
Understanding Fenofibrate Release from Bare and Modified Mesoporous Silica Nanoparticles

 , , ,
, , ,  and
and
Abstract

1. Introduction
2. Materials and Methods
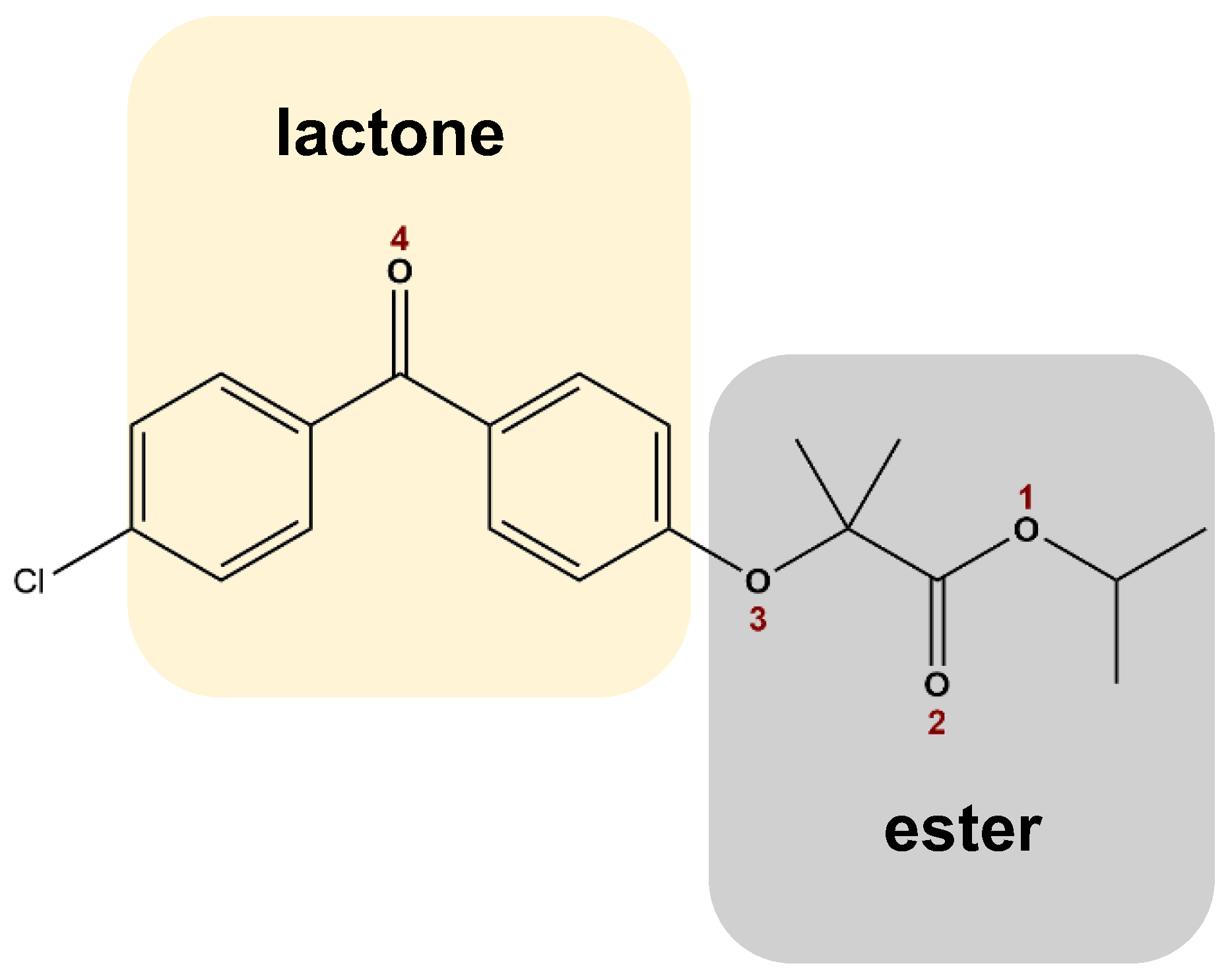
2.1. Materials
2.2. Equipment
2.3. Methods
3. Results
3.1. Structural Characterization of MSNs
3.2. Quantification of Grafted Functional Groups
3.3. FTIR Analysis of MSNs Loaded with FNB
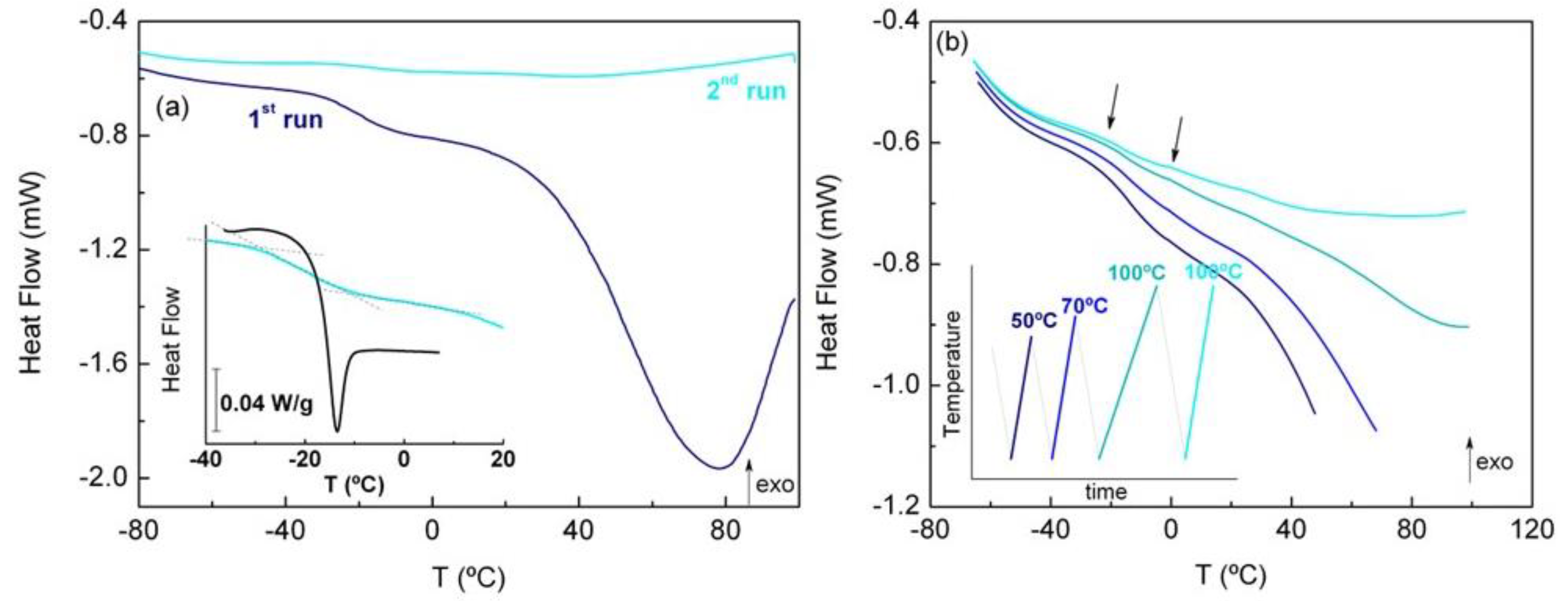
3.4. Differential Scanning Calorimetry (DSC)
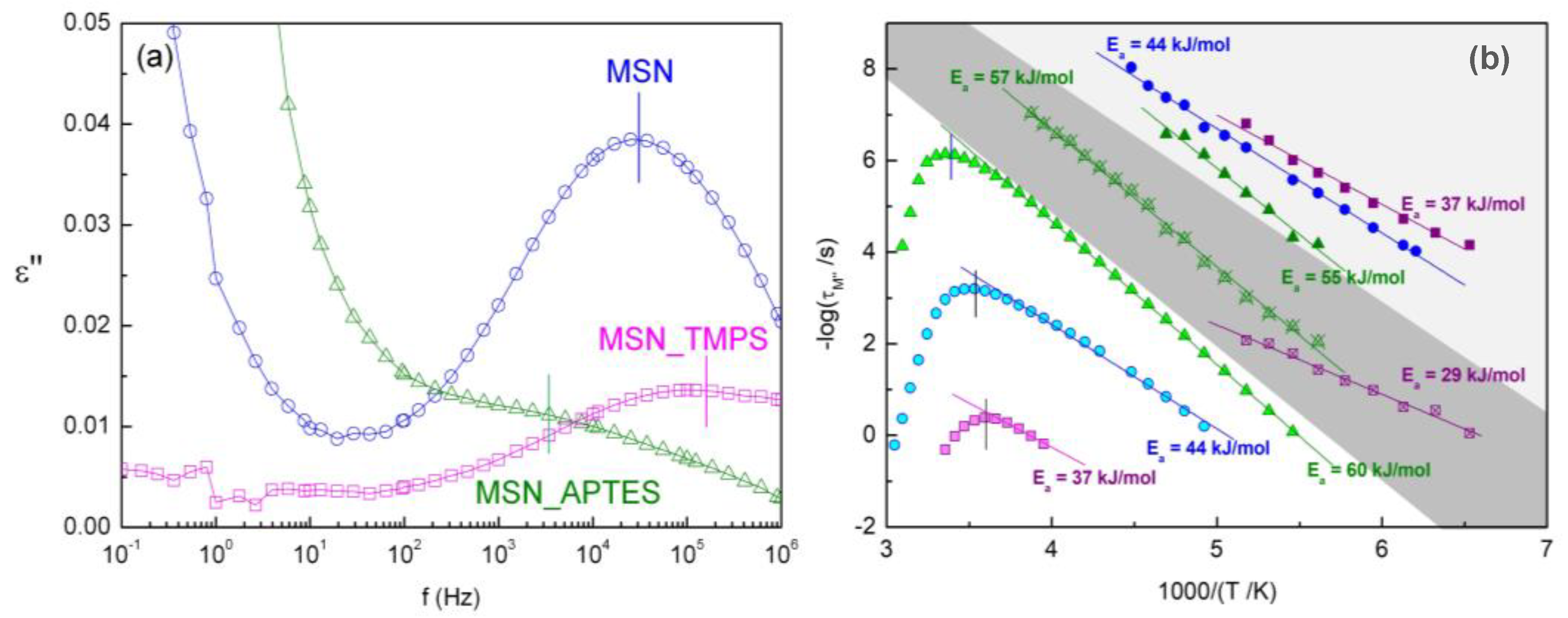
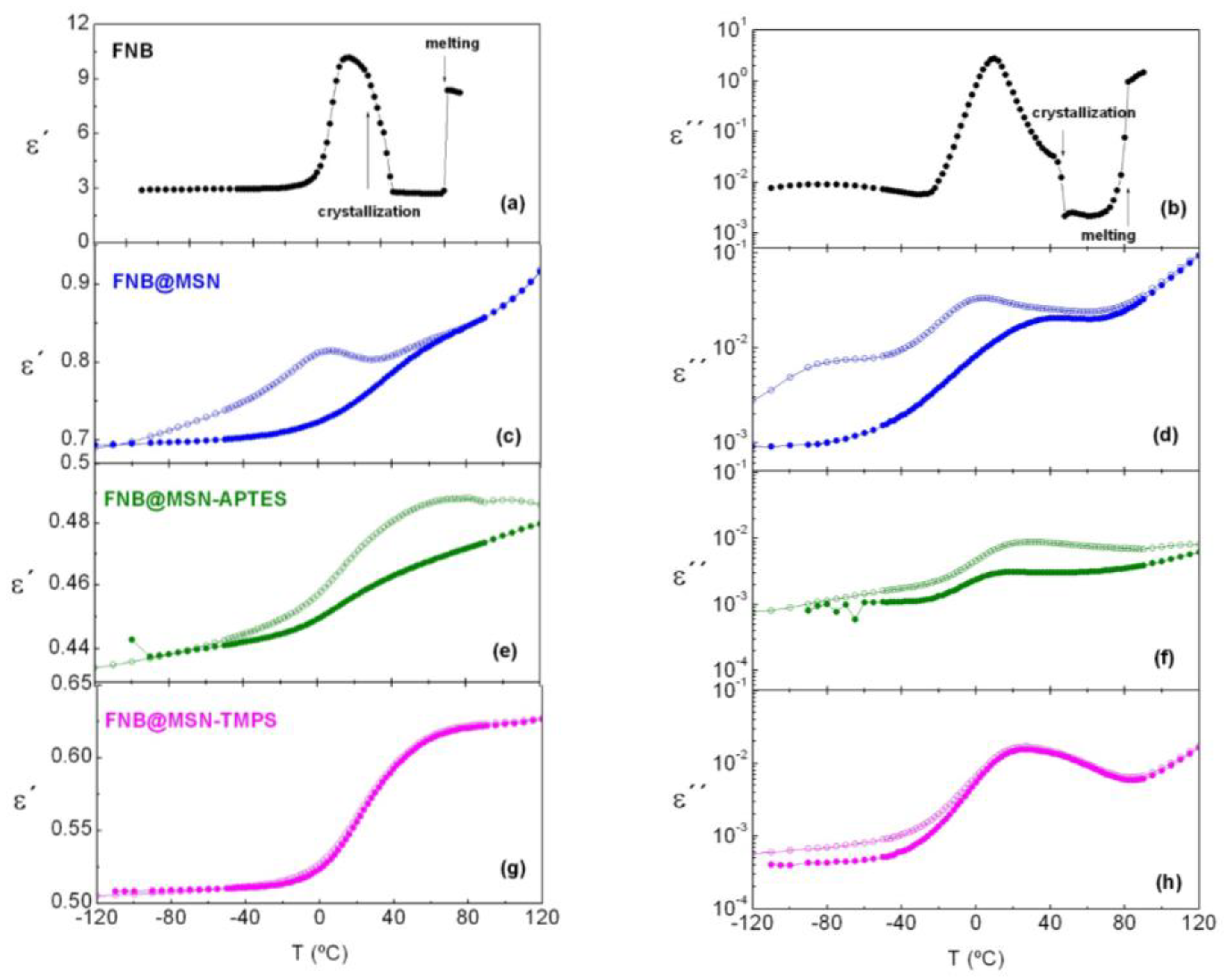
3.5. Dielectric Relaxation Spectroscopy (DRS)
3.5.1. Unloaded Hydrated Matrices
3.5.2. Bulk FNB
3.5.3. FNB-Loaded Matrices
3.5.4. Conductivity of the Loaded Samples
3.6. Release Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grohganz, H.; Priemel, P.A.; Löbmann, K.; Nielsen, L.H.; Laitinen, R.; Mullertz, A.; den Mooter, G.V.; Rades, T. Refining stability and dissolution rate of amorphous drug. Expert Opin. Drug Deliv. 2014, 11, 977–989. [Google Scholar] [CrossRef]
- Beiner, M.; Pankaj, S.; Enke, D.; Steinhart, M. Manipulating the crystalline state of pharmaceuticals by nanoconfinement. Nano Lett. 2007, 7, 1381–1385. [Google Scholar] [CrossRef]
- Brás, A.R.; Fonseca, I.M.; Dionísio, M.; Schönhals, A.; Affouard, F.; Correia, N.T. Influence of nanoscale confinement on the molecular mobility of ibuprofen. J. Phys. Chem. C 2014, 118, 13857–13868. [Google Scholar] [CrossRef]
- Cordeiro, T.; Santos, A.F.M.; Nunes, G.; Cunha, G.; Sotomayor, J.C.; Fonseca, I.M.; Danède, F.; Dias, C.J.; Cardoso, M.M.; Correia, N.T.; et al. Accessing the physical state and molecular mobility of naproxen confined to nanoporous silica matrixes. J. Phys. Chem. C 2016, 120, 14390–14401. [Google Scholar] [CrossRef]
- Graubner, G.; Anders, N.; Sonnenberger, N.; Steinhart, M. Morphology of porous host directs preferred polymorph formation and influences kinetics of solid/solid transition of confined pharmaceuticals. Cryst. Growth Des. 2014, 14, 78–86. [Google Scholar] [CrossRef]
- Khanfar, M.; Fares, M.M.; Qandil, A.M. Mesoporous silica-based macromolecules for dissolution enhancement of irbesartan drug using pre-adjusted pH method. Microporous Mesoporous Mater. 2013, 173, 22–28. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, G.G.Z.; Law, D.; Grant, D.J.W.; Schmitt, E.A. Physical stability of amorphous pharmaceuticals: Importance of configurational thermodynamic quantities and molecular mobility. J. Pharm. Sci. 2002, 91, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Downing, N.S.; Ross, J.S.; Jackevicius, C.A.; Krumholz, H.M. How Abbott’s fenofibrate franchise avoided generic competition. Arch. Intern. Med. 2012, 172, 724–730. [Google Scholar] [CrossRef]
- Ling, H.; Luoma, J.T.; Hilleman, D. A Review of Currently Available Fenofibrate and Fenofibric Acid Formulations. Cardiol. Res. 2013, 4, 47–55. [Google Scholar] [CrossRef]
- Tipduangta, P.; Takieddin, K.; Fabian, L.; Belton, P.; Qi, S. Towards controlling the crystallisation behaviour of fenofibrate melt: Triggers of crystallisation and polymorphic transformation. RSC Adv. 2018, 8, 13153–13525. [Google Scholar] [CrossRef]
- Cao, Y.-N.; Baiyisaiti, A.; Wong, C.-W.; Hsu, S.-H.; Qi, R. Polyurethane nanoparticle-loaded fenofibrate exerts inhibitory effects on nonalcoholic fatty liver disease in mice. Mol. Pharm. 2018, 15, 4550–4557. [Google Scholar] [CrossRef]
- Domenech, T.; Doyle, P.S. High loading capacity nanoencapsulation and release of hydrophobic drug nanocrystals from microgel particles. Chem. Mater. 2020, 32, 498–509. [Google Scholar] [CrossRef]
- Quan, G.; Wu, Q.; Zhang, Z.; Zhan, Z.; Zhou, C.; Chen, B.; Zhang, Z.; Li, G.; Pan, X.; Wu, C. Enhancing in vitro dissolution and in vivo bioavailability of fenofibrate by solid self-emulsifying matrix combined with SBA-15. Colloids Surf. B Biointerfaces 2017, 141, 476–482. [Google Scholar] [CrossRef]
- Jia, Z.; Lin, P.; Xiang, Y.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. A novel nanomatrix system consisted of colloidal silica and pH-sensitive polymethylacrylate improves the oral bioavailability of fenofibrate. Eur. J. Pharm. Biopharm. 2011, 79, 126–134. [Google Scholar] [CrossRef]
- Varache, M.; Bezverkhyy, I.; Saviot, L.; Bouyer, F.; Baras, F.; Bouyer, F. Optimization of MCM-41 type silica nanoparticles for biological applications: Control of size and absence of aggregation and cell cytotoxicity. J. Non-Cryst. Solids 2015, 408, 87–97. [Google Scholar] [CrossRef]
- Skwira, A.; Szewczyk, A.; Konopacka, A.; Górska, M.; Majda, D.; Sądej, R.; Prokopowicz, M. Silica-polymer composites as the novel antibiotic delivery systems for bone tissue infection. Pharmaceutics 2020, 12, 28. [Google Scholar] [CrossRef]
- Alharthi, S.; Ziora, Z.M.; Janjua, T.; Popat, A.; Moyle, P.M. Formulation and biological evaluation of mesoporous silica nanoparticles loaded with combinations of sortase A inhibitors and antimicrobial peptides. Pharmaceutics 2022, 14, 986. [Google Scholar] [CrossRef]
- Sanganwar, G.P.; Gupta, R.B. Dissolution-rate enhancement of fenofibrate by adsorption onto silica using supercritical carbon dioxide. Int. J. Pharm. 2008, 360, 213–218. [Google Scholar] [CrossRef]
- Szklarz, G.; Adrjanowicz, K.; Tarnacka, M.; Pionteck, J.; Paluch, M. Confinement-induced changes in the glassy dynamics and crystallization behavior of supercooled fenofibrate. J. Phys. Chem. C 2018, 122, 1384–1395. [Google Scholar] [CrossRef]
- Bukara, K.; Schueller, L.; Rosier, J.; Martens, M.A.; Daems, T.; Verheyden, L.; Eelen, S.; Van Speybroeck, M.; Libanati, C.; Martens, J.A.; et al. Ordered mesoporous silica to enhance the bioavailability of poorly water-soluble drugs: Proof of concept in man. Eur. J. Pharm. Biopharm. 2016, 108, 220–225. [Google Scholar] [CrossRef]
- Bukara, K.; Schueller, L.; Rosier, J.; Daems, T.; Verheyden, L.; Eelen, S.; Martens, J.A.; Van den Mooter, G.; Bugarski, B.; Kiekens, F. In vivo performance of fenofibrate formulated with ordered mesoporous silica versus 2-marketed formulations: A comparative bioavailability study in beagle dogs. J. Pharm. Sci. 2016, 105, 2381–2385. [Google Scholar] [CrossRef] [PubMed]
- Sailaja, U.; Thayyil, M.S.; Kumar, N.S.K.; Govindaraj, G. Molecular dynamics of amorphous pharmaceutical fenofibrate studied by broadband dielectric spectroscopy. J. Pharm. Anal. 2016, 6, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Diogo, H.P.; Viciosa, M.T.; Ramos, J.J.M. Differential scanning calorimetry and thermally stimulated depolarization currents study on the molecular dynamics in amorphous fenofibrate. Thermochim. Acta 2016, 623, 29–35. [Google Scholar] [CrossRef]
- Schrader, A.M.; Monroe, J.I.; Sheil, R.; Dobbs, H.A.; Keller, T.J.; Li, Y.; Jain, S.; Shella, M.S.; Israelachvili, J.N.; Han, S. Surface chemical heterogeneity modulates silica surface hydration. Proc. Natl. Acad. Sci. USA 2018, 115, 2890–2895. [Google Scholar] [CrossRef]
- Havriliak, S.; Negami, S. A complex plane representation of dielectric and mechanical relaxation processes in some polymers. Polymer 1967, 8, 161–210. [Google Scholar] [CrossRef]
- Havriliak, S.; Negami, S. A complex plane analysis of α-dispersions in some polymer systems. J. Polym. Sci. Part C. 1966, 14, 99–117. [Google Scholar] [CrossRef]
- Kremer, F.; Schönhals, A. Broadband Dielectric Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2003; ISBN 9783540434078. [Google Scholar]
- Vogel, D.H. Das Temperaturabhaengigkeitsgesetz der Viskositaet von Fluessigkeiten. Phys. Z. 1921, 22, 645. [Google Scholar]
- Fulcher, G.S. Analysis of recent measurements of the viscosity of glasses. J. Am. Ceram. Soc. 1925, 8, 339. [Google Scholar] [CrossRef]
- Tammann, G.; Hesse, G.Z. Die Abhängigkeit der Viscosität von der Temperatur bie unterkühlten Flüssigkeiten. Anorg. Allg. Chem. 1926, 156, 245. [Google Scholar] [CrossRef]
- Calderón, S.V.; Ribeiro, T.; Farinha, J.P.S.; Baleizão, C.; Ferreira, P.J. On the structure of amorphous mesoporous silica nanoparticles by aberration-corrected STEM. Small 2018, 14, 1802180. [Google Scholar] [CrossRef]
- Ribeiro, T.; Rodrigues, A.S.; Calderón, S.; Fidalgo, A.; Gonçalves, J.L.M.; André, V.; Duarte, M.T.; Ferreira, P.J.; Farinha, J.P.S.; Baleizão, C. Silica nanocarriers with user-defined precise diameters by controlled template self-assembly. J. Colloid Interface Sci. 2020, 561, 609–619. [Google Scholar] [CrossRef]
- Limnell, T.; Riikonen, J.; Salonen, J.; Kaukonen, A.M.; Laitinen, L.; Hirvonen, J.; Lehto, V.P. Surface chemistry and pore size affect carrier properties of mesoporous silicon microparticles. Int. J. Pharm. 2007, 343, 141–147. [Google Scholar] [CrossRef]
- Hong, S.; Shen, S.; Tan, D.C.T.; Ng, W.K.; Liu, X.; Chia, L.S.O.; Irwan, A.W.; Tan, R.; Nowak, S.A.; Marsh, K.; et al. High drug load, stable, manufacturable and bioavailable fenofibrate formulations in mesoporous silica: A comparison of spray drying versus solvent impregnation methods. Drug Deliv. 2016, 23, 316–327. [Google Scholar] [CrossRef]
- Inocêncio, S.; Cordeiro, T.; Matos, I.; Danède, F.; Sotomayor, J.C.; Fonseca, I.M.; Correia, N.T.; Corvo, M.C.; Dionísio, M. Ibuprofen incorporated into unmodified and modified mesoporous silica: From matrix synthesis to drug release. Microporous Mesoporous Mater. 2021, 310, 110541. [Google Scholar] [CrossRef]
- Ribeiro, T.; Coutinho, E.; Rodrigues, A.S.; Baleizão, C.; Farinha, J.P.S. Hybrid mesoporous silica nanocarriers with thermovalve-regulated controlled release. Nanoscale 2017, 9, 13485–13494. [Google Scholar] [CrossRef]
- Watterson, S.; Hudson, S.; Svard, M.; Rasmuson, A.C. Thermodynamics of fenofibrate and solubility in pure organic solvents. Fluid Phase Equilibria 2014, 367, 143–150. [Google Scholar] [CrossRef]
- Crucho, C.I.C.; Baleizão, C.; Farinha, J.P.S. Functional group coverage and conversion quantification in nanostructured silica by 1H-NMR. Anal. Chem. 2017, 89, 681–687. [Google Scholar] [CrossRef]
- Zhuravlev, L.T.; Potapov, V.V. Density of silanol groups on the surface of silica precipitated from a hydrothermal solution. Russ. J. Phys. Chem. 2006, 80, 1119–1128. [Google Scholar] [CrossRef]
- Avval, T.G.; Průša, S.; Cushman, C.V.; Hodges, G.T.; Fearn, S.; Kim, S.H.; Čechal, J.; Vaníčkov, E.; Bábík, P.; Šikola, T.; et al. A tag-and-count approach for quantifying surface silanol densities on fused silica based on atomic layer deposition and high-sensitivity low-energy ion scattering. Appl. Surf. Sci. 2023, 607, 154551. [Google Scholar] [CrossRef]
- Puibasset, J.; Pellenq, R.J.M. Grand canonical Monte Carlo simulation study of water structure on hydrophilic mesoporous and plane silica substrates. J. Chem. Phys. 2003, 119, 9226–9232. [Google Scholar] [CrossRef]
- Acres, R.G.; Ellis, A.V.; Alvino, J.; Lenahan, C.E.; Khodakov, D.A.; Metha, G.F.; Andersson, G.G. Molecular structure of 3-Aminopropyltriethoxysilane layers formed on silanol-terminated silicon surfaces. Phys. Chem. C 2012, 116, 6289–6297. [Google Scholar] [CrossRef]
- Liu, H.; Li, H.; Ding, Z.; Fu, A.; Wang, H.; Guo, P.; Yu, J.; Wang, C.; Zhao, X.S. Preparation of porous hollow SiO2 spheres by a modified Stöber process using MF microspheres as templates. J. Clust. Sci. 2012, 23, 273–285. [Google Scholar] [CrossRef]
- DWood, L.; Rabinovich, E.M.; Johnson, D.W.; MacChesney, J.B.; Vogel, E.M. Preparation of High-Silica Glasses from Colloidal Gels: III, Infrared Spectrophotometric Studies. J. Am. Ceram. Soc. 1983, 66, 693–699. [Google Scholar]
- Murphy, D.; de Pinho, M.N. An ATR-FTIR study of water in cellulose acetate membranes prepared by phase inversion. J. Membr. Sci. 1995, 106, 245–257. [Google Scholar] [CrossRef]
- Ahmadi, E.; Dehghannejad, N.; Hashemikia, S.; Ghasemnejad, M.; Tabebordbar, H. Synthesis and surface modification of mesoporous silica nanoparticles and its application as carriers for sustained drug delivery. Drug Deliv. 2014, 21, 164–172. [Google Scholar] [CrossRef] [PubMed]
- She, X.; Chen, L.; Li, C.; He, C.; He, L.; Kong, L. Functionalization of hollow mesoporous silica nanoparticles for improved 5-FU loading. J. Nanomater. 2015, 16, 108. [Google Scholar] [CrossRef]
- Li, Y.-S.; Wang, Y.; Ceesay, S. Vibrational spectra of phenyltriethoxysilane, phenyltrimethoxysilane and their sol–gels. Spectrochem. Acta Part A 2009, 71, 1819–1824. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Devi, T.S.R.; Sakthivel, P.S. Qualitative and quantitative analysis on fibrates—A spectroscopic study. Asian J. Chem. 2008, 20, 4249–4268. [Google Scholar]
- Coates, J. Interpretation of infrared spectra, a practical approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., McKelvy, M.L., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2006. [Google Scholar]
- Shi, X.; Shao, Y.; Sheng, X. A New Polymorph of fenofibrate prepared by polymer- mediated crystallization. J. Cryst. Growth 2018, 498, 93–102. [Google Scholar] [CrossRef]
- Heinz, A.; Gordon, K.C.; McGoverin, C.M.; Rades, T.; Strachan, C.J. Understanding the solid state forms of fenofibrate—A spectroscopic and computational study. Eur. J. Pharm. Biopharm. 2009, 71, 100–108. [Google Scholar] [CrossRef]
- Tipduangta, T.; Takieddin, K.; Fabián, L.; Belton, P.; Qi, S. A new low melting-point polymorph of fenofibrate prepared via talc induced heterogeneous nucleation. Cryst. Growth Des. 2015, 15, 5011–5020. [Google Scholar] [CrossRef]
- Ngo, D.; Liu, H.; Chen, Z.; Kaya, H.; Zimudzi, T.J.; Gin, S.; Mahadevan, T.; Du, J.; Kim, S.H. Hydrogen bonding interactions of H2O and SiOH on a boroaluminosilicate glass corroded in aqueous solution. npj Mater. Degrad. 2020, 4, 1. [Google Scholar] [CrossRef]
- Laurson, P.; Raudsepp, P.; Kaldmäe, H.; Kikas, A.; Mäeorg, U. The deconvolution of FTIR-ATR spectra to five Gaussians for detection of small changes in plant–water clusters. AIP Adv. 2020, 10, 085214. [Google Scholar] [CrossRef]
- Salama, F.M.M.; Nassar, M.W.I.; El-Din, M.M.K.S.; Attia, K.A.M.; Kaddah, M.Y. Determination of fenofibrate and the degradation product using simultaneous UV-derivative spectrometric method and HPLC. Am. J. Anal. Chem. 2011, 2, 332–343. [Google Scholar] [CrossRef]
- Duong, T.V.; Reekmans, G.; Venkatesham, A.; Aerschot, A.V.; Adriaensens, P.; Humbeeck, J.V.; Mooter, G.V. Spectroscopic investigation of the formation and disruption of hydrogen bonds in pharmaceutical semicrystalline dispersions. Mol. Pharm. 2017, 14, 1726–1741. [Google Scholar] [CrossRef]
- Di Martino, P.; Palmieri, G.F.; Martelli, S. Evidence of a metastable form of fenofibrate. Pharmazie 2000, 55, 625–626. [Google Scholar]
- Surana, R.; Randall, L.; Pyne, A.; Vemuri, N.M.; Suyanarayanan, R. Determination of glass transition temperature and in situ study of the plasticizing effect of water by inverse gas chromatography. Pharm. Res. 2003, 20, 1647–1654. [Google Scholar] [CrossRef]
- Gupta, J.; Nunes, C.; Jonnalagadda, S. A molecular dynamics approach for predicting the glass transition temperature and plasticization effect in amorphous pharmaceuticals. Mol. Pharm. 2013, 10, 4136–4145. [Google Scholar] [CrossRef]
- Mauro, J.C.; Smedskjaer, M.M. Statistical mechanics of glass. J. Non-Cryst. Solids 2014, 396, 41–53. [Google Scholar] [CrossRef]
- Antonino, R.S.C.M.Q.; Ruggiero, M.; Song, Z.; Nascimento, T.L.; Lima, E.M.; Bohr, A.; Knopp, M.M.; Löbmann, K. Impact of drug loading in mesoporous silica-amorphous formulations on thephysical stability of drugs with high recrystallization tendency. Int. J. Pharm. 2019, 1, 100026. [Google Scholar]
- Johari, G.P.; Whalley, E. The dielectric properties of ice Ih in the range 272–133 K. J. Chem. Phys. 1981, 75, 1333–1340. [Google Scholar] [CrossRef]
- Ryabov, Y.; Gutina, A.; Arkhipov, V.; Feldman, Y. Dielectric relaxation of water adsorbed in porous glass. J. Phys. Chem. B 2001, 105, 1845–1850. [Google Scholar] [CrossRef]
- Cerveny, S.; Schwartz, G.A.; Otegui, J.; Colmenero, J.; Loichen, J.; Westermann, S. Dielectric study of hydration water in silica nanoparticles. J. Phys. Chem. C 2012, 116, 24340–24349. [Google Scholar] [CrossRef]
- Correia, N.T.; Ramos, J.J.M. On the cooperativity of the β-relaxation: A discussion based on dielectric relaxation and thermally stimulated depolarization currents data. Phys. Chem. Chem. Phys. 2000, 2, 5712–5715. [Google Scholar] [CrossRef]
- Gutina, A.; Antropova, T.; Rysiakiewicz-Pasek, E.; Virnik, K.; Feldman, Y. Dielectric relaxation in porous glasses. Microporous Mesoporous Mater. 2003, 58, 237–254. [Google Scholar] [CrossRef]
- Frunza, L.; Schönhals, A.; Kosslick, H.; Frunza, S. Relaxation processes of water confined to AlMCM-41 molecular sieves. Influence of the hydroxyl groups of the pore surface. Eur. Phys. J. E 2008, 26, 379–386. [Google Scholar] [CrossRef]
- Vasilyeva, M.A.; Gusev, Y.A.; Shtyrlin, V.G.; Gutina, A.G.; Puzenko, A.; Ishai, P.B.; Feldman, Y. Dielectric relaxation of water in clay minerals. Clays Clay Miner. 2014, 62, 62–73. [Google Scholar] [CrossRef]
- Afzal, A.; Thayyil, M.S.; Sivaramakrishnan, P.A.; Urpayilc, S.; Capaccioli, S. Molecular dynamics in the supercooled liquid and glassy states of bezafibrate and binary mixture of fenofibrate. J. Non-Cryst. Solids 2020, 550, 120407. [Google Scholar] [CrossRef]
- Szklarz, G.; Adrjanowicz, K.; Dulski, M.; Knapik, J.; Paluch, M. Dielectric relaxation study at ambient and elevated pressure of the modeled lipophilic drug fenofibrate. J. Phys. Chem. B 2016, 120, 11298–11306. [Google Scholar] [CrossRef]
- Stalinska, J.; Zimolag, E.; Pianovich, N.A.; Zapata, A.; Lassak, A.; Rak, M.; Dean, M.; Ucar-Bilyeu, D.; Wyczechowska, D.; Culicchia, F.; et al. Chemically modified variants of Fenofibrate with antiglioblastoma potential. Transl. Oncol. 2019, 12, 895–907. [Google Scholar] [CrossRef]
- Dib, E.; Costa, I.M.; Vayssilov, G.N.; Aleksandrov, H.A.; Mintova, S. Complex H-bonded silanol network in zeolites revealed by IR and NMR spectroscopy combined with DFT calculations. J. Mater. Chem. A 2021, 9, 27347–27352. [Google Scholar] [CrossRef]
- Brás, A.R.; Dionísio, M.; Schönhals, A. Confinement and surface effects on the molecular dynamics of a nematic mixture investigated by dielectric relaxation spectroscopy. J. Phys. Chem. B 2008, 112, 8227–8235. [Google Scholar] [CrossRef]
- Leys, J.; Glorieux, C.; Thoen, J. Confinement effects on strongly polar alkylcyanobiphenyl liquid crystals probed by dielectric spectroscopy. J. Phys. Condens. Matter 2008, 20, 244111. [Google Scholar] [CrossRef]
- Kremer, F. (Ed.) Dynamics in Geometrical Confinement; Springer: Berlin/Heidelberg, Germany, 2014; ISBN 978-3-319-06099-6. [Google Scholar]
- Adam, G.; Gibbs, J.H. On the temperature dependence of cooperative relaxation properties in glass-forming liquids. J. Chem. Phys. 1965, 43, 139–146. [Google Scholar] [CrossRef]
- Uhl, M.; Fischer, J.K.H.; Sippel, P.; Bunzen, H.; Lunkenheimer, P.; Volkmer, D.; Loidl, A. Glycerol confined in zeolitic imidazolate frameworks: The temperature-dependent cooperativity length scale of glassy freezing. J. Chem. Phys. 2019, 150, 024504. [Google Scholar] [CrossRef]
- Böhmer, R.; Ngai, K.L.; Angell, C.A.; Plazek, D.J. Nonexponential relaxations in strong and fragile glass formers. J. Chem. Phys. 1993, 99, 4201–4209. [Google Scholar] [CrossRef]
- Cordeiro, T.; Matos, I.; Danède, F.; Sotomayor, J.C.; Fonseca, I.M.; Corvo, M.C.; Dionísio, M.; Viciosa, M.T.; Affouard, F.; Correia, N.T. Evidence of Strong Guest-Host Interactions in Simvastatin Loaded in Mesoporous Silica MCM-4. Pharmaceutics 2023, 15, 1320. [Google Scholar] [CrossRef]
- Samet, M.; Levchenko, V.; Boiteux, G.; Seytre, G.; Kallel, A.; Serghei, A. Electrode polarization vs. Maxwell-Wagner-Sillars interfacial polarization in dielectric spectra of materials: Characteristic frequencies and scaling laws. J. Chem. Phys. 2015, 142, 194703. [Google Scholar] [CrossRef]
- Hammami, H.; Arous, M.; Lagache, M.; Kallel, A. Study of the interfacial MWS relaxation by dielectric spectroscopy in unidirectional PZT fibres/epoxy resin composites. J. Alloy. Compd. 2007, 430, 1–8. [Google Scholar] [CrossRef]
- Jonscher, A.K. The “universal” dielectric response. Nature 1977, 267, 673–679. [Google Scholar] [CrossRef]
- Murzin, D.Y.; Heikkilä, T. Modeling of drug dissolution kinetics with sigmoidal behavior from ordered mesoporous silica. Chem. Eng. Commun. 2014, 201, 579–592. [Google Scholar] [CrossRef]
- Johari, G.P.; Goldstein, M. Viscous liquids and the glass transition. II. Secondary relaxations in glasses of rigid molecules. J. Chem. Phys. 1970, 53, 2372–2388. [Google Scholar] [CrossRef]
- Ngai, K.L.; Kamińska, E.; Sekuła, M.; Paluch, M. Primary and secondary relaxations in bis-5-hydroxypentylphthalate revisited. J. Chem. Phys. 2005, 123, 204507. [Google Scholar] [CrossRef] [PubMed]
- Ngai, K.L. An extended coupling model description of the evolution of dynamics with time in supercooled liquids and ionic conductors. J. Phys. Condens. Matter 2003, 15, S1107. [Google Scholar] [CrossRef]
- Ngai, K.L.; Paluch, M. Classification of secondary relaxation in glass-formers based on dynamic properties. J. Chem. Phys. 2004, 120, 857–873. [Google Scholar] [CrossRef]
- Alvarez, F.; Alegra, A.; Colmenero, J. Relationship between the time-domain Kohlrausch-Williams-Watts and frequency-domain Havriliak-Negami relaxation functions. Phys. Rev. B 1991, 44, 7306. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | FNB Loaded (% w/w) * | Available Vp (cm3) | VFNB (cm3) ** | Pore Volume Occupied by FNB (% v/v) |
|---|---|---|---|---|
| MSN | 22.8 | 0.095 | 0.035 | 37 |
| MSN_APTES | 22.8 | 0.096 | 0.036 | 37 |
| MSN_TMPS | 33.3 | 0.081 | 0.050 | 62 |
| Sample | mmol g−1 MSNs | Modifying Molecules/nm−2 |
|---|---|---|
| MSN_APTES | 3.14 | 1.93 |
| MSN_TMPS | 0.98 | 0.60 |
| Sample | Tg-on (°C) Heating 1 Tend-1 = 50 °C | Tg-on (°C) Heating 2 Tend-2 = 70 °C | Tg-on (°C) Heating 3 Tend-3 = 100 °C | Water Content (%) |
|---|---|---|---|---|
| FNB@MSNs | −23.5 | −21.5 | −21.5 | 4.1 |
| FNB@MSNs_APTES | −20.9 | −18.6 | −19.8 | 5.7 |
| FNB@MSNs_TMPS | −14.7 | −12.5 | −9.4 | 2.0 |
| FNB | Tg-on (°C) = −17.7 | |||
| Process I | Process I* | Process II | |||||
|---|---|---|---|---|---|---|---|
| Ea (kJ mol−1) | τ∞ (s) | Ea (kJ mol−1) | τ∞ (s) | Ea (kJ mol−1) | τ∞ (s) | Tmax * (°C) | |
| MSN | 44 | 8.0 × 10−19 | -- | -- | 44 | 2.9 × 10−12 | 9.0 |
| MSN_APTES | 55 | 4.7 × 10−21 | 57 | 2.1 × 10−19 | 60 | 6.3 × 10−18 | 21.8 |
| MSN_TMPS | 37 | 1.8 × 10−17 | 29 | 1.1 × 10−10 | n.a. | n.a. | 4.2 |
| Sample | Process | Function | τ0 (s) | Ea (kJ mol−1) | B (K) | T0 (K) |
|---|---|---|---|---|---|---|
| FNB | γ | Arrhenius | 3.2 × 10−14 | 28.7 | ||
| α | VFTH | 4.0 × 10−15 | 1598 | 205 | ||
| σ | VFTH | 1.3 × 10−6 | 635 | 273 | ||
| FNB@MSN | αfast | Arrhenius | 7.7 × 10−19 | 79.4 | ||
| SOH | Arrhenius | 8.2 × 10−24 | 106.5 | |||
| MWS | VFTH | 1.4 × 10−9 | 2395 | 204 | ||
| σ | Arrhenius | 3.6 × 10−15 | 90.5 | |||
| FNB@MSN_APTES | SOH/α | VFTH | 8.7 × 10−13 | 1508.8 | 198.6 | |
| SAPTES | VFTH | 2.1 × 10−11 | 801.8 | 276.4 | ||
| MWS/σ | Arrhenius | 1.5 × 10−14 | 98.8 | |||
| FNB@MSN_TMPS | SOH/α | VFTH | 1.5 × 10−20 | 4978.2 | 151.9 | |
| STMPS | VFTH | 1.7 × 10−23 | 8846.7 | 107.2 | ||
| MWS | Arrhenius | 1.7 × 10−16 | 100.7 | |||
| σ | Arrhenius | 5.1 × 10−14 | 88.5 |
| Sample | msample (g) | FNBloaded (wt%) | mFNB (g) | nFNB (mol) | mMSN (g) | nMSN (mol g−1) | nrel (mmol g−1) | nrel (mol) | nreleased (%) |
|---|---|---|---|---|---|---|---|---|---|
| FNB@MSN | 1.11 × 10−3 | 22.8 | 2.53 × 10−4 | 7.01 × 10−7 | 8.57 × 10−4 | 8.18 × 10−4 | 2.36 × 10−3 | 2.02 × 10−9 | 0.29 |
| FNB@MSN_APTES | 1.13 × 10−3 | 22.8 | 2.58 × 10−4 | 7.14 × 10−7 | 8.72 × 10−4 | 8.18 × 10−4 | 2.17 × 10−2 | 1.89 × 10−8 | 2.65 |
| FNB@MSN_TMPS | 1.16 × 10−3 | 33.3 | 3.86 × 10−4 | 1.07 × 10−6 | 7.74 × 10−4 | 1.38 × 10−3 | 1.60 × 10−2 | 1.24 × 10−8 | 1.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figari, G.; Gonçalves, J.L.M.; Diogo, H.P.; Dionísio, M.; Farinha, J.P.; Viciosa, M.T. Understanding Fenofibrate Release from Bare and Modified Mesoporous Silica Nanoparticles. Pharmaceutics 2023, 15, 1624. https://doi.org/10.3390/pharmaceutics15061624
Figari G, Gonçalves JLM, Diogo HP, Dionísio M, Farinha JP, Viciosa MT. Understanding Fenofibrate Release from Bare and Modified Mesoporous Silica Nanoparticles. Pharmaceutics. 2023; 15(6):1624. https://doi.org/10.3390/pharmaceutics15061624
Chicago/Turabian StyleFigari, Giorgia, José L. M. Gonçalves, Hermínio P. Diogo, Madalena Dionísio, José Paulo Farinha, and María Teresa Viciosa. 2023. "Understanding Fenofibrate Release from Bare and Modified Mesoporous Silica Nanoparticles" Pharmaceutics 15, no. 6: 1624. https://doi.org/10.3390/pharmaceutics15061624
APA StyleFigari, G., Gonçalves, J. L. M., Diogo, H. P., Dionísio, M., Farinha, J. P., & Viciosa, M. T. (2023). Understanding Fenofibrate Release from Bare and Modified Mesoporous Silica Nanoparticles. Pharmaceutics, 15(6), 1624. https://doi.org/10.3390/pharmaceutics15061624