebellar ataxia autosomal recessive 1 (SCAR1), and spinocerebellar ataxia autosomal recessive with axonal neuropathy 2 (SCAN2) | SETX | X | X | X | |||
| Ataxia telangiectasia | ATM | X | X | ||||
| Cockayne syndrome, also known as Neill–Dingwall Syndrome | ERCC6, ERCC8 | X | X | ||||
| Xeroderma Pigmentosum | XPA | X | |||||
| Aminoacidopathies | Homocysteine methylation disorders (cobalamin and methylenetetrahydrofolate reductase) | MMACHC | X | ||||
| MTHFR | [2,3,14] | ||||||
| Serine deficiency | PGDH | [3] | |||||
| Tyrosinaemia type 1 | FAH | X | |||||
| Inflammatory | CD59 deficiency | CD59 | X | ||||
| Aicardi–Goutieres syndrome | RNASEH2A, RNASEH2B, RNASEH2C, TREX1, ADAR1, IFH1, SAMHD1 | [1] | |||||
| Adenosine deaminase 2 deficiency (DADA2) | ADA2 | [133] | |||||
| Vitamin-related disorders | Brown–Vialetto–Van Laere (BVVL), also known as riboflavin transporter deficiency (RTD) | SLC52A2, SLC52A3 | X | X | |||
| Biotinidase deficiency | BTD | [2,3] | |||||
| Abetalipoproteinemia | MTTP | X | X | ||||
| Ataxia with isolated vitamin E deficiency (AVED) | TTPA | X | X | ||||
| Cerebral folate deficiency (CFD) | FOLR1 | [2] | |||||
| Thiamine metabolism dysfunction syndrome 4 (THMD4) | SLC25A19 | X | X | ||||
| Amyloidosis | Familial amyloid polyneuropathy (FAP) type I-II | TTR | X | X | X | ||
| Familial amyloid polyneuropathy (FAP) type III, also known as Van Allen or Iowa type | APOA1 | X | |||||
| Familial amyloid polyneuropathy (FAP) type IV | GSN | X | |||||
| Giant axonal neuropathy | Giant axonal neuropathy 2 | DCAF8 | X | ||||
| Giant axonal neuropathy 1 | GAN | X | X | X | |||
| Other | Tangier | ABCA1 | X | X | |||
| Xanthomatosis cerebrotendinous (XCT) | CYP27A1 | X | X | ||||
| Charlevoix–Saguenay Spastic Ataxia (ARSACS) | SACS | X | X | ||||
| Chorea acanthocytosis, also known as choreoacanthocytosis | VPS13A | X | |||||
| McLeod syndrome | XK | X | |||||
| Spinal and bulbar muscular atrophy, also known as Kennedy disease | AR | [1] | |||||
| X-fragile tremor and ataxia syndrome (FXTAS) | FMR1 | X | |||||
| Cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS) | RFC1 | [1,3,14] | |||||
| Posterior column ataxia and retinitis pigmentosa (PCARP) | FLVCR1 | X | X | X | |||
| Polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract (PHARC) | ABHD12 | X | |||||
| Pelizaeus–Merzbacher | GJC2 | X | |||||
| PLP1 | X | ||||||
| Waardenburg syndrome | SOX10 | X | X | X | |||
| Adult polyglucosan body disease (APBD), also known as glycogenosis type IV, glycogen storage disorder type 4, and Andersen disease | GBE1 | X | |||||
| Neurofibromatosis type II | NF2 | X | |||||
| Agenesis of the corpus callosum with peripheral neuropathy (ACCPN), also known as Andermann syndrome | SLC12A6 | X | X | X | |||
| Triple A syndrome | AAAS | X | |||||
| Congenital disorder of glycosylation type 1A (CDG1A) | PMM2 | X | |||||
| Congenital cataracts, facial dysmorphism, and neuropathy (CCFDN) | CTDP1 | X | X | X | |||
| Leukodystrophy hypomyelination and congenital cataract (HLD5 HCC) | FAM126A | X | X | ||||
| Myopathy congenital, deafness and neuropathy (CMND) | SPTBN4 | X | |||||
| Congenital insensitivity to pain | CLTCL1 | X | X | ||||
| Merosin-deficient congenital muscular dystrophy | LAMA2 | X | |||||
| Neurodegeneration with brain iron accumulation 2A (NBI2A), also known as infantile neuroaxonal dystrophy (INAD) | PLA2G6 | X | |||||
| Familial dysautonomia, also known as hereditary sensory autonomic neuropathy with intellectual disability (HSAN9) and hereditary spastic paraplegia 49 | TECPR2 | [1,14] | |||||
| Hypomyelinating leukodystrophy 6, also known as TUBB4A-related leukodystrophy | TUBB4A | [1] | |||||
| Pontocerebellar hypoplasia type 1B (PCH1B) | EXOSC3 | [1] | |||||
| Pontocerebellar hypoplasia (PCH9) | AMPD2 | [1] | |||||
| Action myoclonus–renal failure syndrome (AMRF) | SCARB2 | [1,14] | |||||
| Kyphoscoliotic type of Ehlers–Danlos syndrome (EDS 6) | PLOD1 | [1] | |||||
| Familial visceral amyloidosis | B2M | [1] | |||||
Appendix B
Table A2.
Information on inherited neuropathies for which therapies have been tested clinically over the last five years.
Table A2.
Information on inherited neuropathies for which therapies have been tested clinically over the last five years.
| Diseases for Which Trials Were Found | Gene | Inheritance | Physiopathology/Comments | Clinical Aspects | Type of Neuropathy | Standard of Care |
|---|---|---|---|---|---|---|
| Hereditary transthyretin amyloidosis- polyneuropathy (ATTRv-PN) | TTR | AD | Mutation changes the tetrameric structure of the TTR protein, leads to dissociation into misfolded monomer subunits, which then accumulate as amyloid deposits in different tissues including peripheral nerves [134]. | Systemic disease including cardiomyopathy, renal, and ocular involvement [134]. | Sensorimotor predominantly axonal, length-dependent and autonomic neuropathies [134] |
|
| Spinocerebellar ataxia type 38 (SCA 38) | ELOVL5 | AD | More than 40 distinct subtypes have been identified: mechanism depends on the variant [136]. | Starts at around age 40 with cerebellar symptoms. As the disease progresses, patients may present with hyposmia, hearing loss and pes cavus without paresia [114]. | Predominantly sensorimotor axonal polyneuropathy [114] |
|
| Spinocerebellar ataxia type 2 (SCA2) | CAG trinucleotide repeat expansion in the ATXN2 gene | AD | More than 40 distinct subtypes have been identified: mechanism depends on the variant [136]. | Slow saccades, ataxia, tremor, parkinsonism [1]. | Motor-predominant axonal neuropathy [1] |
|
| Familial dysautonomia | ELP-1 | AR | Defect in baroreceptor neurons in cranial nerves IX and X [137]. | Variability in blood pressure among other dysautonomia symptoms [137]. | Small-fibre neuropathy [137] |
|
| Hereditary sensory and autonomic neuropathy type 1 (HSAN1) | SPTLC1 and SPTLC2 | AD | Mutation reduces the affinity of the serine palmitoyltransferase enzyme for its normal substrate, serine and increases affinity for alanine and glycine, leading to the production of abnormal neurotoxic 1-deoxysphingolipids [138]. | Dysautonomia symptoms, skin ulcers, muscle weakness, sensory loss, and neuropathic pain [139]. | Small-fibre and sensory axonal neuropathy [139] |
|
| Ataxia-Telangiectasia (A-T) | ATM | AR | Mutation of ATM gene impairs DNA repair [140]. | Ataxia and oculomotor apraxia beginning in early infancy, dystonia and/or chorea, conjunctival telangiectasia, susceptibility to infections and malignancies [1]. | Sensory axonal neuropathy [1,141] |
|
| Charcot–Marie–Tooth 1A (CMT1A) | PMP22 | AD | Duplication of the PMP22 gene, leading to over-expression of PMP22 protein, and gain-of-function phenotype [142,143]. | Neuropathy is the sole or predominant clinical feature of the disease [4]. | Demyelinating sensorimotor neuropathy [4] |
|
| Metachromatic leukodystrophy (MLD) | ASA | AR | Deficient activity of the lysosomal enzyme arylsulfatase A, results in accumulation of sulfatide in cells of both peripheral and central nervous systems [144]. | Three forms are commonly described depending on age of onset: late-infantile, around 2 years; juvenile, between 3 and 16 years; and adult, after 16 years. The younger onsets are the most severe forms, with clinical regression. Adults present with optic atrophia, cognitive impairment, ataxia, and paresia [144]. | Demyelinating neuropathy [1] |
|
| X-linked adrenoleukodystrophy (X-ALD) | ABCD1 | X-linked | Mutation in protein involved in the transmembrane transport of very-long-chain fatty acids leads to accumulation of these fatty acids in different tissues [147]. | Very broad spectrum. Symptoms range from adrenocortical insufficiency only to cerebral adrenoleukodystrophy to adrenomyeloneuropathy (AMN). Most adult patients present with AMN, which involves myelopathy, neuropathy, and possible cerebral involvement [147]. | Sensorimotor axonal (sometimes demyelinating) neuropathy [1] |
|
| Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy disease | Expansion of CAG repeat in the AR gene | X-linked | Mutation of the receptor for androgen results in degeneration of lower motor neurons and skeletal muscles [148]. | Muscle weakness, atrophy, fasciculations, occasionally androgen insensitivity [1,148]. | Motor neuropathy [1] |
|
| Adult polyglucosan body disease (APBD) | GBE1 | AR | Mutation of GBE1 results in glycogen branching enzyme deficiency, which causes polyglucosan body accumulation in various tissues including peripheral nerves and cerebral white matter [149]. | Cognitive impairment, spasticity, bladder dysfunction [149]. | Sensorimotor axonal-predominant neuropathy [1,3]. |
|
| Fabry disease (FD) | GLA | X-linked | Mutation of GLA results in α-galactosidase A activity deficiency, leading to accumulation of globotriaosylceramide (Gb3) and its deacylated form globotriaosylsphingosine (lyso-Gb3) in plasma and different cell types. The severity of phenotype depends on the level of galactosidase A activity [150]. | Multisystemic disease including cardiomyopathy, renal failure, and angiokeratoma [150]. | Sensory axonal and small-fibre neuropathy [1,3] |
|
| Acid sphingomyelinase deficiency (ASMD) | SMPD1 | AR | Mutation results in lysosomal enzyme acid sphingomyelinase activity deficiency, leading to sphingomyelin accumulation in various tissues, including the macrophage–monocyte system [152]. | Phenotypic spectrum ranges from severe infantile presentation with early death to subacute/chronic neurovisceral forms. Patients present with hepatosplenomegaly, thrombocytopenia, and interstitial lung disease [152]. | Mostly demyelinating polyneuropathy [127,128] |
|
| Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) | MT-TL1 | Maternal inheritance | Mutation results in mitochondrial dysfunction [153]. | Stroke-like episodes [153]. | Subclinical polyneuropathy, predominantly sensory axonal [1,154] |
|
| Gaucher disease type 1 (GD1) | GBA1 | AR | Mutation results in β-glucocerebrosidase enzyme activity deficiency, leading to lysosomal glucocerebroside accumulation in various tissues [155]. | Hepatosplenomegaly, thrombocytopenia, anaemia, bone disease, growth failure [155] | Sensorimotor axonal neuropathy [14,118] |
|
| Acute intermittent porphyria (AIP) | HMBS | AD | Porphobilinogen deaminase enzyme activity deficiency results from HMBS mutation, leading to depletion of free heme. This results in up-regulation of the delta-aminolevulinic acid synthase and overproduction of toxic heme intermediates that are thought to cause disease manifestations [156]. | Acute neurovisceral attacks characterized by abdominal pain and mental status changes (seizures, psychosis) [157]. | Autonomic neuropathy and acute motor axonal neuropathy resembling Guillain–Barré syndrome during attacks. Subjects not treated promptly or with recurrent attacks may develop chronic neuropathies, typically motor axonal polyneuropathies [1,158]. |
|
| Friedreich ataxia (FRDA) | Trinucleotide (GAA) repeat expansions in the FXN gene | AR | Reduction in the amount of functional mitochondrial frataxin protein results from repeat expansion in FXN, leading to mitochondrial dysfunction and increased sensitivity to oxidative stress [160]. | Ataxia, cardiomyopathy, diabetes, and loss of visual and sensorineural hearing function [161]. | Sensory axonal neuropathy [1,3] |
|
| Bile acid synthesis disorder (BASD) | Several causative genes | Depends on condition | Deficiency or lack of activity in enzymes that catalyse the conversion of cholesterol to bile acids (i.e., single enzyme defects) or defects in oxidation and shortening of the cholesterol side chain caused by generalized peroxisomal dysfunction (Zellweger spectrum disorder) [162]. | Liver dysfunction [162] | Neuropathies are seen in single-enzyme defects such as in patients with cerebrotendinous xanthomatosis (predominantly sensory axonal neuropathy), with alpha-methylacyl-CoA racemase deficiency (axonal or demyelinating neuropathy) [1] and in subjects with Zellweger spectrum disorder who survive into adulthood (mostly demyelinating neuropathies) [163]. |
|
| Fragile-X associated tremor/ataxia syndrome (FXTAS) | Premutation expansion (59-199 CGG) in the FMR1 gene | X-linked | Expansion results in overproduction of FMR1 mRNA [164] | Late-onset intention tremor, ataxia, parkinsonism, cognitive decline [164] | Sensory axonal neuropathy [1,3,164] |
|
Abbreviations: TTR, transthyretin; AD, autosomal dominant; AR, autosomal recessive; ERT, enzyme replacement therapy; SRT, substrate reduction therapy; FDA, US Food and Drug Administration.
Appendix C
Table A3.
Risk of bias.
Table A3.
Risk of bias.
| Reference of Study | JADAD Score | Reference of Study | JADAD Score | Reference of Study | JADAD Score | Reference of Study | JADAD Score |
|---|---|---|---|---|---|---|---|
| [18] | 1 | [42] | 5 | [65] | 5 | [85] | 1 |
| [19] | 1 | [43] | 5 | [66] | 5 | [87] | 1 |
| [20] | 1 | [44] | 1 | [67] | 1 | [88] | 2 |
| [22] | 5 | [45] | 1 | [68] | 1 | [89] | 5 |
| [23] | 5 | [46] | 1 | [69] | 1 | [90] | 5 |
| [24] | 3 | [48] | 1 | [70] | 1 | [91] | 1 |
| [25] | 1 | [49] | 4 | [71] | 1 | [92] | 1 |
| [26] | 3 | [50] | 5 | [72] | 0 | [93] | 1 |
| [27] | 1 | [51] | 1 | [73] | 1 | ||
| [29] | 1 | [53] | 2 | [74] | 5 | ||
| [31] | 4 | [54] | 1 | [75] | 1 | ||
| [32] | 4 | [55] | 0 | [76] | 0 | ||
| [33] | 4 | [57] | 0 | [77] | 4 | ||
| [34] | 4 | [58] | 1 | [78] | 5 | ||
| [35] | 1 | [59] | 1 | [79] | 5 | ||
| [36] | 1 | [60] | 1 | [80] | 1 | ||
| [37] | 1 | [61] | 1 | [81] | 5 | ||
| [38] | 1 | [62] | 4 | [82] | 5 | ||
| [40] | 1 | [63] | 1 | [83] | 5 | ||
| [41] | 5 | [64] | 0 | [84] | 5 |
References
- Rossor, A.M.; Carr, A.S.; Devine, H.; Chandrashekar, H.; Pelayo-Negro, A.L.; Pareyson, D.; Shy, M.E.; Scherer, S.S.; Reilly, M.M. Peripheral neuropathy in complex inherited diseases: An approach to diagnosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 846–863. [Google Scholar] [CrossRef]
- Fernández-Eulate, G.; Carreau, C.; Benoist, J.-F.; Lamari, F.; Rucheton, B.; Shor, N.; Nadjar, Y. Diagnostic approach in adult-onset neurometabolic diseases. J. Neurol. Neurosurg. Psychiatry 2022, 93, 413–421. [Google Scholar] [CrossRef]
- Masingue, M.; Fernández-Eulate, G.; Debs, R.; Tard, C.; Labeyrie, C.; Leonard-Louis, S.; Dhaenens, C.-M.; Masson, M.; Latour, P.; Stojkovic, T. Strategy for genetic analysis in hereditary neuropathy. Rev. Neurol. 2023, 179, 10–29. [Google Scholar] [CrossRef]
- Bird, T.D. Charcot-Marie-Tooth Hereditary Neuropathy Overview; University of Washington: Seattle, DC, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1358/ (accessed on 28 March 2023).
- Sargiannidou, I.; Kagiava, A.; Kleopa, K.A. Gene therapy approaches targeting Schwann cells for demyelinating neuropathies. Brain Res. 2020, 1728, 146572. [Google Scholar] [CrossRef]
- Timmerman, V.; Strickland, A.V.; Züchner, S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes 2014, 5, 13–32. [Google Scholar] [CrossRef]
- Jennings, M.J.; Lochmüller, A.; Atalaia, A.; Horvath, R. Targeted Therapies for Hereditary Peripheral Neuropathies: Systematic Review and Steps Towards a ‘treatabolome’. J. Neuromuscul. Dis. 2021, 8, 383–400. [Google Scholar] [CrossRef]
- Pisciotta, C.; Saveri, P.; Pareyson, D. Challenges in Treating Charcot-Marie-Tooth Disease and Related Neuropathies: Current Management and Future Perspectives. Brain Sci. 2021, 11, 1447. [Google Scholar] [CrossRef] [PubMed]
- Hereditary Neuropathy (Version 1.462). Available online: https://panelapp.genomicsengland.co.uk/panels/85/ (accessed on 3 April 2023).
- Orphanet: Diagnostic Des Neuropathies Peripheriques Panel. Available online: https://www.orpha.net/consor/cgi-bin/ClinicalLabs_Search.php?lng=FR&data_id=118068&search=ClinicalLabs_Search_Simple&data_type=Test&title=Diagnostic-des-neuropathies-peripheriques--Panel-&MISSING%20CONTENT=Diagnostic-des-neuropathies-peripheriques--Panel- (accessed on 3 April 2023).
- PEPAN—Overview: Comprehensive Peripheral Neuropathy Gene Panel, Varies. Available online: https://www.mayocliniclabs.com/test-catalog/overview/617688#Clinical-and-Interpretive (accessed on 3 April 2023).
- Orphanet: Search a Disease. Available online: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN (accessed on 5 April 2023).
- GeneCards—Human Genes | Gene Database | Gene Search. Available online: https://www.genecards.org/ (accessed on 5 April 2023).
- Finsterer, J.; Löscher, W.N.; Wanschitz, J.; Iglseder, S. Orphan Peripheral Neuropathies. J. Neuromuscul. Dis. 2021, 8, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Ovid: Welcome to Ovid. Available online: https://ovidsp.ovid.com/ (accessed on 5 April 2023).
- Home—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 5 April 2023).
- Brown, D. JADAD Scores. RCEMLearning India. Available online: https://www.rcemlearning.org/modules/critical-appraisal-appraising-a-treatment-early-goal-directed-therapy/lessons/methodology-jadad-scores/topic/jadad-scores/ (accessed on 3 April 2023).
- Waddington Cruz, M.; Amass, L.; Keohane, D.; Schwartz, J.; Li, H.; Gundapaneni, B. Early intervention with tafamidis provides long-term (5.5-year) delay of neurologic progression in transthyretin hereditary amyloid polyneuropathy. Amyloid Int. J. Exp. Clin. Investig. 2016, 23, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Coelho, T.; Waddington Cruz, M.; Li, H.; Stewart, M.; Ebede, B. Evaluation of Mortality During Long-Term Treatment with Tafamidis for Transthyretin Amyloidosis with Polyneuropathy: Clinical Trial Results up to 8.5 Years. Neurol. Ther. 2020, 9, 105–115. [Google Scholar] [CrossRef]
- Gundapaneni, B.K.; Sultan, M.B.; Keohane, D.J.; Schwartz, J.H. Tafamidis delays neurological progression comparably across Val30Met and non-Val30Met genotypes in transthyretin familial amyloid polyneuropathy. Eur. J. Neurol. 2018, 25, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Verma, B.; Patel, P. Tafamidis; StatPearls Publishing: Tampa, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK574508/ (accessed on 28 March 2023).
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- González-Duarte, A.; Berk, J.L.; Quan, D.; Mauermann, M.L.; Schmidt, H.H.; Polydefkis, M.; Waddington-Cruz, M.; Ueda, M.; Conceição, I.M.; Kristen, A.V.; et al. Analysis of autonomic outcomes in APOLLO, a phase III trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. J. Neurol. 2020, 267, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Goel, V.; Attarwala, H.; Sweetser, M.T.; Clausen, V.A.; Robbie, G.J. Patisiran Pharmacokinetics, Pharmacodynamics, and Exposure-Response Analyses in the Phase 3 APOLLO Trial in Patients with Hereditary Transthyretin-Mediated (hATTR) Amyloidosis. J. Clin. Pharmacol. 2020, 60, 37–49. [Google Scholar] [CrossRef]
- Coelho, T.; Adams, D.; Conceição, I.; Waddington-Cruz, M.; Schmidt, H.H.; Buades, J.; Campistol, J.; Berk, J.L.; Polydefkis, M.; Wang, J.J.; et al. A phase II, open-label, extension study of long-term patisiran treatment in patients with hereditary transthyretin-mediated (hATTR) amyloidosis. Orphanet J. Rare Dis. 2020, 15, 179. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Merkel, M.; Hale, C.; Marantz, J.L. Experience of patisiran with transthyretin stabilizers in patients with hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2020, 10, 289–300. [Google Scholar] [CrossRef]
- Schmidt, H.H.; Wixner, J.; Planté-Bordeneuve, V.; Muñoz-Beamud, F.; Lladó, L.; Gillmore, J.D.; Mazzeo, A.; Li, X.; Arum, S.; Jay, P.Y.; et al. Patisiran treatment in patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy after liver transplantation. Am. J. Transplant. 2022, 22, 1646–1657. [Google Scholar] [CrossRef]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef]
- Adams, D.; Tournev, I.L.; Taylor, M.S.; Coelho, T.; Planté-Bordeneuve, V.; Berk, J.L.; González-Duarte, A.; Gillmore, J.D.; Low, S.-C.; Sekijima, Y.; et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: A randomized clinical trial. Amyloid Int. J. Exp. Clin. Investig. 2023, 30, 18–26. [Google Scholar] [CrossRef]
- Keam, S.J. Vutrisiran: First Approval. Drugs 2022, 82, 1419–1425. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Dyck, P.J.B.; Coelho, T.; Cruz, M.W.; Iii, T.H.B.; Khella, S.; Karam, C.; Berk, J.L.; Polydefkis, M.J.; Kincaid, J.C.; Wiesman, J.F.; et al. Neuropathy symptom and change: Inotersen treatment of hereditary transthyretin amyloidosis. Muscle Nerve 2020, 62, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.B.; Kincaid, J.C.; Wiesman, J.F.; Polydefkis, M.; Litchy, W.J.; Mauermann, M.L.; Ackermann, E.J.; Guthrie, S.; Pollock, M.; Jung, S.W.; et al. mNIS+7 and lower limb function in inotersen treatment of hereditary transthyretin-mediated amyloidosis. Muscle Nerve 2020, 62, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Yarlas, A.; Waddington-Cruz, M.; White, M.K.; Kessler, A.S.; Lovley, A.; Pollock, M.; Guthrie, S.; Ackermann, E.J.; Hughes, S.G.; et al. Inotersen preserves or improves quality of life in hereditary transthyretin amyloidosis. J. Neurol. 2020, 267, 1070–1079. [Google Scholar] [CrossRef]
- Brannagan, T.H.; Wang, A.K.; Coelho, T.; Cruz, M.W.; Polydefkis, M.J.; Dyck, P.J.; Plante-Bordeneuve, V.; Berk, J.L.; Barroso, F.; Merlini, G.; et al. Early data on long-term efficacy and safety of inotersen in patients with hereditary transthyretin amyloidosis: A 2-year update from the open-label extension of the NEURO-TTR trial. Eur. J. Neurol. 2020, 27, 1374–1381. [Google Scholar] [CrossRef]
- Brannagan, T.H.; Coelho, T.; Wang, A.K.; Polydefkis, M.J.; Dyck, P.J.; Berk, J.L.; Drachman, B.; Gorevic, P.; Whelan, C.; Conceição, I.; et al. Long-term efficacy and safety of inotersen for hereditary transthyretin amyloidosis: NEURO-TTR open-label extension 3-year update. J. Neurol. 2022, 269, 6416–6427. [Google Scholar] [CrossRef] [PubMed]
- Yarlas, A.; Lovley, A.; McCausland, K.; Brown, D.; Vera-Llonch, M.; Conceição, I.; Karam, C.; Khella, S.; Obici, L.; Waddington-Cruz, M. Early Data on Long-term Impact of Inotersen on Quality-of-Life in Patients with Hereditary Transthyretin Amyloidosis Polyneuropathy: Open-Label Extension of NEURO-TTR. Neurol. Ther. 2021, 10, 865–886. [Google Scholar] [CrossRef]
- Karam, C.; Brown, D.; Yang, M.; Done, N.; Zhu, J.J.; Greatsinger, A.; Bozas, A.; Vera-Llonch, M.; Signorovitch, J. Long-term treatment effects of inotersen on health-related quality of life in patients with hATTR amyloidosis with polyneuropathy: Analysis of the open-label extension of the NEURO-TTR trial. Muscle Nerve 2022, 66, 438–446. [Google Scholar] [CrossRef]
- Mahfouz, M.; Maruyama, R.; Yokota, T. Inotersen for the Treatment of Hereditary Transthyretin Amyloidosis. Methods Mol. Biol. Clifton N. J. 2020, 2176, 87–98. [Google Scholar] [CrossRef]
- Manes, M.; Alberici, A.; Di Gregorio, E.; Boccone, L.; Premi, E.; Mitro, N.; Pasolini, M.P.; Pani, C.; Paghera, B.; Orsi, L.; et al. Long-term efficacy of docosahexaenoic acid (DHA) for Spinocerebellar Ataxia 38 (SCA38) treatment: An open label extension study. Parkinsonism Relat. Disord. 2019, 63, 191–194. [Google Scholar] [CrossRef]
- Norcliffe-Kaufmann, L.; Palma, J.A.; Martinez, J.; Kaufmann, H. Carbidopa for Afferent Baroreflex Failure in Familial Dysautonomia: A Double-Blind Randomized Crossover Clinical Trial. Hypertens. Dallas Tex. 1979 2020, 76, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Fridman, V.; Suriyanarayanan, S.; Novak, P.; David, W.; Macklin, E.A.; McKenna-Yasek, D.; Walsh, K.; Aziz-Bose, R.; Oaklander, A.L.; Brown, R.; et al. Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1. Neurology 2019, 92, e359–e370. [Google Scholar] [CrossRef] [PubMed]
- Attarian, S.; Young, P.; Brannagan, T.H.; Adams, D.; Van Damme, P.; Thomas, F.P.; Casanovas, C.; Tard, C.; Walter, M.C.; Péréon, Y.; et al. A double-blind, placebo-controlled, randomized trial of PXT3003 for the treatment of Charcot-Marie-Tooth type 1A. Orphanet J. Rare Dis. 2021, 16, 433. [Google Scholar] [CrossRef]
- Dali, C.; Groeschel, S.; Moldovan, M.; Farah, M.H.; Krägeloh-Mann, I.; Wasilewski, M.; Li, J.; Barton, N.; Krarup, C. Intravenous arylsulfatase A in metachromatic leukodystrophy: A phase 1/2 study. Ann. Clin. Transl. Neurol. 2021, 8, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Dali, C.; Sevin, C.; Krägeloh-Mann, I.; Giugliani, R.; Sakai, N.; Wu, J.; Wasilewski, M. Safety of intrathecal delivery of recombinant human arylsulfatase A in children with metachromatic leukodystrophy: Results from a phase 1/2 clinical trial. Mol. Genet. Metab. 2020, 131, 235–244. [Google Scholar] [CrossRef]
- Fumagalli, F.; Calbi, V.; Sora, M.G.N.; Sessa, M.; Baldoli, C.; Rancoita, P.M.V.; Ciotti, F.; Sarzana, M.; Fraschini, M.; Zambon, A.A.; et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: Long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 2022, 399, 372–383. [Google Scholar] [CrossRef]
- Public Health—European Commission. Union Register of Medicinal Products. Available online: https://ec.europa.eu/health/documents/community-register/html/h1493.htm (accessed on 3 April 2023).
- Casasnovas, C.; Ruiz, M.; Schlüter, A.; Naudí, A.; Fourcade, S.; Veciana, M.; Castañer, S.; Albertí, A.; Bargalló, N.; Johnson, M.; et al. Biomarker Identification, Safety, and Efficacy of High-Dose Antioxidants for Adrenomyeloneuropathy: A Phase II Pilot Study. Neurother. J. Am. Soc. Exp. Neurother. 2019, 16, 1167–1182. [Google Scholar] [CrossRef]
- Yamada, S.; Hashizume, A.; Hijikata, Y.; Inagaki, T.; Ito, D.; Kishimoto, Y.; Kinoshita, F.; Hirakawa, A.; Shimizu, S.; Nakamura, T.; et al. Mexiletine in spinal and bulbar muscular atrophy: A randomized controlled trial. Ann. Clin. Transl. Neurol. 2022, 9, 1702–1714. [Google Scholar] [CrossRef]
- Schiffmann, R.; Wallace, M.E.; Rinaldi, D.; Ledoux, I.; Luton, M.-P.; Coleman, S.; Akman, H.O.; Martin, K.; Hogrel, J.-Y.; Blankenship, D.; et al. A double-blind, placebo-controlled trial of triheptanoin in adult polyglucosan body disease and open-label, long-term outcome. J. Inherit. Metab. Dis. 2018, 41, 877–883. [Google Scholar] [CrossRef]
- Diep, J.K.; Yu, R.Z.; Viney, N.J.; Schneider, E.; Guo, S.; Henry, S.; Monia, B.; Geary, R.; Wang, Y. Population pharmacokinetic/pharmacodynamic modelling of eplontersen, an antisense oligonucleotide in development for transthyretin amyloidosis. Br. J. Clin. Pharmacol. 2022, 88, 5389–5398. [Google Scholar] [CrossRef]
- FDA Accepts Ionis NDA for Eplontersen in Rare Hereditary Disease | FDAnews. Available online: https://www.fdanews.com/articles/211422-fda-accepts-ionis-nda-for-eplontersen-in-rare-hereditary-disease (accessed on 20 March 2023).
- Gouverneur, C. Topline Results from Phase 3 ATTRibute-CM Study | BridgeBio. bridgebiowp. Published December 27, 2021. Available online: https://bridgebio.com/news/bridgebio-pharma-reports-month-12-topline-results-from-phase-3-attribute-cm-study/ (accessed on 20 March 2023).
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’connell, D.; Walsh, K.R.; Wood, K.; et al. Transthyretin stabilization activity of the catechol-O-methyltransferase inhibitor tolcapone (SOM0226) in hereditary ATTR amyloidosis patients and asymptomatic carriers: Proof-of-concept study. Amyloid Int. J. Exp. Clin. Investig. 2019, 26, 74–84. [Google Scholar] [CrossRef]
- Pinheiro, F.; Varejão, N.; Esperante, S.; Santos, J.; Velázquez-Campoy, A.; Reverter, D.; Pallarès, I.; Ventura, S. Tolcapone, a potent aggregation inhibitor for the treatment of familial leptomeningeal amyloidosis. FEBS J. 2021, 288, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Müntze, J.; Gensler, D.; Maniuc, O.; Liu, D.; Cairns, T.; Oder, D.; Hu, K.; Lorenz, K.; Frantz, S.; Wanner, C.; et al. Oral Chaperone Therapy Migalastat for Treating Fabry Disease: Enzymatic Response and Serum Biomarker Changes After 1 Year. Clin. Pharmacol. Ther. 2019, 105, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Narita, I.; Ohashi, T.; Sakai, N.; Hamazaki, T.; Skuban, N.; Castelli, J.P.; Lagast, H.; Barth, J.A. Efficacy and safety of migalastat in a Japanese population: A subgroup analysis of the ATTRACT study. Clin. Exp. Nephrol. 2020, 24, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in Fabry disease: 30-month results from the open-label extension of the randomized, phase 3 ATTRACT study. Mol. Genet. Metab. 2020, 131, 219–228. [Google Scholar] [CrossRef]
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study. Genet. Med. 2019, 21, 1987–1997. [Google Scholar] [CrossRef]
- Deegan, P.B.; Goker-Alpan, O.; Geberhiwot, T.; Hopkin, R.J.; Lukina, E.; Tylki-Szymanska, A.; Zaher, A.; Sensinger, C.; Gaemers, S.J.; Modur, V.; et al. Venglustat, an orally administered glucosylceramide synthase inhibitor: Assessment over 3 years in adult males with classic Fabry disease in an open-label phase 2 study and its extension study. Mol. Genet. Metab. 2023, 138, 106963. [Google Scholar] [CrossRef]
- Ramaswami, U.; Bichet, D.G.; Clarke, L.A.; Dostalova, G.; Fainboim, A.; Fellgiebel, A.; Forcelini, C.M.; Haack, K.A.; Hopkin, R.J.; Mauer, M.; et al. Low-dose agalsidase beta treatment in male pediatric patients with Fabry disease: A 5-year randomized controlled trial. Mol. Genet. Metab. 2019, 127, 86–94. [Google Scholar] [CrossRef]
- Hwang, S.M.; Lee, B.H.M.; Kim, W.-S.M.; Kim, D.-S.M.; Cheon, C.K.M.; Lee, C.H.M.; Choi, Y.; Choi, J.-H.M.; Kim, J.H.M.; Yoo, H.-W.M. A phase II, multicenter, open-label trial to evaluate the safety and efficacy of ISU303 (Agalsidase beta) in patients with Fabry disease. Medicine 2022, 101, e30345. [Google Scholar] [CrossRef]
- Schiffmann, R.; Goker-Alpan, O.; Holida, M.; Giraldo, P.; Barisoni, L.; Colvin, R.B.; Jennette, C.J.; Maegawa, G.; Boyadjiev, S.A.; Gonzalez, D.; et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: A 1-year Phase 1/2 clinical trial. J. Inherit. Metab. Dis. 2019, 42, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Coarelli, G.; Heinzmann, A.; Ewenczyk, C.; Fischer, C.; Chupin, M.; Monin, M.-L.; Hurmic, H.; Calvas, F.; Calvas, P.; Goizet, C.; et al. Safety and efficacy of riluzole in spinocerebellar ataxia type 2 in France (ATRIL): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2022, 21, 225–233. [Google Scholar] [CrossRef]
- Rodriguez-Labrada, R.; Ortega-Sanchez, R.; Casaña, P.H.; Morales, O.S.; Padrón-Estupiñan, M.D.C.; Batista-Nuñez, M.; Rodríguez, D.J.; Canales-Ochoa, N.; Acosta, A.P.; Montero, J.M.; et al. Erythropoietin in Spinocerebellar Ataxia Type 2: Feasibility and Proof-of-Principle Issues from a Randomized Controlled Study. Mov. Disord. 2022, 37, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Povalko, N.; Inoue, E.; Nakamura, H.; Ishii, A.; Suzuki, Y.; Yoneda, M.; Kanda, F.; Kubota, M.; Okada, H.; et al. Therapeutic regimen of L-arginine for MELAS: 9-year, prospective, multicenter, clinical research. J. Neurol. 2018, 265, 2861–2874. [Google Scholar] [CrossRef]
- Guerrero-Molina, M.P.; Morales-Conejo, M.; Delmiro, A.; Morán, M.; Domínguez-González, C.; Arranz-Canales, E.; Ramos-González, A.; Arenas, J.; Martín, M.A.; de la Aleja, J.G. High-dose oral glutamine supplementation reduces elevated glutamate levels in cerebrospinal fluid in patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome. Eur. J. Neurol. 2023, 30, 538–547. [Google Scholar] [CrossRef]
- Ohsawa, Y.; Hagiwara, H.; Nishimatsu, S.-I.; Hirakawa, A.; Kamimura, N.; Ohtsubo, H.; Fukai, Y.; Murakami, T.; Koga, Y.; Goto, Y.-I.; et al. Taurine supplementation for prevention of stroke-like episodes in MELAS: A multicentre, open-label, 52-week phase III trial. J. Neurol. Neurosurg. Psychiatry 2019, 90, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Lukina, E.; Ben Turkia, H.; Shankar, S.P.; Feldman, H.B.; Ghosn, M.; Mehta, A.; Packman, S.; Lau, H.; Petakov, M.; et al. Clinical outcomes after 4.5 years of eliglustat therapy for Gaucher disease type 1: Phase 3 ENGAGE trial final results. Am. J. Hematol. 2021, 96, 1156–1165. [Google Scholar] [CrossRef]
- Lukina, E.; Watman, N.; Dragosky, M.; Lau, H.; Arreguin, E.A.; Rosenbaum, H.; Zimran, A.; Foster, M.C.; Gaemers, S.J.M.; Peterschmitt, M.J. Outcomes after 8 years of eliglustat therapy for Gaucher disease type 1: Final results from the Phase 2 trial. Am. J. Hematol. 2019, 94, 29–38. [Google Scholar] [CrossRef]
- Zimran, A.; Gonzalez-Rodriguez, D.E.; Abrahamov, A.; Cooper, P.A.; Varughese, S.; Giraldo, P.; Petakov, M.; Tan, E.S.; Chertkoff, R. Long-term safety and efficacy of taliglucerase alfa in pediatric Gaucher disease patients who were treatment-naive or previously treated with imiglucerase. Blood Cells Mol. Dis. 2018, 68, 163–172. [Google Scholar] [CrossRef]
- Kuter, D.J.; Wajnrajch, M.; Hernandez, B.; Wang, R.; Chertkoff, R.; Zimran, A. Open-label, expanded access study of taliglucerase alfa in patients with Gaucher disease requiring enzyme replacement therapy. Blood Cells Mol. Dis. 2020, 82, 102418. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Ventura, P.; Bonkovsky, H.L.; Gouya, L.; Aguilera-Peiró, P.; Bissell, D.M.; Stein, P.E.; Balwani, M.; Anderson, D.K.E.; Parker, C.; Kuter, D.J.; et al. Efficacy and safety of givosiran for acute hepatic porphyria: 24-month interim analysis of the randomized phase 3 ENVISION study. ENVISION Investigators, ed. Liver Int. 2022, 42, 161–172. [Google Scholar] [CrossRef]
- Wang, B.; Ventura, P.; Takase, K.-I.; Thapar, M.; Cassiman, D.; Kubisch, I.; Liu, S.; Sweetser, M.T.; Balwani, M. Disease burden in patients with acute hepatic porphyria: Experience from the phase 3 ENVISION study. Orphanet J. Rare Dis. 2022, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Sardh, E.; Harper, P.; Balwani, M.; Stein, P.; Rees, D.; Bissell, D.M.; Desnick, R.; Parker, C.; Phillips, J.; Bonkovsky, H.L.; et al. Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N. Engl. J. Med. 2019, 380, 549–558. [Google Scholar] [CrossRef]
- Lynch, D.R.; Hauser, L.; McCormick, A.; Wells, M.; Na Dong, Y.; McCormack, S.; Schadt, K.; Perlman, S.; Subramony, S.H.; Mathews, K.D.; et al. Randomized, double-blind, placebo-controlled study of interferon-gamma 1b in Friedreich Ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.; Salemi, J.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef]
- Qureshi, M.Y.; Patterson, M.C.; Clark, V.; Johnson, J.N.; Moutvic, M.A.; Driscoll, S.W.; Kemppainen, J.L.; Huston, J.; Anderson, J.R.; Badley, A.D.; et al. Safety and efficacy of (+)-epicatechin in subjects with Friedreich’s ataxia: A phase II, open-label, prospective study. J. Inherit. Metab. Dis. 2021, 44, 502–514. [Google Scholar] [CrossRef]
- Wang, H.; Norton, J.; Xu, L.; DeMartinis, N.; Sen, R.; Shah, A.; Farmer, J.; Lynch, D. Results of a randomized double-blind study evaluating luvadaxistat in adults with Friedreich ataxia. Ann. Clin. Transl. Neurol. 2021, 8, 1343–1352. [Google Scholar] [CrossRef]
- Lynch, D.R.; Chin, M.P.; Delatycki, M.B.; Subramony, S.H.; Corti, M.; Hoyle, J.C.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Mathews, K.D.; et al. Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann. Neurol. 2021, 89, 212–225. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26. [Google Scholar] [CrossRef]
- Zesiewicz, T.; Heerinckx, F.; De Jager, R.; Omidvar, O.; Kilpatrick, M.; Shaw, J.; Shchepinov, M.S. Randomized, clinical trial of RT001: Early signals of efficacy in Friedreich’s ataxia. Mov. Disord. 2018, 33, 1000–1005. [Google Scholar] [CrossRef]
- Heubi, J.E.; Setchell, K.D.R. Open-label Phase 3 Continuation Study of Cholic Acid in Patients with Inborn Errors of Bile Acid Synthesis. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 423–429. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Approves Cholbam for the Treatment of Rare Bile Acid Synthesis Disorders and Grants Rare Pediatric Disease Priority Review Voucher | Travere Therapeutics, Inc. Available online: https://ir.travere.com/news-releases/news-release-details/us-food-and-drug-administration-approves-cholbam-treatment-rare (accessed on 28 March 2023).
- Hasegawa, S.; Kumada, S.; Tanuma, N.; Tsuji-Hosokawa, A.; Kashimada, A.; Mizuno, T.; Moriyama, K.; Sugawara, Y.; Shirai, I.; Miyata, Y.; et al. Long-Term Evaluation of Low-Dose Betamethasone for Ataxia Telangiectasia. Pediatr. Neurol. 2019, 100, 60–66. [Google Scholar] [CrossRef] [PubMed]
- EryDel Announces Top-Line Results from Phase3 ATTeST Trial Demonstrating Significant Clinical Benefit of EryDex in Ataxia Telangiectasia. Available online: https://assobiotec.federchimica.it/en/news/detail/2021/07/12/erydel-announces-top-line-results-from-phase3-attest-trial-demonstrating-significant-clinical-benefit-of-erydex-in-ataxia-telangiectasia (accessed on 28 March 2023).
- Köhler, W.; Engelen, M.; Eichler, F.; Lachmann, R.; Fatemi, A.; Sampson, J.; Salsano, E.; Gamez, J.; Molnar, M.J.; Pascual, S.; et al. Safety and efficacy of leriglitazone for preventing disease progression in men with adrenomyeloneuropathy (ADVANCE): A randomised, double-blind, multi-centre, placebo-controlled phase 2-3 trial. Lancet Neurol. 2023, 22, 127–136. [Google Scholar] [CrossRef]
- Grunseich, C.; Miller, R.; Swan, T.; Glass, D.J.; El Mouelhi, M.; Fornaro, M.; Petricoul, O.; Vostiar, I.; Roubenoff, R.; Meriggioli, M.N.; et al. Safety, tolerability, and preliminary efficacy of an IGF-1 mimetic in patients with spinal and bulbar muscular atrophy: A randomised, placebo-controlled trial. Lancet Neurol. 2018, 17, 1043–1052. [Google Scholar] [CrossRef]
- Diaz, G.A.; Jones, S.A.; Scarpa, M.; Mengel, K.E.; Giugliani, R.; Guffon, N.; Batsu, I.; Fraser, P.A.; Li, J.; Zhang, Q.; et al. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet. Med. 2021, 23, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Diaz, G.A.; Lachmann, R.H.; Jouvin, M.-H.; Nandy, I.; Ji, A.J.; Puga, A.C. Olipudase alfa for treatment of acid sphingomyelinase deficiency (ASMD): Safety and efficacy in adults treated for 30 months. J. Inherit. Metab. Dis. 2018, 41, 829–838. [Google Scholar] [CrossRef]
- Hall, D.A.; Robertson, E.E.; Leehey, M.; McAsey, A.; Ouyang, B.; Berry-Kravis, E.; O’keefe, J.A. Open-label pilot clinical trial of citicoline for fragile X-associated tremor/ataxia syndrome (FXTAS). PloS ONE 2020, 15, e0225191. [Google Scholar] [CrossRef]
- Attarian, S.; Vallat, J.-M.; Magy, L.; Funalot, B.; Gonnaud, P.-M.; Lacour, A.; Péréon, Y.; Dubourg, O.; Pouget, J.; Micallef, J.; et al. An exploratory randomised double-blind and placebo-controlled phase 2 study of a combination of baclofen, naltrexone and sorbitol (PXT3003) in patients with Charcot-Marie-Tooth disease type 1A. Orphanet J. Rare Dis. 2014, 9, 199. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maia, L.F.; Da Silva, A.M.; Cruz, M.W.; Planté-Bordeneuve, V.; Suhr, O.B.; Conceiçao, I.; Schmidt, H.H.-J.; Trigo, P.; Kelly, J.W.; et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J. Neurol. 2013, 260, 2802–2814. [Google Scholar] [CrossRef]
- Carroll, A.; Dyck, P.J.; de Carvalho, M.; Kennerson, M.; Reilly, M.M.; Kiernan, M.C.; Vucic, S. Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 668–678. [Google Scholar] [CrossRef]
- Cantone, A.; Sanguettoli, F.; Dal Passo, B.; Serenelli, M.; Rapezzi, C. The treatment of amyloidosis is being refined. Eur. Heart J. Suppl. J. Eur. Soc. Cardiol. 2022, 24 (Suppl. SI), I131–I138. [Google Scholar] [CrossRef]
- Narayanan, P.; Curtis, B.R.; Shen, L.; Schneider, E.; Tami, J.A.; Paz, S.; Burel, S.A.; Tai, L.-J.; Machemer, T.; Kwoh, T.J.; et al. Underlying Immune Disorder May Predispose Some Transthyretin Amyloidosis Subjects to Inotersen-Mediated Thrombocytopenia. Nucleic Acid. Ther. 2020, 30, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Gorevic, P.; Franklin, J.; Chen, J.; Sajeev, G.; Wang, J.C.H.; Lin, H. Indirect treatment comparison of the efficacy of patisiran and inotersen for hereditary transthyretin-mediated amyloidosis with polyneuropathy. Expert. Opin. Pharmacother. 2021, 22, 121–129. [Google Scholar] [CrossRef]
- Planté-Bordeneuve, V.; Lin, H.; Gollob, J.; Agarwal, S.; Betts, M.; Fahrbach, K.; Chitnis, M.; Polydefkis, M. An indirect treatment comparison of the efficacy of patisiran and tafamidis for the treatment of hereditary transthyretin-mediated amyloidosis with polyneuropathy. Expert. Opin. Pharmacother. 2019, 20, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Nakanishi, H.; Nakamura, T.; Atsuta, N.; Yamada, S.; Hijikata, Y.; Hashizume, A.; Suzuki, K.; Katsuno, M.; Sobue, G. Myotonia-like symptoms in a patient with spinal and bulbar muscular atrophy. Neuromuscul. Disord. NMD 2015, 25, 913–915. [Google Scholar] [CrossRef]
- Dainese, L.; Monin, M.-L.; Demeret, S.; Brochier, G.; Froissart, R.; Spraul, A.; Schiffmann, R.; Seilhean, D.; Mochel, F. Abnormal glycogen in astrocytes is sufficient to cause adult polyglucosan body disease. Gene 2013, 515, 376–379. [Google Scholar] [CrossRef]
- Godel, T.; Bäumer, P.; Pham, M.; Köhn, A.; Muschol, N.; Kronlage, M.; Kollmer, J.; Heiland, S.; Bendszus, M.; Mautner, V.-F. Human dorsal root ganglion in vivo morphometry and perfusion in Fabry painful neuropathy. Neurology 2017, 89, 1274–1282. [Google Scholar] [CrossRef]
- Hofmann, L.; Hose, D.; Grießhammer, A.; Blum, R.; Döring, F.; Dib-Hajj, S.; Waxman, S.; Sommer, C.; Wischmeyer, E.; Üçeyler, N. Characterization of small fiber pathology in a mouse model of Fabry disease. eLife 2018, 7, e39300. [Google Scholar] [CrossRef]
- Waltz, T.B.; Burand, A.J.; Sadler, K.E.; Stucky, C.L. Sensory-specific peripheral nerve pathology in a rat model of Fabry disease. Neurobiol. Pain. 2021, 10, 100074. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Schiffmann, R.; Floeter, M.K.; Dambrosia, J.M.; Gupta, S.; Moore, D.F.; Sharabi, Y.; Khurana, R.K.; Brady, R.O. Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve 2003, 28, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Hilz, M.J.; Brys, M.; Marthol, H.; Stemper, B.; Dütsch, M. Enzyme replacement therapy improves function of C-, Adelta-, and Abeta-nerve fibers in Fabry neuropathy. Neurology 2004, 62, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Kopp, J.B.; Iii, H.A.A.; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in Fabry disease: A randomized controlled trial. JAMA 2001, 285, 2743–2749. [Google Scholar] [CrossRef]
- Wu, X.; Katz, E.; Della Valle, M.C.; Mascioli, K.; Flanagan, J.J.; Castelli, J.P.; Schiffmann, R.; Boudes, P.; Lockhart, D.J.; Valenzano, K.J.; et al. A pharmacogenetic approach to identify mutant forms of α-galactosidase A that respond to a pharmacological chaperone for Fabry disease. Hum. Mutat. 2011, 32, 965–977. [Google Scholar] [CrossRef] [PubMed]
- EMA. Givlaari. European Medicines Agency. Published January 29, 2020. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/givlaari (accessed on 29 March 2023).
- Bonkowsky, H.L.; Schady, W. Neurologic Manifestations of Acute Porphyria. Semin. Liver Dis. 1982, 2, 108–124. [Google Scholar] [CrossRef]
- Di Gregorio, E.; Borroni, B.; Giorgio, E.; Lacerenza, D.; Ferrero, M.; Buono, N.L.; Ragusa, N.; Mancini, C.; Gaussen, M.; Calcia, A.; et al. ELOVL5 Mutations Cause Spinocerebellar Ataxia 38. Am. J. Hum. Genet. 2014, 95, 209–217. [Google Scholar] [CrossRef]
- Borroni, B.; Di Gregorio, E.; Orsi, L.; Vaula, G.; Costanzi, C.; Tempia, F.; Mitro, N.; Caruso, D.; Manes, M.; Pinessi, L.; et al. Clinical and neuroradiological features of spinocerebellar ataxia 38 (SCA38). Park. Relat. Disord. 2016, 28, 80–86. [Google Scholar] [CrossRef]
- Romano, S.; Coarelli, G.; Marcotulli, C.; Leonardi, L.; Piccolo, F.; Spadaro, M.; Frontali, M.; Ferraldeschi, M.; Vulpiani, M.C.; Ponzelli, F.; et al. Riluzole in patients with hereditary cerebellar ataxia: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015, 14, 985–991. [Google Scholar] [CrossRef]
- Velázquez-Pérez, L.; Rodríguez-Labrada, R.; García-Rodríguez, J.C.; Almaguer-Mederos, L.E.; Cruz-Mariño, T.; Laffita-Mesa, J.M. A comprehensive review of spinocerebellar ataxia type 2 in Cuba. Cerebellum 2011, 10, 184–198. [Google Scholar] [CrossRef]
- Rey, F.; Balsari, A.; Giallongo, T.; Ottolenghi, S.; Di Giulio, A.M.; Samaja, M.; Carelli, S. Erythropoietin as a Neuroprotective Molecule: An Overview of Its Therapeutic Potential in Neurodegenerative Diseases. ASN Neuro 2019, 11, 1759091419871420. [Google Scholar] [CrossRef]
- Biegstraaten, M.; Mengel, E.; Maródi, L.; Petakov, M.; Niederau, C.; Giraldo, P.; Hughes, D.; Mrsic, M.; Mehta, A.; Hollak, C.E.M.; et al. Peripheral neuropathy in adult type 1 Gaucher disease: A 2-year prospective observational study. Brain 2010, 133, 2909–2919. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Pelled, D.; Riebeling, C.; Bodennec, J.; De-Morgan, A.; Waller, H.; Schiffmann, R.; Futerman, A.H. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J. Biol. Chem. 2003, 278, 23594–23599. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Szer, J.; Mehta, A.; Zimran, A. How we manage Gaucher Disease in the era of choices. Br. J. Haematol. 2018, 182, 467–480. [Google Scholar] [CrossRef]
- Dyck, P.J.B.; González-Duarte, A.; Obici, L.; Polydefkis, M.; Wiesman, J.; Antonino, I.; Litchy, W. Development of measures of polyneuropathy impairment in hATTR amyloidosis: From NIS to mNIS + 7. J. Neurol. Sci. 2019, 405, 116424. [Google Scholar] [CrossRef]
- Corrà, M.F.; Warmerdam, E.; Vila-Chã, N.; Maetzler, W.; Maia, L. Wearable Health Technology to Quantify the Functional Impact of Peripheral Neuropathy on Mobility in Parkinson’s Disease: A Systematic Review. Sensors 2020, 20, 6627. [Google Scholar] [CrossRef] [PubMed]
- Brognara, L.; Mazzotti, A.; Di Martino, A.; Faldini, C.; Cauli, O. Wearable Sensor for Assessing Gait and Postural Alterations in Patients with Diabetes: A Scoping Review. Medicina 2021, 57, 1145. [Google Scholar] [CrossRef]
- Abdelnaby, R.; Elgenidy, A.; Sonbol, Y.T.; Dardeer, K.T.; Ebrahim, M.A.; Maallem, I.; Youssef, M.W.; Moawad, M.H.E.D.; Hassan, Y.G.; Rabie, S.A.; et al. Nerve Sonography in Charcot–Marie–Tooth Disease: A Systematic Review and Meta-analysis of 6061 Measured Nerves. Ultrasound Med. Biol. 2022, 48, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.M.; Evans, M.R.; Grider, T.; Sinclair, C.D.; Thedens, D.; Shah, S.; Yousry, T.A.; Hanna, M.G.; Nopoulos, P.; Thornton, J.S.; et al. Validation of MRC Centre MRI calf muscle fat fraction protocol as an outcome measure in CMT1A. Neurology 2018, 91, e1125–e1129. [Google Scholar] [CrossRef] [PubMed]
- Sandelius, Å.; Zetterberg, H.; Blennow, K.; Adiutori, R.; Malaspina, A.; Laura, M.; Reilly, M.M.; Rossor, A.M. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology 2018, 90, e518–e524. [Google Scholar] [CrossRef]
- Landrieu, P.; Saïd, G. Peripheral neuropathy in type A Niemann-Pick disease. Acta Neuropathol. 1984, 63, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Gumbinas, M.; Larsen, M.; Liu, H.M. Peripheral neuropathy in classic Niemann-Pick disease: Ultrastructure of nerves and skeletal muscles. Neurology 1975, 25, 107. [Google Scholar] [CrossRef]
- Steinman, L.; Tharp, B.R.; Dorfman, L.J.; Forno, L.S.; Sogg, R.L.; Kelts, K.A.; O’Brien, J.S. Peripheral neuropathy in the cherry-red spot-myoclonus syndrome (sialidosis type I). Ann. Neurol. 1980, 7, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Varho, T.; Jääskeläinen, S.; Tolonen, U.; Sonninen, P.; Vainionpaa, L.; Aula, P.; Sillanpaa, M. Central and peripheral nervous system dysfunction in the clinical variation of Salla disease. Neurology 2000, 55, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 121. [Google Scholar] [CrossRef]
- Ahrens-Nicklas, R.; Schlotawa, L.; Ballabio, A.; Brunetti-Pierri, N.; De Castro, M.; Dierks, T.; Eichler, F.; Ficicioglu, C.; Finglas, A.; Gaertner, J.; et al. Complex care of individuals with Multiple Sulfatase Deficiency: Clinical cases and consensus statement. Mol. Genet. Metab. 2018, 123, 337–346. [Google Scholar] [CrossRef]
- Carneiro, D.R.; Rebelo, O.; Matos, A.; Baldeiras, I.; Almendra, L.; Fernandes, C.; Negrão, L.; Almeida, M.R.; Matias, F.; Brás, J.; et al. Vasculitic peripheral neuropathy in deficiency of adenosine deaminase 2. Neuromuscul. Disord. NMD 2021, 31, 891–895. [Google Scholar] [CrossRef]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.-G.; Ikeda, S.-i.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef]
- Adams, D. Recent advances in the treatment of familial amyloid polyneuropathy. Ther. Adv. Neurol. Disord. 2013, 6, 129–139. [Google Scholar] [CrossRef]
- Bhandari, J.; Thada, P.K.; Samanta, D. Spinocerebellar Ataxia; StatPearls Publishing: Tampa, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557816/ (accessed on 21 March 2023).
- Kaufmann, H.; Norcliffe-Kaufmann, L.; Palma, J.A. Baroreflex Dysfunction. N. Engl. J. Med. 2020, 382, 163–178. [Google Scholar] [CrossRef]
- Penno, A.; Reilly, M.M.; Houlden, H.; Laurá, M.; Rentsch, K.; Niederkofler, V.; Stoeckli, E.T.; Nicholson, G.; Eichler, F.; Brown, R.H.; et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J. Biol. Chem. 2010, 285, 11178–11187. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; King, R.; Blake, J.; Groves, M.; Love, S.; Woodward, C.; Hammans, S.; Nicoll, J.; Lennox, G.; O’Donovan, D.G.; et al. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain J. Neurol. 2006, 129 Pt. 2, 411–425. [Google Scholar] [CrossRef]
- Ambrose, M.; Gatti, R.A. Pathogenesis of ataxia-telangiectasia: The next generation of ATM functions. Blood 2013, 121, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Storey, E. Genetic Cerebellar Ataxias. Semin. Neurol. 2014, 34, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Rossor, A.; Tomaselli, P.; Reilly, M. Recent advances in the genetic neuropathies. Curr. Opin. Neurol. 2016, 29, 537–548. [Google Scholar] [CrossRef]
- Li, J.; Parker, B.; Martyn, C.; Natarajan, C.; Guo, J. The PMP22 Gene and Its Related Diseases. Mol. Neurobiol. 2013, 47, 673–698. [Google Scholar] [CrossRef]
- Gieselmann, V. Metachromatic leukodystrophy: Genetics, pathogenesis and therapeutic options. Acta Paediatr. 2008, 97 (Suppl. S457), 15–21. [Google Scholar] [CrossRef]
- Van den Broek, B.T.A.; Page, K.; Paviglianiti, A.; Hol, J.; Allewelt, H.; Volt, F.; Michel, G.; Diaz, M.A.; Bordon, V.; O’Brien, T.; et al. Early and late outcomes after cord blood transplantation for pediatric patients with inherited leukodystrophies. Blood Adv. 2018, 2, 49–60. [Google Scholar] [CrossRef]
- Boucher, A.A.; Miller, W.; Shanley, R.; Ziegler, R.; Lund, T.; Raymond, G.; Orchard, P.J. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: The largest single-institution cohort report. Orphanet J. Rare Dis. 2015, 10, 94. [Google Scholar] [CrossRef]
- Engelen, M.; Kemp, S.; De Visser, M.; Van Geel, B.M.; Wanders, R.J.A.; Aubourg, P. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef]
- Katsuno, M.; Tanaka, F.; Adachi, H.; Banno, H.; Suzuki, K.; Watanabe, H.; Sobue, G. Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA). Prog. Neurobiol. 2012, 99, 246–256. [Google Scholar] [CrossRef]
- Akman, H.O.; Lossos, A.; Kakhlon, O. GBE1 Adult Polyglucosan Body Disease; University of Washington: Seattle, DC, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK5300/ (accessed on 28 March 2023).
- Desnick, R.J.; Brady, R.; Barranger, J.; Collins, A.J.; Germain, D.P.; Goldman, M.; Grabowski, G.; Packman, S.; Wilcox, W.R. Fabry Disease, an Under-Recognized Multisystemic Disorder: Expert Recommendations for Diagnosis, Management, and Enzyme Replacement Therapy. Ann. Intern. Med. 2003, 138, 338–346. [Google Scholar] [CrossRef]
- Levine, H. Treatment Options for Fabry Disease. WebMD. Available online: https://www.webmd.com/children/fabry-disease-treat (accessed on 29 March 2023).
- Wasserstein, M.P.; Aron, A.; Brodie, S.E.; Simonaro, C.; Desnick, R.J.; McGovern, M.M. Acid sphingomyelinase deficiency: Prevalence and characterization of an intermediate phenotype of Niemann-Pick disease. J. Pediatr. 2006, 149, 554–559. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Momoi, M.Y.; Tominaga, K.; Momoi, T.; Nihei, K.; Yanagisawa, M.; Kagawa, Y.; Ohta, S. A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem. Biophys. Res. Commun. 1990, 173, 816–822. [Google Scholar] [CrossRef]
- Luigetti, M.; Sauchelli, D.; Primiano, G.; Cuccagna, C.; Bernardo, D.; Monaco, M.L.; Servidei, S. Peripheral neuropathy is a common manifestation of mitochondrial diseases: A single-centre experience. Eur. J. Neurol. 2016, 23, 1020–1027. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Szer, J.; Zimran, A. Gaucher Disease and Related Lysosomal Storage Diseases. In Williams Hematology, 10th ed.; Kaushansky, K., Prchal, J.T., Burns, L.J., Lichtman, M.A., Levi, M., Linch, D.C., Eds.; McGraw-Hill Education: New York, NY, USA, 2021. [Google Scholar]
- Bissell, D.M.; Lai, J.C.; Meister, R.K.; Blanc, P.D. Role of delta-aminolevulinic acid in the symptoms of acute porphyria. Am. J. Med. 2015, 128, 313–317. [Google Scholar] [CrossRef]
- Silva, C.D.; Mateus, J.E.; Teles, C.; Vaio, T. Acute intermittent porphyria: Analgesia can be dangerous. BMJ Case Rep. 2019, 12, e231133. [Google Scholar] [CrossRef]
- Tracy, J.A.; Dyck, P.J.B. Porphyria and its neurologic manifestations. Handb. Clin. Neurol. 2014, 120, 839–849. [Google Scholar] [CrossRef]
- Stein, P.E.; Badminton, M.N.; Rees, D.C. Update review of the acute porphyrias. Br. J. Haematol. 2017, 176, 527–538. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Bidichandani, S.I. Friedreich ataxia- pathogenesis and implications for therapies. Neurobiol. Dis. 2019, 132, 104606. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, J.M.; Balcer, L.J.; Wilson, R.B. Friedreich ataxia: Effects of genetic understanding on clinical evaluation and therapy. Arch. Neurol. 2002, 59, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.S.; Bove, K.E.; Lovell, M.A.; Sokol, R.J. Mechanisms of disease: Inborn errors of bile acid synthesis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2008, 5, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Berendse, K.; Engelen, M.; Ferdinandusse, S.; Majoie, C.B.L.M.; Waterham, H.R.; Vaz, F.; Koelman, J.H.T.M.; Barth, P.G.; Wanders, R.J.A.; Poll-The, B.T. Zellweger spectrum disorders: Clinical manifestations in patients surviving into adulthood. J. Inherit. Metab. Dis. 2016, 39, 93–106. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile, X. Neurology 2001, 57, 127–130. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).