

Improved Metal-Free Approach for the Synthesis of Protected Thiol Containing Thymidine Nucleoside Phosphoramidite and Its Application for the Synthesis of Ligatable Oligonucleotide Conjugates
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. General Remarks
2.2. Synthesis of Thiol-Modified Monomer
2.3. Oligonucleotide Synthesis
2.4. Peptide Synthesis, Purification, and Analysis
2.5. Preparation of Conjugates in Solution Phase
2.6. Preparation of Conjugates on Solid Phase
2.7. Templated Ligation
2.8. Denaturing DNA Gel Electrophoresis
2.9. Stability of Thiosuccinimide Bond under Ligation Conditions
3. Results and Discussion
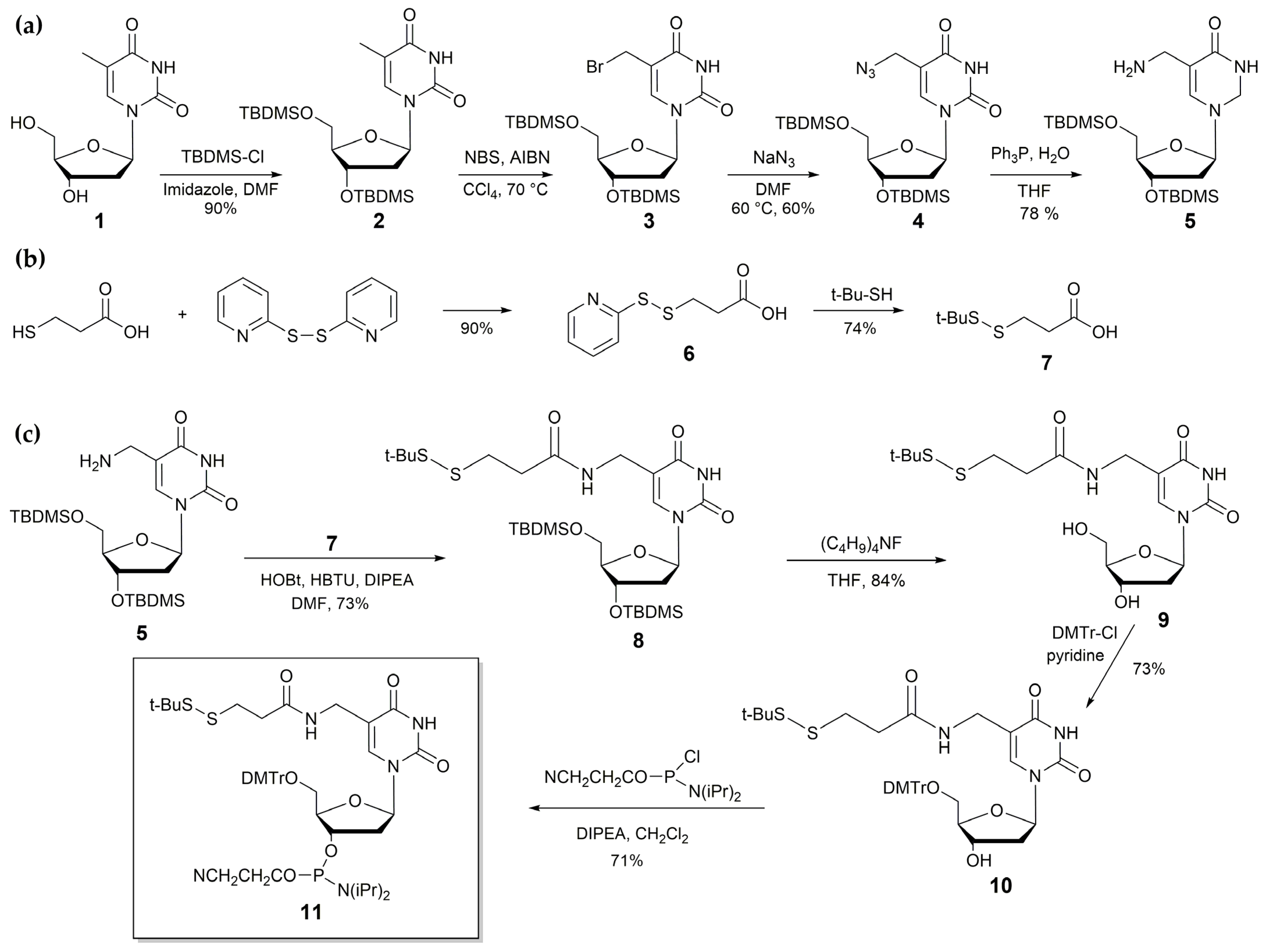
3.1. Synthesis of Thiol-Containing Thymidine Nucleoside and Its Incorporation into Oligonucleotide Sequences
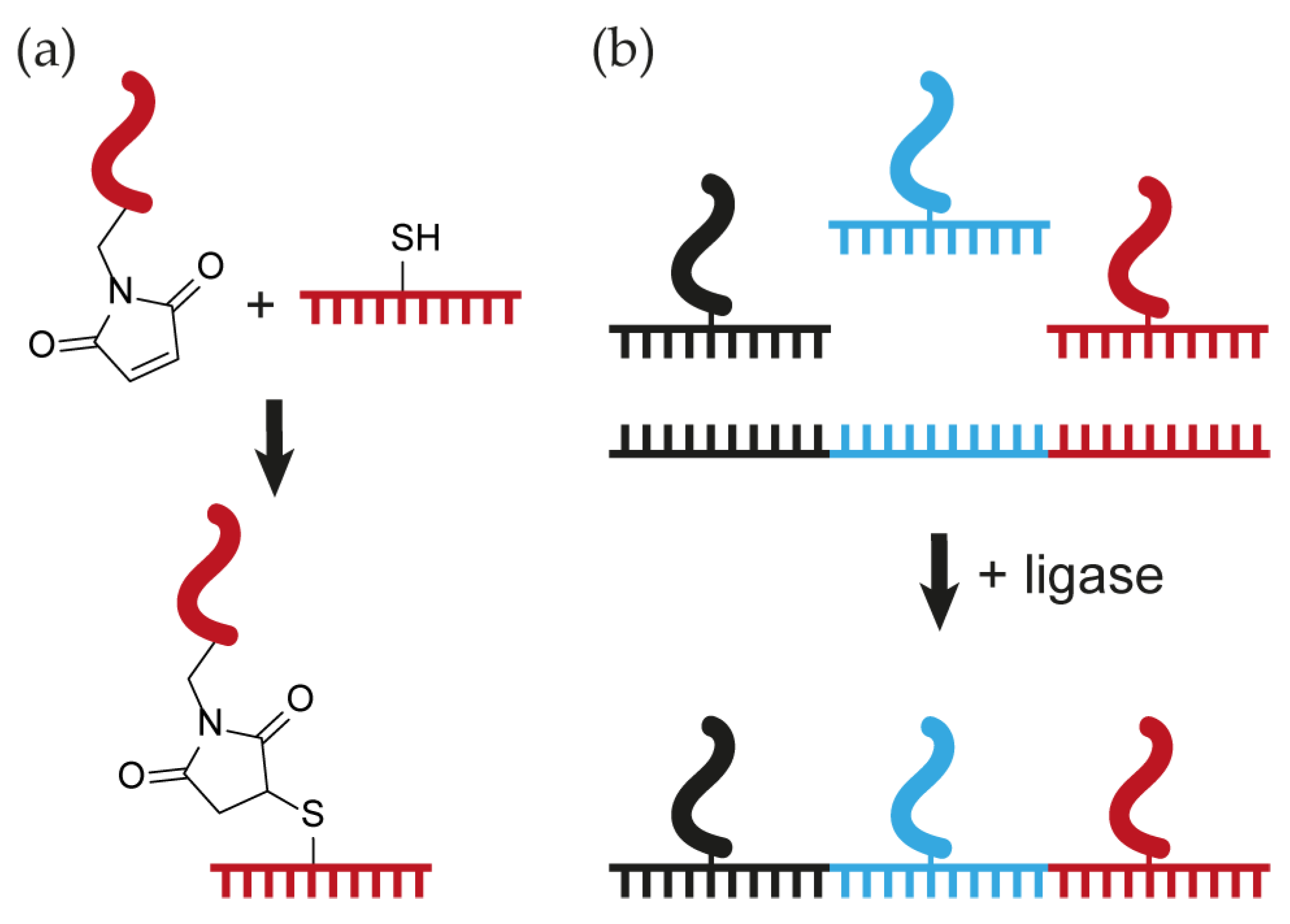
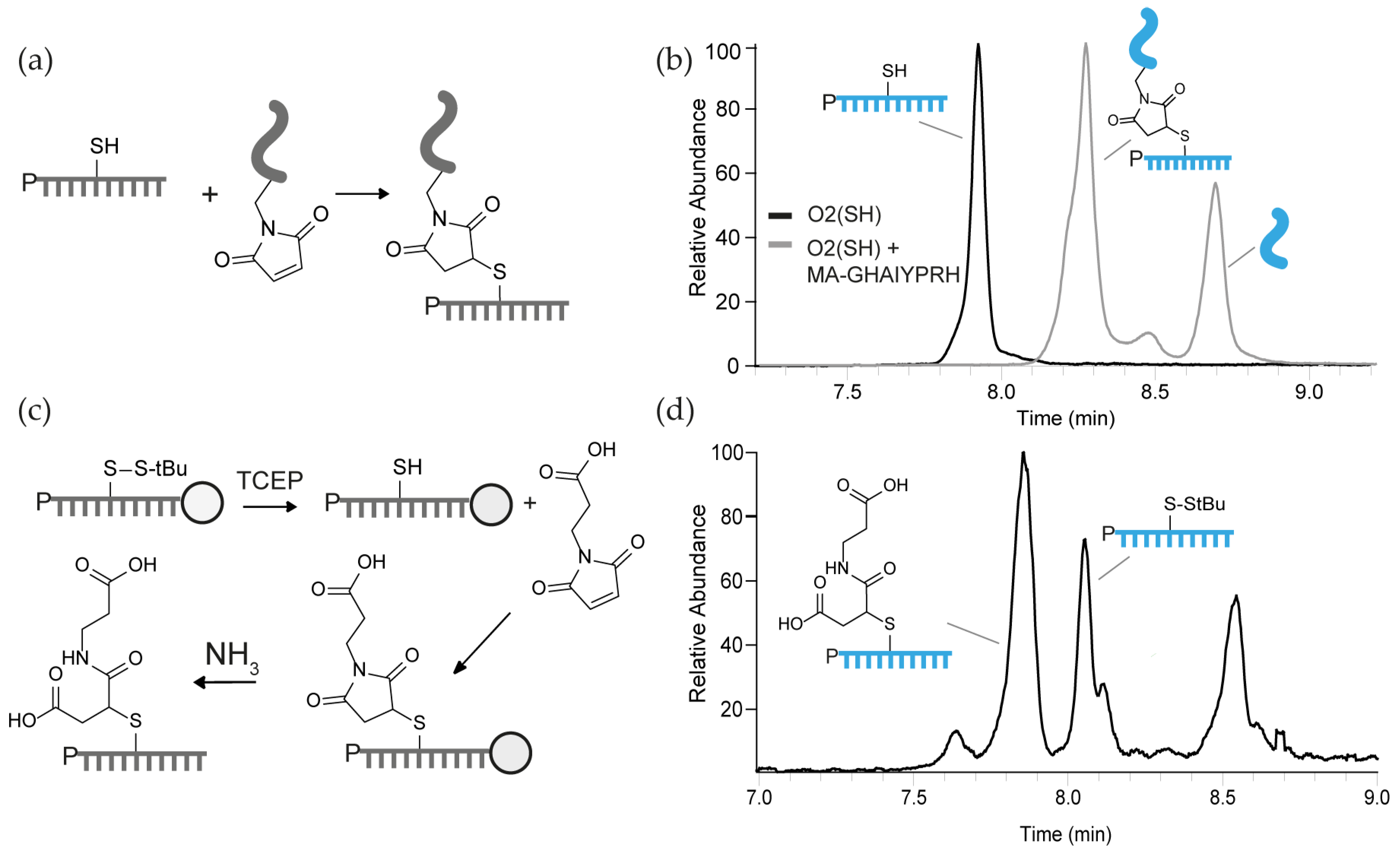
3.2. Thiol-Maleimide Conjugation
3.3. Templated Ligation of Peptide–Oligonucleotide Conjugates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Craig, K.; Abrams, M.; Amiji, M. Recent Preclinical and Clinical Advances in Oligonucleotide Conjugates. Expert Opin. Drug Deliv. 2018, 15, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthélémy, P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjug. Chem. 2019, 30, 366–383. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Dovgan, I.; Koniev, O.; Kolodych, S.; Wagner, A. Antibody-Oligonucleotide Conjugates as Therapeutic, Imaging, and Detection Agents. Bioconjug. Chem. 2019, 30, 2483–2501. [Google Scholar] [CrossRef] [PubMed]
- Diezmann, F.; Seitz, O. DNA-Guided Display of Proteins and Protein Ligands for the Interrogation of Biology. Chem. Soc. Rev. 2011, 40, 5789–5801. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.A.R.; Diehnelt, C.W.; Belcher, P.; Greving, M.; Woodbury, N.W.; Johnston, S.A.; Chaput, J.C. Creating Protein Affinity Reagents by Combining Peptide Ligands on Synthetic DNA Scaffolds. J. Am. Chem. Soc. 2009, 131, 17233–17241. [Google Scholar] [CrossRef]
- Guo, C.; Kong, D.; Lei, Y.; Hili, R. Expanding the Chemical Diversity of DNA. Synlett 2018, 29, 1405–1414. [Google Scholar] [CrossRef]
- O’Reilly, R.K.; Turberfield, A.J.; Wilks, T.R. The Evolution of DNA-Templated Synthesis as a Tool for Materials Discovery. Acc. Chem. Res. 2017, 50, 2496–2509. [Google Scholar] [CrossRef]
- Usanov, D.L.; Chan, A.I.; Maianti, J.P.; Liu, D.R. Second-Generation DNA-Templated Macrocycle Libraries for the Discovery of Bioactive Small Molecules. Nat. Chem. 2018, 10, 704–714. [Google Scholar] [CrossRef]
- Sun, H.; Yang, L.; Thompson, M.P.; Schara, S.; Cao, W.; Choi, W.; Hu, Z.; Zang, N.; Tan, W.; Gianneschi, N.C. Recent Advances in Amphiphilic Polymer-Oligonucleotide Nanomaterials via Living/Controlled Polymerization Technologies. Bioconjug. Chem. 2019, 30, 1889–1904. [Google Scholar] [CrossRef]
- Klabenkova, K.; Fokina, A.; Stetsenko, D. Chemistry of Peptide-Oligonucleotide Conjugates: A Review. Molecules 2021, 26, 5420. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Murat, P.; Defrancq, E. Recent Developments in Oligonucleotide Conjugation. Chem. Soc. Rev. 2010, 39, 2054–2070. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ming, X.; Nakagawa, O. The Chemistry and Biology of Oligonucleotide Conjugates. Acc. Chem. Res. 2012, 45, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, H.; Diezmann, F.; Seitz, O. DNA as a Molecular Ruler: Interrogation of a Tandem SH2 Domain with Self-Assembled, Bivalent DNA-Peptide Complexes. Angew. Chem. Int. Ed. 2011, 50, 4146–4150. [Google Scholar] [CrossRef] [PubMed]
- Gottschling, D.; Seliger, H.; Tarrasón, G.; Piulats, J.; Eritja, R. Synthesis of Oligodeoxynucleotides Containing N4-Mercaptoethylcytosine and Their Use in the Preparation of Oligonucleotide−Peptide Conjugates Carrying c-Myc Tag-Sequence. Bioconjug. Chem. 1998, 9, 831–837. [Google Scholar] [CrossRef]
- Pujari, S.S.; Zhang, Y.; Ji, S.; Distefano, M.D.; Tretyakova, N.Y. Site-Specific Cross-Linking of Proteins to DNA via a New Bioorthogonal Approach Employing Oxime Ligation. Chem. Comm. 2018, 54, 6296–6299. [Google Scholar] [CrossRef]
- Graham, D.; Grondin, A.; Mchugh, C.; Fruk, L.; Smith, W.E. Internal Labeling of Oligonucleotide Probes by Diels-Alder Cycloaddition. Tetrahedron Lett. 2002, 43, 4785–4788. [Google Scholar] [CrossRef]
- Astakhova, I.K.; Hansen, L.H.; Vester, B.; Wengel, J. Peptide–LNA Oligonucleotide Conjugates. Org. Biomol. Chem 2013, 11, 4240–4249. [Google Scholar] [CrossRef]
- Gramlich, P.M.E.; Warncke, S.; Gierlich, J.; Carell, T. Click-Click-Click: Single to Triple Modification of DNA. Angew. Chem. Int. Ed. 2008, 47, 3442–3444. [Google Scholar] [CrossRef]
- Hili, R.; Niu, J.; Liu, D.R. DNA Ligase-Mediated Translation of DNA into Densely Functionalized Nucleic Acid Polymers. J. Am. Chem. Soc. 2013, 135, 98–101. [Google Scholar] [CrossRef]
- Guo, C.; Watkins, C.P.; Hili, R. Sequence-Defined Scaffolding of Peptides on Nucleic Acid Polymers. J. Am. Chem. Soc. 2015, 137, 11191–11196. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Movahedi, M.; Mahdavi-Amiri, Y.; Yeung, W.; Tiburcio, T.; Chen, D.; Hili, R. Evolutionary Outcomes of Diversely Functionalized Aptamers Isolated from in Vitro Evolution. ACS Synth. Biol. 2020, 9, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Yeung, W.; Hili, R. In Vitro Selection of Diversely Functionalized Aptamers. J. Am. Chem. Soc. 2017, 139, 13977–13980. [Google Scholar] [CrossRef] [PubMed]
- Stenzel, M.H. Bioconjugation Using Thiols: Old Chemistry Rediscovered to Connect Polymers with Nature’s Building Blocks. ACS Macro Lett. 2013, 2, 14–18. [Google Scholar] [CrossRef]
- Stasińska, A.R.; Putaj, P.; Chmielewski, M.K. Disulfide Bridge as a Linker in Nucleic Acids’ Bioconjugation. Part I: An Overview of Synthetic Strategies. Bioorg. Chem. 2019, 92, 103223. [Google Scholar] [CrossRef]
- Stasińska, A.R.; Putaj, P.; Chmielewski, M.K. Disulfide Bridge as a Linker in Nucleic Acids’ Bioconjugation. Part II: A Summary of Practical Applications. Bioorg. Chem. 2020, 95, 103518. [Google Scholar] [CrossRef]
- The Glen Report 24.12 New Product—S-Bz-Thiol-Modifier C6-DT 2012. Available online: https://www.glenresearch.com/reports/gr24-12 (accessed on 12 December 2022).
- Far, S.; Gouyette, C.; Melnyk, O. A Novel Phosphoramidite for the Synthesis of A-Oxo Aldehyde-Modified Oligodeoxynucleotides. Tetrahedron 2005, 61, 6138–6142. [Google Scholar] [CrossRef]
- Goodwin, J.T.; Glick, G.D. Synthesis of a Disulfide Stabilized RNA Hairpin. Tetrahedron Lett. 1994, 35, 1647–1650. [Google Scholar] [CrossRef]
- Hou, X.; Wang, G.; Gaffney, B.L.; Jones, R.A. Synthesis of Guanosine and Deoxyguanosine Phosphoramidites with Cross-Linkable Thioalkyl Tethers for Direct Incorporation into RNA and DNA. Nucleosides Nucleotides Nucleic Acids 2009, 28, 1076–1094. [Google Scholar] [CrossRef]
- Pérez-Rentero, S.; Garibotti, A.V.; Eritja, R. Solid-Phase Synthesis of Oligodeoxynucleotides Containing N4-2-(t-Butyldisulfanyl)Ethyl-5-Methylcytosine Moieties. Molecules 2010, 15, 5692–5707. [Google Scholar] [CrossRef]
- Sun, S.; Tang, X.-Q.; Merchant, A.; Anjaneyulu, P.S.R.; Piccirilli, J.A. Efficient Synthesis of 5-(Thioalkyl)Uridines via Ring Opening of α-Ureidomethylene Thiolactones. J. Org. Chem. 1996, 61, 5708–5709. [Google Scholar] [CrossRef]
- Osborne, S.E.; Ellington, A.D. Incorporating Disulfide Cross-Links at the Terminus of Oligonucleotides via Solid-Phase Nucleic Acid Synthesis. Bioorg. Med. Chem. Lett. 1996, 6, 2339–2342. [Google Scholar] [CrossRef]
- Diezmann, F.; von Kleist, L.; Haucke, V.; Seitz, O. Probing Heterobivalent Binding to the Endocytic AP-2 Adaptor Complex by DNA-Based Spatial Screening. Org. Biomol. Chem. 2015, 13, 8008–8015. [Google Scholar] [CrossRef] [PubMed]
- Diezmann, F.; Eberhard, H.; Seitz, O. Native Chemical Ligation in the Synthesis of Internally Modified Oligonucleotide–Peptide Conjugates. Pept. Sci. 2010, 94, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Krim, J.; Taourirte, M.; Grünewald, C.; Krstic, I.; Engels, J. Microwave-Assisted Click Chemistry for Nucleoside Functionalization: Useful Derivatives for Analytical and Biological Applications. Synthesis 2013, 45, 396–405. [Google Scholar] [CrossRef]
- Navath, R.S.; Kurtoglu, Y.E.; Wang, B.; Kannan, S.; Romero, R.; Kannan, R.M. Dendrimer-Drug Conjugates for Tailored Intracellular Drug Release Based on Glutathione Levels. Bioconjug. Chem. 2008, 19, 2446–2455. [Google Scholar] [CrossRef]
- Gubu, A.; Li, L.; Ning, Y.; Zhang, X.; Lee, S.; Feng, M.; Li, Q.; Lei, X.; Jo, K.; Tang, X. Bioorthogonal Metabolic DNA Labelling Using Vinyl Thioether-Modified Thymidine and o-Quinolinone Quinone Methide. Chem. Eur. J. 2018, 24, 5895–5900. [Google Scholar] [CrossRef]
- Heilmann, T.; Ackermann, D.; Lopez, J. Refractive Index to Monitor Solid-Phase Oligonucleotide Synthesis. Org. Process Res. Dev. 2022. [Google Scholar] [CrossRef]
- Sobkowski, M.; Kraszewski, A.; Stawiński, J. The Reactions of H-Phosphonates with Bifunctional Reagents. Part, V. Functionalization of Support-Bound Oligonucleotides and Synthesis of Non-Radioactive Hybridization Probes. Nucleosides Nucleotides 1998, 17, 253–267. [Google Scholar] [CrossRef]
- Chang, C.A.; Horn, T. An Improved Deprotection Procedure of Amine-Containing Oligonucleotides from Acrylonitrile Modification. Nucleosides Nucleotides 2006, 18, 1205–1206. [Google Scholar] [CrossRef]
- Aubert, Y.; Bourgerie, S.; Meunier, L.; Mayer, R.; Roche, A.C.; Monsigny, M.; Thuong, N.T.; Asseline, U. Optimized Synthesis of Phosphorothioate Oligodeoxyribonucleotides Substituted with a 5′-Protected Thiol Function and a 3′-Amino Group. Nucleic Acids Res. 2000, 28, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, W.H.A.; van Boeckel, C.A.A. A New Strategy for the Solid-Phase Synthesis of 5′-Thiolated Oligodeoxynucleotides. Tetrahedron 1993, 49, 10931–10944. [Google Scholar] [CrossRef]
- Battersby, T.R.; Ang, D.N.; Burgstaller, P.; Jurczyk, S.C.; Bowser, M.T.; Buchanan, D.D.; Kennedy, R.T.; Benner, S.A. Quantitative Analysis of Receptors for Adenosine Nucleotides Obtained via In Vitro Selection from a Library Incorporating a Cationic Nucleotide Analog. J. Am. Chem. Soc. 1999, 121, 9781–9789. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, I.; Mujahid, A.; Rasool, N.; Rizwan, K.; Malik, A.; Ahmad, G.; Shah, S.A.A.; Rashid, U.; Nasir, N.M. Palladium and Copper Catalyzed Sonogashira Cross Coupling an Excellent Methodology for C-C Bond Formation over 17 Years: A Review. Catalysts 2020, 10, 443. [Google Scholar] [CrossRef]
- Garg, N.K.; Woodroofe, C.C.; Lacenere, C.J.; Quake, S.R.; Stoltz, B.M. A Ligand-Free Solid-Supported System for Sonogashira Couplings: Applications in Nucleoside Chemistry. Chem. Comm. 2005, 4551–4553. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.T.; Glick, G.D. Incorporation of Alkylthiol Chains at C-5 of Deoxyuridine. Tetrahedron Lett. 1993, 34, 5549–5552. [Google Scholar] [CrossRef]
- Held, H.A.; Benner, S.A. Challenging Artificial Genetic Systems: Thymidine Analogs with 5-Position Sulfur Functionality. Nucleic Acids Res. 2002, 30, 3857–3869. [Google Scholar] [CrossRef]
- Held, H.A.; Roychowdhury, A.; Benner, S.A. C-5 Modified Nucleosides: Direct Insertion of Alkynyl-Thio Functionality in Pyrimidines. Nucleosides Nucleotides Nucleic Acids 2003, 22, 391–404. [Google Scholar] [CrossRef]
- Sun, C.-L.; Shi, Z.-J. Transition-Metal-Free Coupling Reactions. Chem. Rev. 2014, 114, 9219–9280. [Google Scholar] [CrossRef]
- Pérez-Rentero, S.; Grijalvo, S.; Ferreira, R.; Eritja, R. Synthesis of Oligonucleotides Carrying Thiol Groups Using a Simple Reagent Derived from Threoninol. Molecules 2012, 17, 10026–10045. [Google Scholar] [CrossRef]
- Burns, J.A.; Butler, J.C.; Moran, J.; Whitesides, G.M. Selective Reduction of Disulfides by Tris(2-Carboxyethyl)Phosphine. J. Org. Chem. 1991, 56, 2648–2650. [Google Scholar] [CrossRef]
- Lee, J.H.; Engler, J.A.; Collawn, J.F.; Moore, B.A. Receptor Mediated Uptake of Peptides That Bind the Human Transferrin Receptor. Eur. J. Biochem. 2001, 268, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, R.; Li, J.; Liu, Y.; Fu, Y.; Zhou, J.; Yang, G.; Shan, Y. Revealing the Dynamic Mechanism by Which Transferrin Promotes the Cellular Uptake of HAIYPRH Peptide-Conjugated Nanostructures by Force Tracing. Mol. Pharm. 2021, 18, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.; Schönzart, L.; Israel, I.; Beutner, R.; Scharnweber, D.; Worch, H.; Hempel, U.; Schwenzer, B. Oligonucleotide−RGD Peptide Conjugates for Surface Modification of Titanium Implants and Improvement of Osteoblast Adhesion. Bioconjug. Chem. 2009, 20, 710–718. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J.A. A Fusion Peptide Directs Enhanced Systemic Dystrophin Exon Skipping and Functional Restoration in Dystrophin-Deficient Mdx Mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef]
- Samoylova, T.I.; Smith, B.F. Elucidation of Muscle-Binding Peptides by Phage Display Screening. Muscle Nerve 1999, 22, 460–466. [Google Scholar] [CrossRef]
- Kantner, T.; Watts, A.G. Characterization of Reactions between Water-Soluble Trialkylphosphines and Thiol Alkylating Reagents: Implications for Protein-Conjugation Reactions. Bioconjug. Chem. 2016, 27, 2400–2406. [Google Scholar] [CrossRef]
- Guo, C.; Hili, R. Fidelity of the DNA Ligase-Catalyzed Scaffolding of Peptide Fragments on Nucleic Acid Polymers. Bioconjug. Chem. 2017, 28, 314–318. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′–3′) 1 |
|---|---|
| O1 | p-TGTCTGAACC |
| O2 | p-TCACTCTTGC |
| O3 | p-ACTTTCGCAC |
| O1(SH) | p-TGTCT(SH)GAACC |
| O2(SH) | p-TCACT(SH)CTTGC |
| O3(SH) | p-ACTTT(SH)CGCAC |
| O4 | p-CTTTTATCACGGCCC |
| Flu-O5 | Flu-AGAATGCTGGGCAAT |
| O6 | GGGCCGTGATAAAAGGGTTCAGACAGCAAGAGTGAGTGCGAAAGTATTGCCCAGCATTCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kupihár, Z.; Ferenc, G.; Petrovicz, V.L.; Fáy, V.R.; Kovács, L.; Martinek, T.A.; Hegedüs, Z. Improved Metal-Free Approach for the Synthesis of Protected Thiol Containing Thymidine Nucleoside Phosphoramidite and Its Application for the Synthesis of Ligatable Oligonucleotide Conjugates. Pharmaceutics 2023, 15, 248. https://doi.org/10.3390/pharmaceutics15010248
Kupihár Z, Ferenc G, Petrovicz VL, Fáy VR, Kovács L, Martinek TA, Hegedüs Z. Improved Metal-Free Approach for the Synthesis of Protected Thiol Containing Thymidine Nucleoside Phosphoramidite and Its Application for the Synthesis of Ligatable Oligonucleotide Conjugates. Pharmaceutics. 2023; 15(1):248. https://doi.org/10.3390/pharmaceutics15010248
Chicago/Turabian StyleKupihár, Zoltán, Györgyi Ferenc, Vencel L. Petrovicz, Viktória R. Fáy, Lajos Kovács, Tamás A. Martinek, and Zsófia Hegedüs. 2023. "Improved Metal-Free Approach for the Synthesis of Protected Thiol Containing Thymidine Nucleoside Phosphoramidite and Its Application for the Synthesis of Ligatable Oligonucleotide Conjugates" Pharmaceutics 15, no. 1: 248. https://doi.org/10.3390/pharmaceutics15010248
APA StyleKupihár, Z., Ferenc, G., Petrovicz, V. L., Fáy, V. R., Kovács, L., Martinek, T. A., & Hegedüs, Z. (2023). Improved Metal-Free Approach for the Synthesis of Protected Thiol Containing Thymidine Nucleoside Phosphoramidite and Its Application for the Synthesis of Ligatable Oligonucleotide Conjugates. Pharmaceutics, 15(1), 248. https://doi.org/10.3390/pharmaceutics15010248