
Amorphous Solid Dispersions: Role of the Polymer and Its Importance in Physical Stability and In Vitro Performance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of the Polymer in Physical Stability
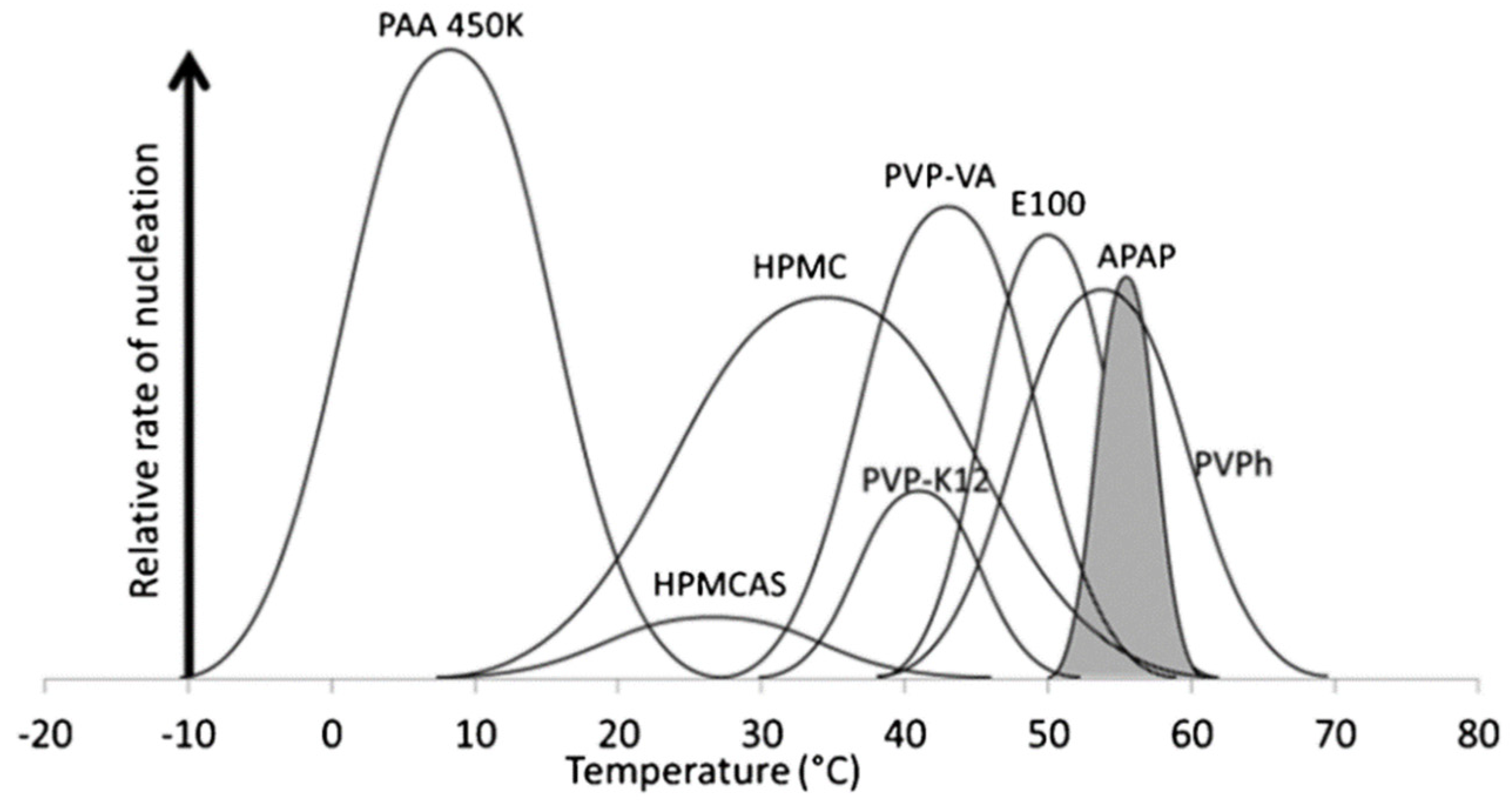
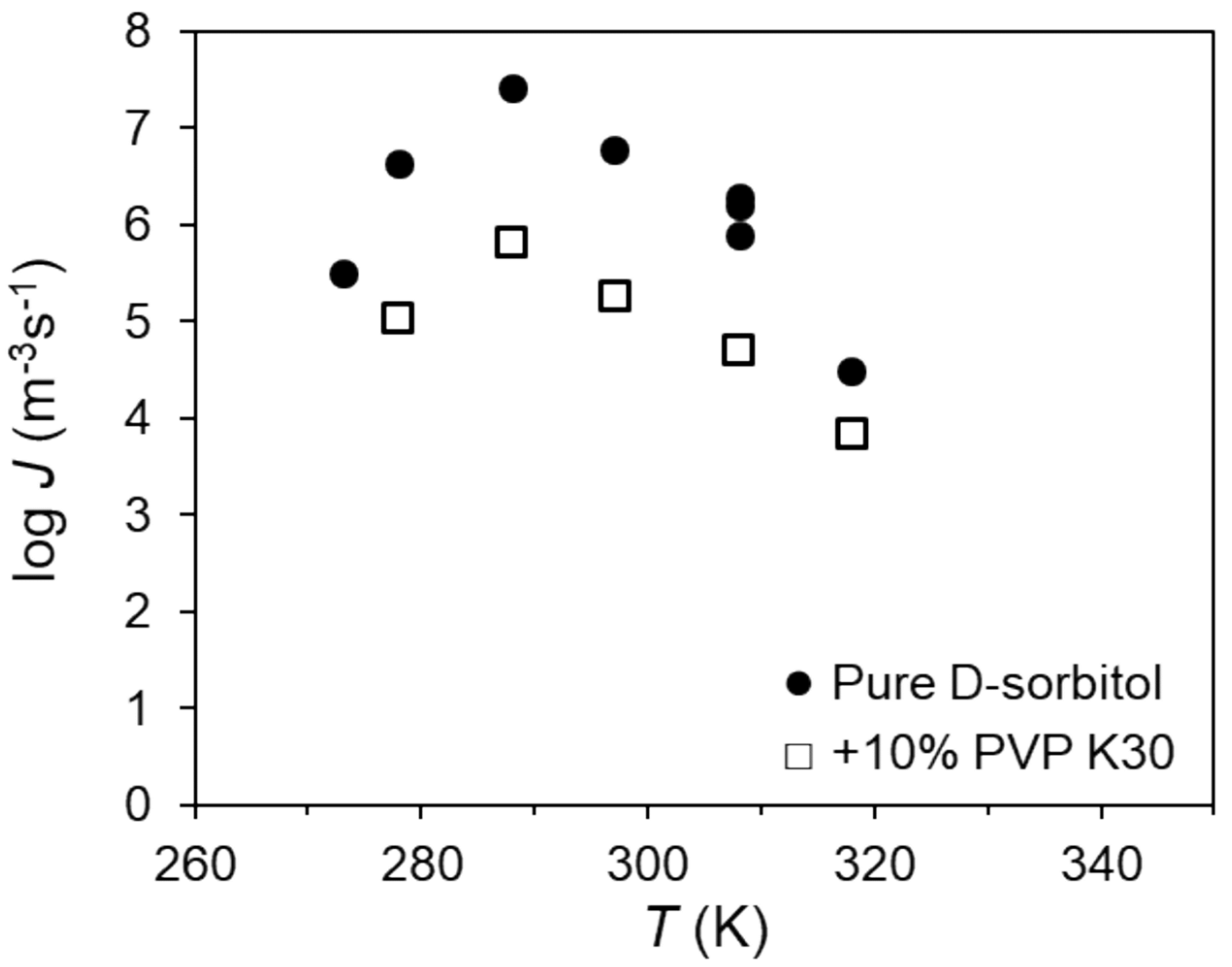
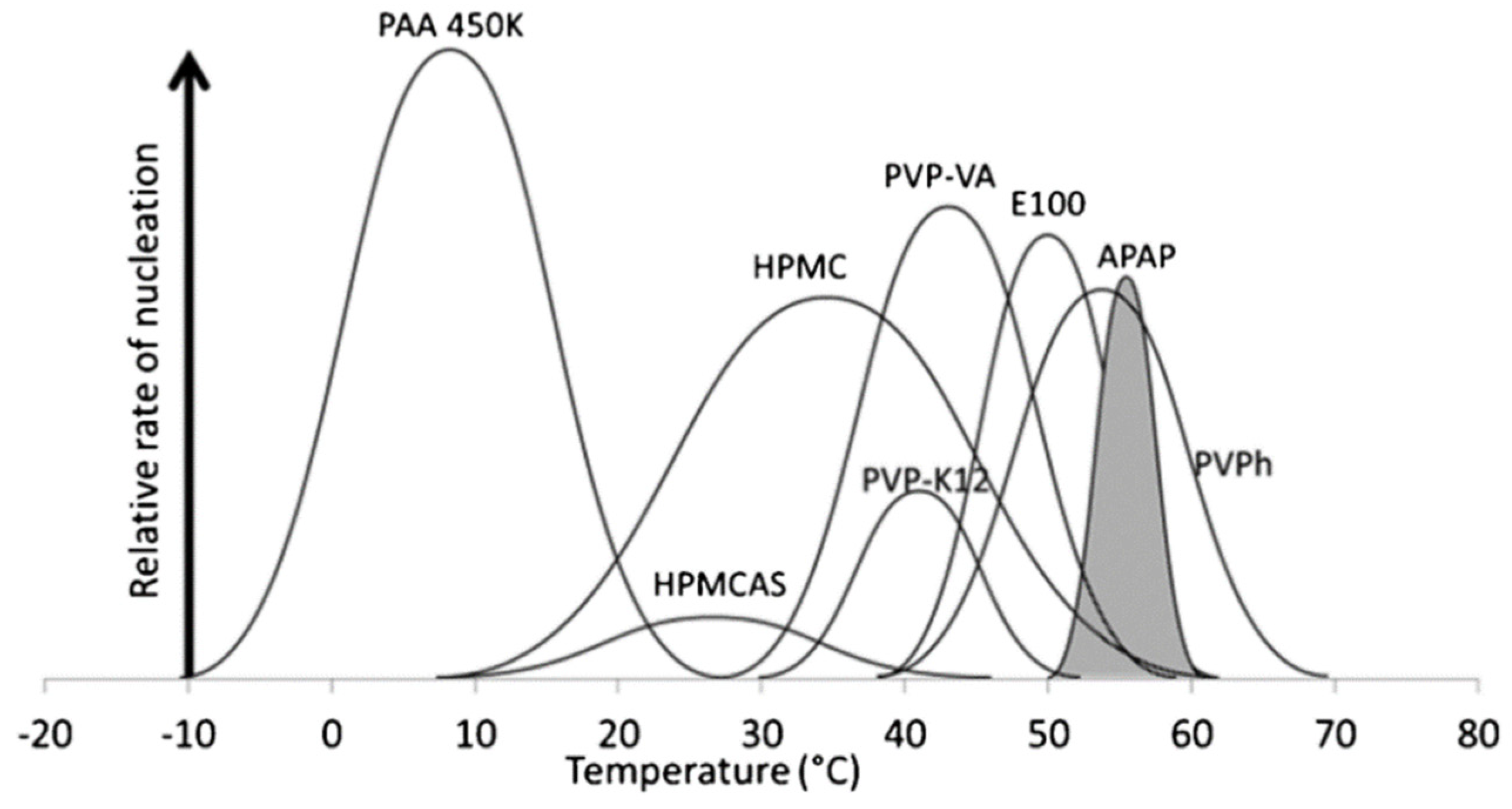
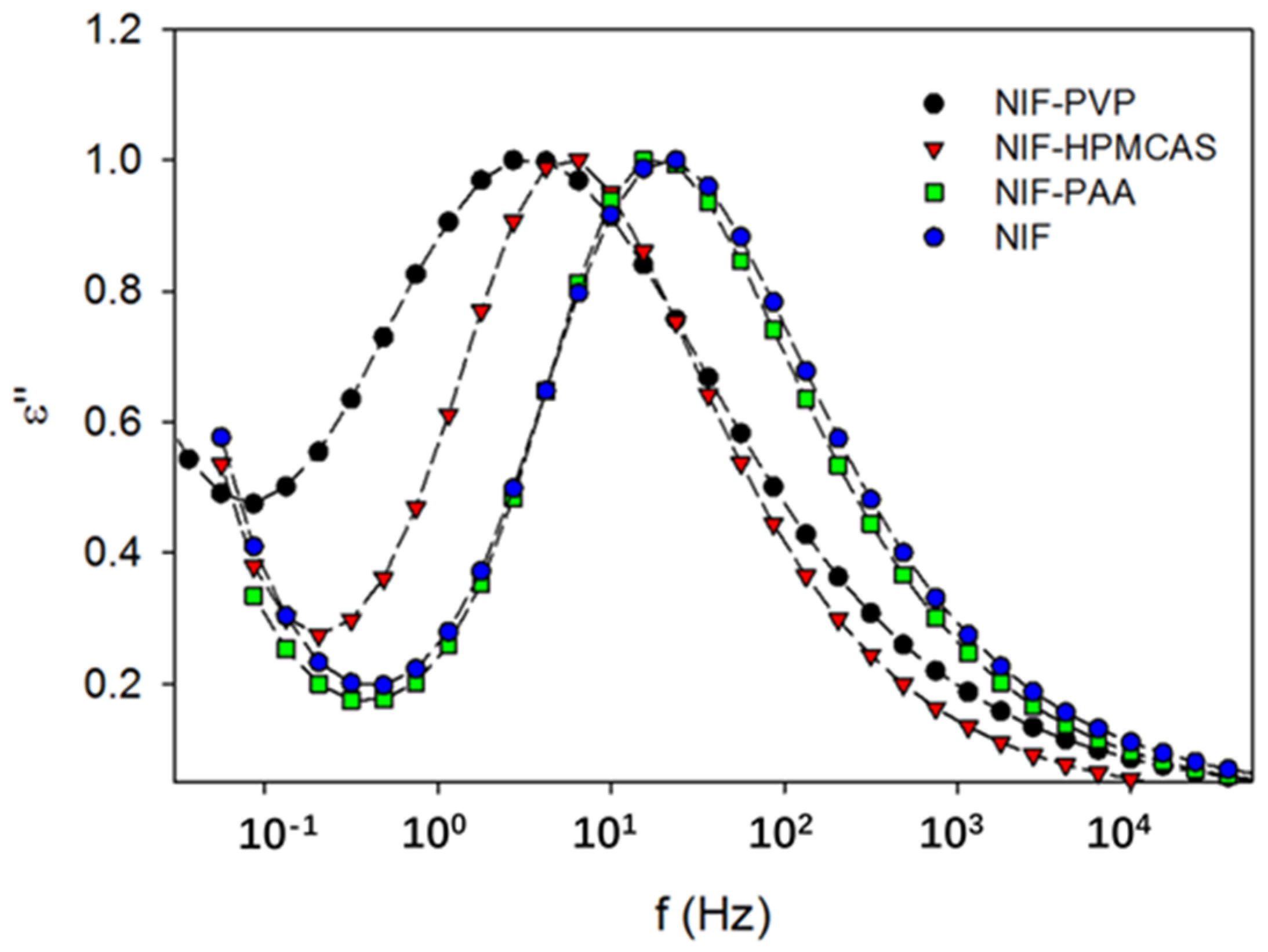
2.1. Effects of the Polymer on Nucleation
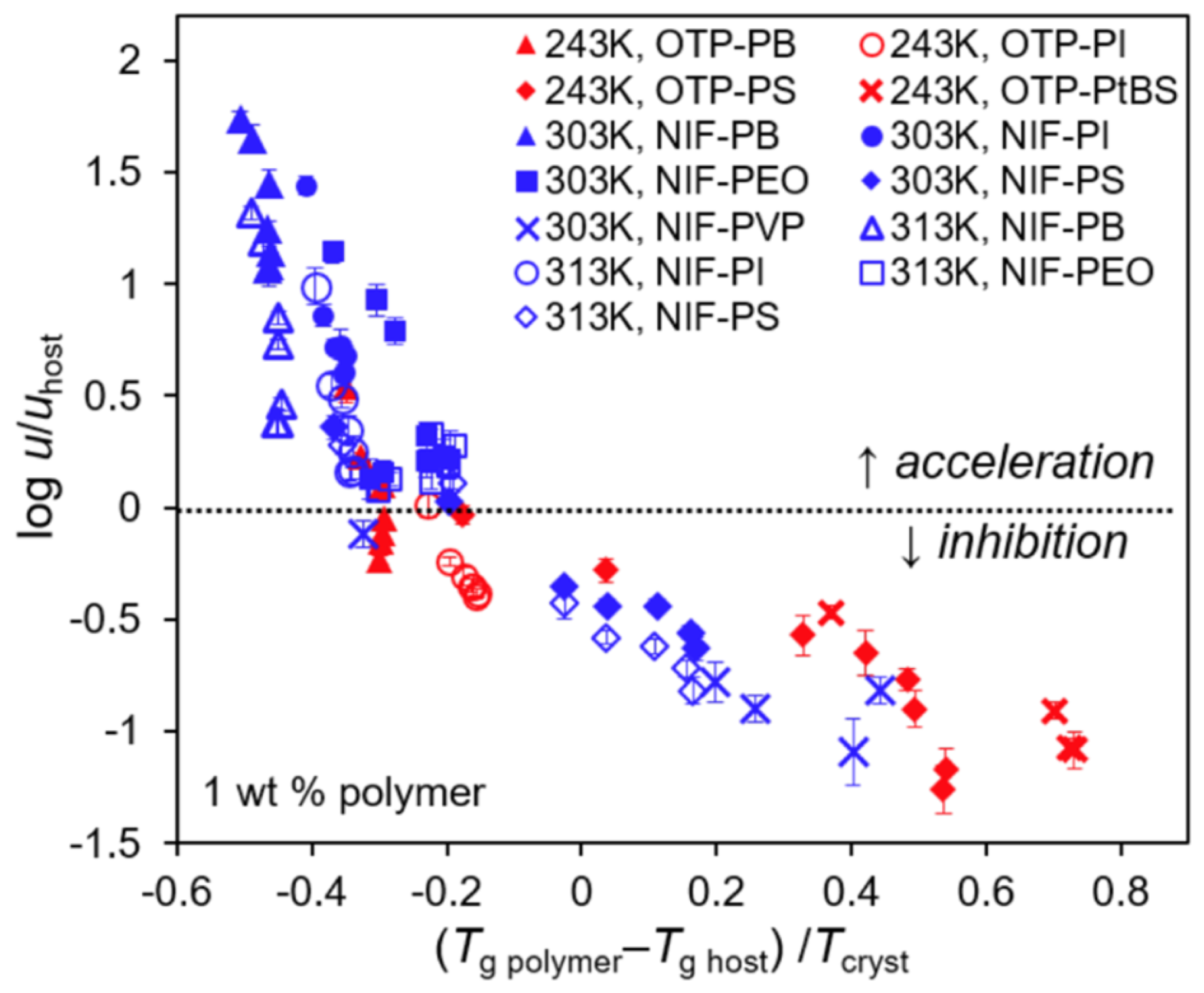
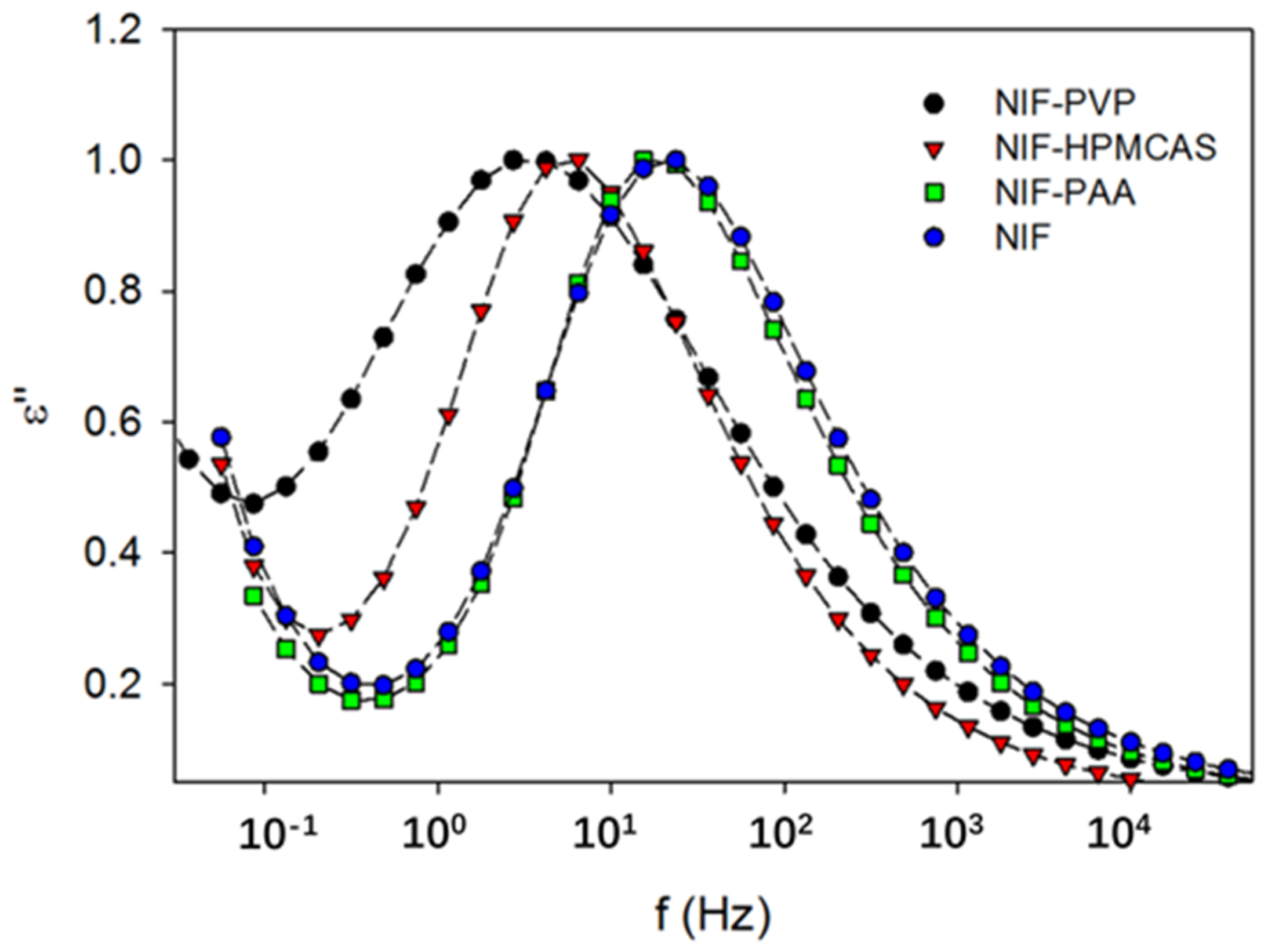
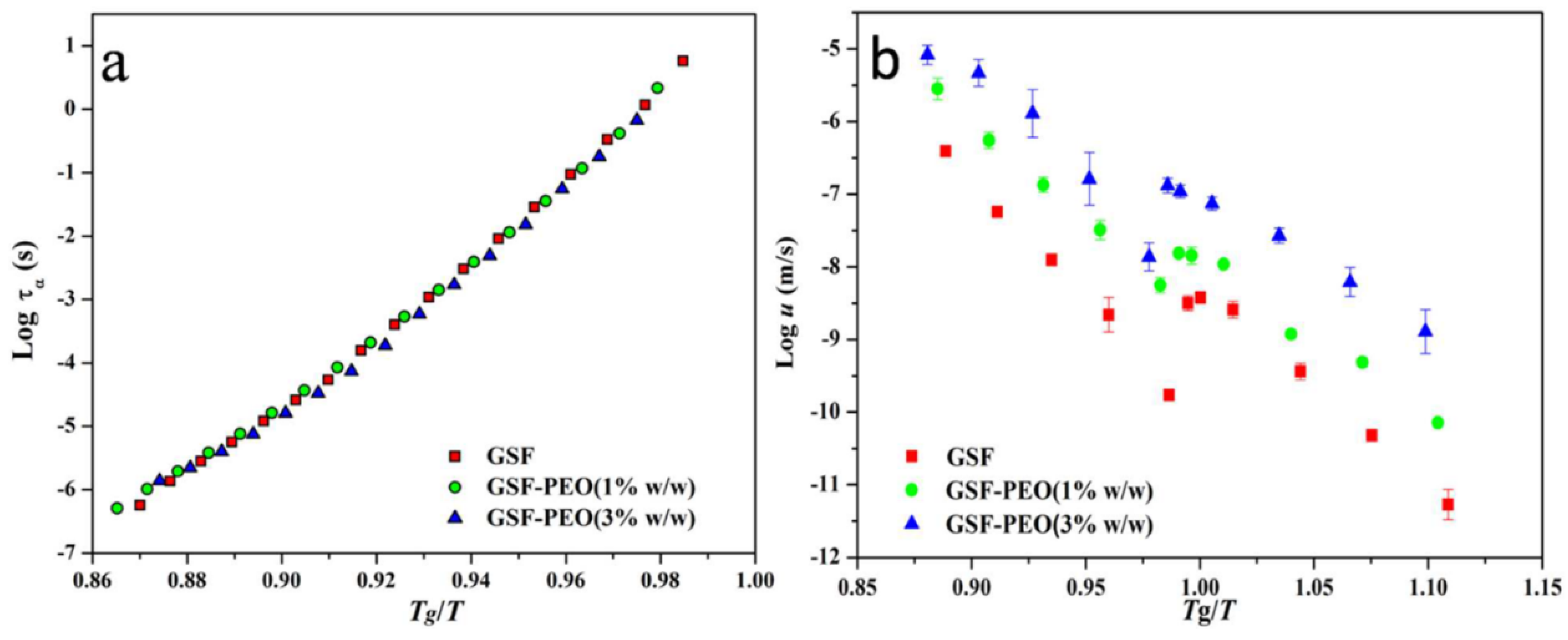
2.2. Effect of the Polymer on Crystal Growth
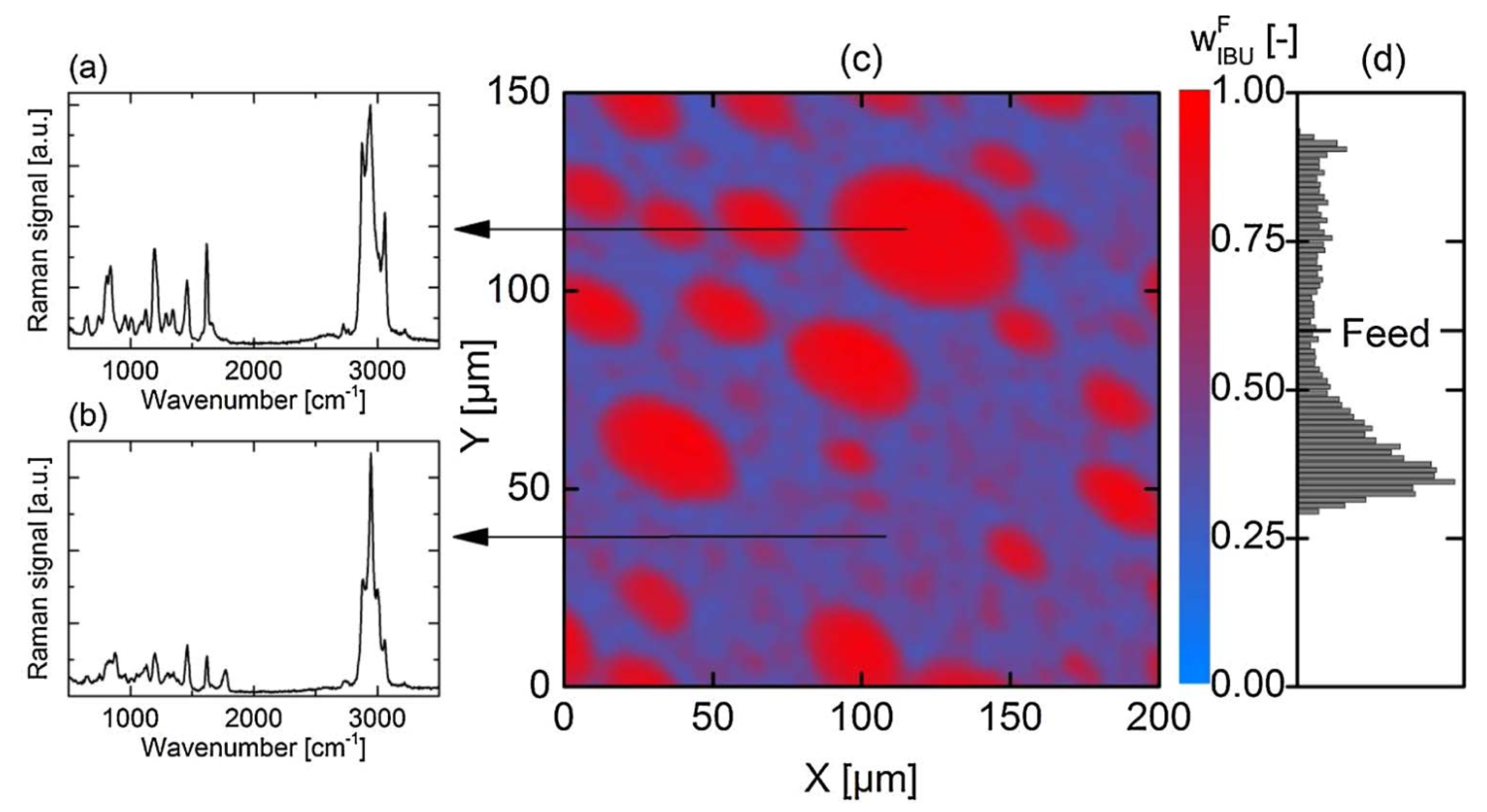
2.3. Effects of Polymers on Miscibility and Phase Separation
3. Role of the Polymer on Dissolution and Supersaturation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Shi, Q.; Moinuddin, S.M.; Cai, T. Advances in coamorphous drug delivery systems. Acta Pharm. Sin. B 2019, 9, 19–35. [Google Scholar] [CrossRef]
- Taylor, L.S.; Zhang, G.G. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv. Drug Deliv. Rev. 2016, 101, 122–142. [Google Scholar] [CrossRef]
- Shi, Q.; Li, F.; Yeh, S.; Moinuddin, S.M.; Xin, J.; Xu, J.; Chen, H.; Ling, B. Recent Advances in Enhancement of Dissolution and Supersaturation of Poorly Water-Soluble Drug in Amorphous Pharmaceutical Solids: A Review. AAPS PharmSciTech 2021, 23, 16. [Google Scholar] [CrossRef]
- Shi, Q.; Li, F.; Yeh, S.; Wang, Y.; Xin, J. Physical stability of amorphous pharmaceutical solids: Nucleation, crystal growth, phase separation and effects of the polymers. Int. J. Pharm. 2020, 590, 119925. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [Green Version]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.S.; Kumar, S.; Zhou, Q. Pharmaceutical amorphous solid dispersion: A review of manufacturing strategies. Acta Pharm. Sin. B 2021, 11, 2505–2536. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.; Wu, H.; Cai, T. Effect of polymeric excipients on nucleation and crystal growth kinetics of amorphous fluconazole. Biomater. Sci. 2021, 9, 4308–4316. [Google Scholar] [CrossRef]
- Lin, X.; Hu, Y.; Liu, L.; Su, L.; Li, N.; Yu, J.; Tang, B.; Yang, Z. Physical Stability of Amorphous Solid Dispersions: A Physicochemical Perspective with Thermodynamic, Kinetic and Environmental Aspects. Pharm. Res. 2018, 35, 125. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Bartels, M.; Degenhardt, M.; Sadowski, G. Physical stability of API/polymer-blend amorphous solid dispersions. Eur. J. Pharm. Biopharm. 2018, 124, 147–157. [Google Scholar] [CrossRef]
- Sahoo, A.; Kumar, N.K.; Suryanarayanan, R. Crosslinking: An avenue to develop stable amorphous solid dispersion with high drug loading and tailored physical stability. J. Control. Release 2019, 311–312, 212–224. [Google Scholar] [CrossRef]
- Yao, X.; Neusaenger, A.L.; Yu, L. Amorphous Drug-Polymer Salts. Pharmaceutics 2021, 13, 1271. [Google Scholar] [CrossRef]
- Huang, C.; Powell, C.T.; Sun, Y.; Cai, T.; Yu, L. Effect of Low-Concentration Polymers on Crystal Growth in Molecular Glasses: A Controlling Role for Polymer Segmental Mobility Relative to Host Dynamics. J. Phys. Chem. B 2017, 121, 1963–1971. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, L.; Wu, T.; Cai, T.; Gunn, E.M.; Yu, L. Stability of Amorphous Pharmaceutical Solids: Crystal Growth Mechanisms and Effect of Polymer Additives. AAPS J. 2012, 14, 380–388. [Google Scholar] [CrossRef]
- Powell, C.T.; Cai, T.; Hasebe, M.; Gunn, E.M.; Gao, P.; Zhang, G.; Gong, Y.; Yu, L. Low-Concentration Polymers Inhibit and Accelerate Crystal Growth in Organic Glasses in Correlation with Segmental Mobility. J. Phys. Chem. B 2013, 117, 10334–10341. [Google Scholar] [CrossRef]
- Trasi, N.S.; Taylor, L.S. Effect of Additives on Crystal Growth and Nucleation of Amorphous Flutamide. Cryst. Growth Des. 2012, 12, 3221–3230. [Google Scholar] [CrossRef]
- Dahan, A.; Beig, A.; Lindley, D.; Miller, J.M. The solubility–permeability interplay and oral drug formulation design: Two heads are better than one. Adv. Drug Deliv. Rev. 2016, 101, 99–107. [Google Scholar] [CrossRef]
- Andronis, V.; Zografi, G. Crystal nucleation and growth of indomethacin polymorphs from the amorphous state. J. Non-Cryst. Solids 2000, 271, 236–248. [Google Scholar] [CrossRef]
- Anwar, J.; Khan, S.; Lindfors, L. Secondary Crystal Nucleation: Nuclei Breeding Factory Uncovered. Angew. Chem. Int. Ed. 2015, 54, 14681–14684. [Google Scholar] [CrossRef]
- Xu, S.; Hou, Z.; Chuai, X.; Wang, Y. Overview of Secondary Nucleation: From Fundamentals to Application. Ind. Eng. Chem. Res. 2020, 59, 18335–18356. [Google Scholar] [CrossRef]
- Yao, X.; Liu, Q.; Wang, B.; Yu, J.; Aristov, M.M.; Shi, C.; Zhang, G.G.Z.; Yu, L. Anisotropic Molecular Organization at a Liquid/Vapor Interface Promotes Crystal Nucleation with Polymorph Selection. J. Am. Chem. Soc. 2022, 144, 11638–11645. [Google Scholar] [CrossRef]
- Vekilov, P.G. Dense Liquid Precursor for the Nucleation of Ordered Solid Phases from Solution. Cryst. Growth Des. 2004, 4, 671–685. [Google Scholar] [CrossRef]
- Alonzo, D.E.; Raina, S.; Zhou, D.; Gao, Y.; Zhang, G.G.Z.; Taylor, L.S. Characterizing the Impact of Hydroxypropylmethyl Cellulose on the Growth and Nucleation Kinetics of Felodipine from Supersaturated Solutions. Cryst. Growth Des. 2012, 12, 1538–1547. [Google Scholar] [CrossRef]
- Trasi, N.S.; Taylor, L.S. Effect of polymers on nucleation and crystal growth of amorphous acetaminophen. CrystEngComm 2012, 14, 5188–5197. [Google Scholar] [CrossRef]
- Yao, X.; Huang, C.; Benson, E.G.; Shi, C.; Zhang, G.G.Z.; Yu, L. Effect of Polymers on Crystallization in Glass-Forming Molecular Liquids: Equal Suppression of Nucleation and Growth and Master Curve for Prediction. Cryst. Growth Des. 2019, 20, 237–244. [Google Scholar] [CrossRef]
- Miyazaki, T.; Aso, Y.; Yoshioka, S.; Kawanishi, T. Differences in crystallization rate of nitrendipine enantiomers in amorphous solid dispersions with HPMC and HPMCP. Int. J. Pharm. 2011, 407, 111–118. [Google Scholar] [CrossRef]
- Konno, H.; Taylor, L.S. Influence of Different Polymers on the Crystallization Tendency of Molecularly Dispersed Amorphous Felodipine. J. Pharm. Sci. 2006, 95, 2692–2705. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Cheng, J.; Chen, H.; Xu, J.; Liu, Z.; Shi, Q.; Zhang, C. Recent Advances in the Application of Characterization Techniques for Studying Physical Stability of Amorphous Pharmaceutical Solids. Crystals 2021, 11, 1440. [Google Scholar] [CrossRef]
- Huang, C.; Chen, Z.; Gui, Y.; Shi, C.; Zhang, G.G.Z.; Yu, L. Crystal nucleation rates in glass-forming molecular liquids: D-sorbitol, D-arabitol, D-xylitol, and glycerol. J. Chem. Phys. 2018, 149, 054503. [Google Scholar] [CrossRef]
- Gui, Y.; Huang, C.; Shi, C.; Stelzer, T.; Zhang, G.G.Z.; Yu, L. Polymorphic selectivity in crystal nucleation. J. Chem. Phys. 2022, 156, 144504. [Google Scholar] [CrossRef]
- Yao, X.; Benson, E.G.; Gui, Y.; Stelzer, T.; Zhang, G.G.Z.; Yu, L. Surfactants Accelerate Crystallization of Amorphous Nifedipine by Similar Enhancement of Nucleation and Growth Independent of Hydrophilic–Lipophilic Balance. Mol. Pharm. 2022, 19, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Mapes, M.K.; Swallen, S.F.; Ediger, M.D. Self-Diffusion of Supercooled o-Terphenyl near the Glass Transition Temperature. J. Phys. Chem. B 2006, 110, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Swallen, S.F.; Ediger, M.D. Self-diffusion of the amorphous pharmaceutical indomethacin near Tg. Soft Matter 2011, 7, 10339–10344. [Google Scholar] [CrossRef]
- Shi, Q.; Cai, T. Fast Crystal Growth of Amorphous Griseofulvin: Relations between Bulk and Surface Growth Modes. Cryst. Growth Des. 2016, 16, 3279–3286. [Google Scholar] [CrossRef]
- Shi, Q.; Wang, Y.; Xu, J.; Liu, Z.; Chin, C.-Y. Fast crystal growth of amorphous nimesulide: Implication of surface effects. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2022, 78, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. Surface mobility of molecular glasses and its importance in physical stability. Adv. Drug Deliv. Rev. 2016, 100, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhu, L.; Kearns, K.L.; Ediger, M.D.; Yu, L. Glasses crystallize rapidly at free surfaces by growing crystals upward. Proc. Natl. Acad. Sci. USA 2011, 108, 5990–5995. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Sun, C.C. Crystal Growth of Celecoxib from Amorphous State: Polymorphism, Growth Mechanism, and Kinetics. Cryst. Growth Des. 2019, 19, 3592–3600. [Google Scholar] [CrossRef]
- Kestur, U.S.; Taylor, L.S. Role of polymer chemistry in influencing crystal growth rates from amorphous felodipine. CrystEngComm 2010, 12, 2390–2397. [Google Scholar] [CrossRef]
- Taylor, L.S.; Zografi, G. Spectroscopic Characterization of Interactions Between PVP and Indomethacin in Amorphous Molecular Dispersions. Pharm. Res. 1997, 14, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Kestur, U.S.; Lee, H.; Santiago, D.; Rinaldi, C.; Won, Y.-Y.; Taylor, L.S. Effects of the Molecular Weight and Concentration of Polymer Additives, and Temperature on the Melt Crystallization Kinetics of a Small Drug Molecule. Cryst. Growth Des. 2010, 10, 3585–3595. [Google Scholar] [CrossRef]
- Sato, T.; Taylor, L.S. Acceleration of the crystal growth rate of low molecular weight organic compounds in supercooled liquids in the presence of polyhydroxybutyrate. CrystEngComm 2017, 19, 80–87. [Google Scholar] [CrossRef]
- Kestur, U.S.; Taylor, L.S. Evaluation of the Crystal Growth Rate of Felodipine Polymorphs in the Presence and Absence of Additives As a Function of Temperature. Cryst. Growth Des. 2013, 13, 4349–4354. [Google Scholar] [CrossRef]
- Zhang, S.; Britten, J.F.; Chow, A.H.L.; Lee, T.W.Y. Impact of Crystal Structure and Polymer Excipients on the Melt Crystallization Kinetics of Itraconazole Polymorphs. Cryst. Growth Des. 2017, 17, 3433–3442. [Google Scholar] [CrossRef]
- Shi, Q.; Zhang, J.; Zhang, C.; Jiang, J.; Tao, J.; Zhou, D.; Cai, T. Selective Acceleration of Crystal Growth of Indomethacin Polymorphs by Low-Concentration Poly(ethylene oxide). Mol. Pharm. 2017, 14, 4694–4704. [Google Scholar] [CrossRef]
- Tian, B.; Gao, W.; Tao, X.; Tang, X.; Taylor, L.S. Impact of Polymers on the Melt Crystal Growth Rate of Indomethacin Polymorphs. Cryst. Growth Des. 2017, 17, 6467–6476. [Google Scholar] [CrossRef]
- Madejczyk, O.; Kaminska, E.; Tarnacka, M.; Dulski, M.; Jurkiewicz, K.; Kaminski, K.; Paluch, M. Studying the Crystallization of Various Polymorphic Forms of Nifedipine from Binary Mixtures with the Use of Different Experimental Techniques. Mol. Pharm. 2017, 14, 2116–2125. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. The Role of Drug–Polymer Hydrogen Bonding Interactions on the Molecular Mobility and Physical Stability of Nifedipine Solid Dispersions. Mol. Pharm. 2014, 12, 162–170. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. The Role of Polymer Concentration on the Molecular Mobility and Physical Stability of Nifedipine Solid Dispersions. Mol. Pharm. 2015, 12, 1477–1484. [Google Scholar] [CrossRef]
- Mistry, P.; Suryanarayanan, R. Strength of Drug–Polymer Interactions: Implications for Crystallization in Dispersions. Cryst. Growth Des. 2016, 16, 5141–5149. [Google Scholar] [CrossRef]
- Mohapatra, S.; Samanta, S.; Kothari, K.; Mistry, P.; Suryanarayanan, R. Effect of Polymer Molecular Weight on the Crystallization Behavior of Indomethacin Amorphous Solid Dispersions. Cryst. Growth Des. 2017, 17, 3142–3150. [Google Scholar] [CrossRef]
- Mistry, P.; Mohapatra, S.; Gopinath, T.; Vogt, F.G.; Suryanarayanan, R. Role of the Strength of Drug–Polymer Interactions on the Molecular Mobility and Crystallization Inhibition in Ketoconazole Solid Dispersions. Mol. Pharm. 2015, 12, 3339–3350. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhang, C.; Su, Y.; Zhang, J.; Zhou, D.; Cai, T. Acceleration of Crystal Growth of Amorphous Griseofulvin by Low-Concentration Poly(ethylene oxide): Aspects of Crystallization Kinetics and Molecular Mobility. Mol. Pharm. 2017, 14, 2262–2272. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Q.; Tao, J.; Peng, Y.; Cai, T. Impact of Polymer Enrichment at the Crystal–Liquid Interface on Crystallization Kinetics of Amorphous Solid Dispersions. Mol. Pharm. 2019, 16, 1385–1396. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Q.; Guo, M.; Liu, Z.; Cai, T. Melt Crystallization of Indomethacin Polymorphs in the Presence of Poly(ethylene oxide): Selective Enrichment of the Polymer at the Crystal–Liquid Interface. Mol. Pharm. 2020, 17, 2064–2071. [Google Scholar] [CrossRef]
- Shi, Q.; Cheng, J.; Li, F.; Xu, J.; Zhang, C. Molecular Mobility and Crystal Growth in Amorphous Binary Drug Delivery Systems: Effects of Low-Concentration Poly(Ethylene Oxide). AAPS PharmSciTech 2020, 21, 317. [Google Scholar] [CrossRef]
- Su, Y.; Yu, L.; Cai, T. Enhanced Crystal Nucleation in Glass-Forming Liquids by Tensile Fracture in the Glassy State. Cryst. Growth Des. 2018, 19, 40–44. [Google Scholar] [CrossRef]
- Powell, C.T.; Xi, H.; Sun, Y.; Gunn, E.; Chen, Y.; Ediger, M.D.; Yu, L. Fast Crystal Growth in o-Terphenyl Glasses: A Possible Role for Fracture and Surface Mobility. J. Phys. Chem. B 2015, 119, 10124–10130. [Google Scholar] [CrossRef]
- Shi, Q.; Tao, J.; Zhang, J.; Su, Y.; Cai, T. Crack- and Bubble-Induced Fast Crystal Growth of Amorphous Griseofulvin. Cryst. Growth Des. 2020, 20, 24–28. [Google Scholar] [CrossRef]
- Powell, C.T.; Chen, Y.; Yu, L. Fracture of molecular glasses under tension and increasing their fracture resistance with polymer additives. J. Non-Cryst. Solids 2015, 429, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Thakore, S.D.; Akhtar, J.; Jain, R.; Paudel, A.; Bansal, A.K. Analytical and Computational Methods for the Determination of Drug-Polymer Solubility and Miscibility. Mol. Pharm. 2021, 18, 2835–2866. [Google Scholar] [CrossRef]
- Duan, P.; Lamm, M.S.; Yang, F.; Xu, W.; Skomski, D.; Su, Y.; Schmidt-Rohr, K. Quantifying Molecular Mixing and Heterogeneity in Pharmaceutical Dispersions at Sub-100 nm Resolution by Spin Diffusion NMR. Mol. Pharm. 2020, 17, 3567–3580. [Google Scholar] [CrossRef] [PubMed]
- Vasanthavada, M.; Tong, W.-Q.; Joshi, Y.; Kislalioglu, M.S. Phase Behavior of Amorphous Molecular Dispersions II: Role of Hydrogen Bonding in Solid Solubility and Phase Separation Kinetics. Pharm. Res. 2005, 22, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Marsac, P.J.; Shamblin, S.L.; Taylor, L.S. Theoretical and Practical Approaches for Prediction of Drug–Polymer Miscibility and Solubility. Pharm. Res. 2006, 23, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Marsac, P.J.; Li, T.; Taylor, L.S. Estimation of Drug–Polymer Miscibility and Solubility in Amorphous Solid Dispersions Using Experimentally Determined Interaction Parameters. Pharm. Res. 2009, 26, 139–151. [Google Scholar] [CrossRef]
- Tao, J.; Sun, Y.; Zhang, G.G.Z.; Yu, L. Solubility of Small-Molecule Crystals in Polymers: D-Mannitol in PVP, Indomethacin in PVP/VA, and Nifedipine in PVP/VA. Pharm. Res. 2009, 26, 855–864. [Google Scholar] [CrossRef]
- Sun, Y.; Tao, J.; Zhang, G.G.Z.; Yu, L. Solubilities of Crystalline Drugs in Polymers: An Improved Analytical Method and Comparison of Solubilities of Indomethacin and Nifedipine in PVP, PVP/VA, and PVAc. J. Pharm. Sci. 2010, 99, 4023–4031. [Google Scholar] [CrossRef]
- Tian, Y.; Booth, J.; Meehan, E.; Jones, D.S.; Li, S.; Andrews, G.P. Construction of Drug–Polymer Thermodynamic Phase Diagrams Using Flory–Huggins Interaction Theory: Identifying the Relevance of Temperature and Drug Weight Fraction to Phase Separation within Solid Dispersions. Mol. Pharm. 2013, 10, 236–248. [Google Scholar] [CrossRef]
- Pekamwar, S.; Kulkarni1, D.; Gadade, D. Accidental formation of eutectics during crystal engineering of lamotrigine with solubility advantage and drug release efficiency. Asian J. Pharm. 2021, 15, 60–67. [Google Scholar]
- Panzade, P.; Shendarkar, G.; Kulkarni, D.; Shelke, S. Solid State Characterization and Dissolution Enhancement of Nevirapine Cocrystals. Adv. Pharm. Bull. 2021, 11, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Knopp, M.M.; Tajber, L.; Tian, Y.; Olesen, N.E.; Jones, D.S.; Kozyra, A.; Löbmann, K.; Paluch, K.J.; Brennan, C.M.; Holm, R.; et al. Comparative Study of Different Methods for the Prediction of Drug–Polymer Solubility. Mol. Pharm. 2015, 12, 3408–3419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, C.B.; Davis, M.T.; Albadarin, A.B.; Walker, G.M. Investigation of the Dependence of the Flory–Huggins Interaction Parameter on Temperature and Composition in a Drug–Polymer System. Mol. Pharm. 2018, 15, 5327–5335. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Jacobs, E.; Jones, D.S.; McCoy, C.P.; Wu, H.; Andrews, G.P. The design and development of high drug loading amorphous solid dispersion for hot-melt extrusion platform. Int. J. Pharm. 2020, 586, 119545. [Google Scholar] [CrossRef]
- Panzade, P.S.; Shendarkar, G.R.; Kulkarni, D.A. Hot Melt Extrusion: An Emerging Green Technique for the Synthesis of High-Quality Pharmaceutical Cocrystals. J. Pharm. Innov. 2022, 17, 283–293. [Google Scholar] [CrossRef]
- Anderson, B.D. Predicting Solubility/Miscibility in Amorphous Dispersions: It Is Time to Move Beyond Regular Solution Theories. J. Pharm. Sci. 2018, 107, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Xiang, T.-X.; Anderson, B.D. Molecular Dynamics Simulation of Amorphous Indomethacin-Poly(Vinylpyrrolidone) Glasses: Solubility and Hydrogen Bonding Interactions. J. Pharm. Sci. 2013, 102, 876–891. [Google Scholar] [CrossRef]
- Xiang, T.-X.; Anderson, B.D. Molecular Dynamics Simulation of Amorphous Hydroxypropylmethylcellulose and Its Mixtures With Felodipine and Water. J. Pharm. Sci. 2017, 106, 803–816. [Google Scholar] [CrossRef]
- Xiang, T.-X.; Anderson, B.D. Effects of Molecular Interactions on Miscibility and Mobility of Ibuprofen in Amorphous Solid Dispersions With Various Polymers. J. Pharm. Sci. 2019, 108, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Gumaste, S.G.; Gupta, S.S.; Serajuddin, A.T.M. Investigation of Polymer-Surfactant and Polymer-Drug-Surfactant Miscibility for Solid Dispersion. AAPS J. 2016, 18, 1131–1143. [Google Scholar] [CrossRef]
- Tian, B.; Tang, X.; Taylor, L.S. Investigating the Correlation between Miscibility and Physical Stability of Amorphous Solid Dispersions Using Fluorescence-Based Techniques. Mol. Pharm. 2016, 13, 3988–4000. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Singh, B.; Agrawal, A.K.; Bansal, A.K. Correlationship of drug-polymer miscibility, molecular relaxation and phase behavior of sipyridamole amorphous solid dispersions. J. Pharm. Sci. 2021, 110, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Medarevi’c, D.; Djuris, J.; Barmpalexis, P.; Kachrimanis, K.; Ibri´c, S. Analytical and computational methods for the estimation of drug-polymer solubility and miscibility in solid dispersions development. Pharmaceutics 2019, 11, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iemtsev, A.; Hassouna, F.; Mathers, A.; Klajmon, M.; Dendisová, M.; Malinová, L.; Školáková, T.; Fulem, M. Physical stability of hydroxypropyl methylcellulose-based amorphous solid dispersions: Experimental and computational study. Int. J. Pharm. 2020, 589, 119845. [Google Scholar] [CrossRef] [PubMed]
- Bochmann, E.S.; Neumann, D.; Gryczke, A.; Wagner, K.G. Micro-scale solubility assessments and prediction models for active pharmaceutical ingredients in polymeric matrices. Eur. J. Pharm. Biopharm. 2019, 141, 111–120. [Google Scholar] [CrossRef]
- Huang, Y.; Dai, W.-G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm. Sin. B 2014, 4, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Purohit, H.S.; Taylor, L.S. Miscibility of Itraconazole–Hydroxypropyl Methylcellulose Blends: Insights with High Resolution Analytical Methodologies. Mol. Pharm. 2015, 12, 4542–4553. [Google Scholar] [CrossRef]
- Luebbert, C.; Huxoll, F.; Sadowski, G. Amorphous-Amorphous Phase Separation in API/Polymer Formulations. Molecules 2017, 22, 296. [Google Scholar] [CrossRef]
- Qian, F.; Huang, J.; Zhu, Q.; Haddadin, R.; Gawel, J.; Garmise, R.; Hussain, M. Is a distinctive single Tg a reliable indicator for the homogeneity of amorphous solid dispersion? Int. J. Pharm. 2010, 395, 232–235. [Google Scholar] [CrossRef]
- Lodge, T.P.; Wood, E.R.; Haley, J.C. Two calorimetric glass transitions do not necessarily indicate immiscibility: The case of PEO/PMMA. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 756–763. [Google Scholar] [CrossRef]
- Li, N.; Taylor, L.S. Nanoscale infrared, thermal, and mechanical characterization of telaprevir-polymer miscibility in amorphous solid dispersions prepared by solvent evaporation. Mol. Pharm. 2016, 13, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Luebbert, C.; Klanke, C.; Sadowski, G. Investigating phase separation in amorphous solid dispersions via Raman mapping. Int. J. Pharm. 2018, 535, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhang, G.G.; Kjoller, K.; Dillon, E.; Purohit, H.S.; Taylor, L.S. Phase separation in surfactant-containing amorphous solid dispersions: Orthogonal analytical methods to probe the effects of surfactants on morphology and phase composition. Int. J. Pharm. 2022, 619, 121708. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Handa, T.; Alonzo, D.E.; Taylor, L.S. Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur. J. Pharm. Biopharm. 2008, 70, 493–499. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Impact of Polymers on Crystal Growth Rate of Structurally Diverse Compounds from Aqueous Solution. Mol. Pharm. 2013, 10, 2381–2393. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Understanding Polymer Properties Important for Crystal Growth Inhibition—Impact of Chemically Diverse Polymers on Solution Crystal Growth of Ritonavir. Cryst. Growth Des. 2012, 12, 3133–3143. [Google Scholar] [CrossRef]
- Alonzo, D.E.; Gao, Y.; Zhou, D.; Mo, H.; Zhang, G.G.; Taylor, L.S. Dissolution and Precipitation Behavior of Amorphous Solid Dispersions. J. Pharm. Sci. 2011, 100, 3316–3331. [Google Scholar] [CrossRef]
- Jackson, M.J.; Kestur, U.S.; Hussain, M.A.; Taylor, L.S. Dissolution of Danazol Amorphous Solid Dispersions: Supersaturation and Phase Behavior as a Function of Drug Loading and Polymer Type. Mol. Pharm. 2016, 13, 223–231. [Google Scholar] [CrossRef]
- Saboo, S.; Moseson, D.E.; Kestur, U.S.; Taylor, L.S. Patterns of drug release as a function of drug loading from amorphous solid dispersions: A comparison of five different polymers. Eur. J. Pharm. Sci. 2020, 155, 105514. [Google Scholar] [CrossRef]
- Nunes, P.D.; Pinto, J.F.; Henriques, J.; Paiva, A.M. Insights into the Release Mechanisms of ITZ:HPMCAS Amorphous Solid Dispersions: The Role of Drug-Rich Colloids. Mol. Pharm. 2022, 19, 51–66. [Google Scholar] [CrossRef]
- Ting, J.; Tale, S.; Purchel, A.A.; Jones, S.D.; Widanapathirana, L.; Tolstyka, Z.P.; Guo, L.; Guillaudeu, S.; Bates, F.S.; Reineke, T.M. High-Throughput Excipient Discovery Enables Oral Delivery of Poorly Soluble Pharmaceuticals. ACS Cent. Sci. 2016, 2, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.M.; Porter III, W.W.; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in polymer design for enhancing oral drug solubility and delivery. Bioconjugate Chem. 2018, 29, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Amponsah-Efah, K.K.; Mistry, P.; Eisenhart, R.; Suryanarayanan, R. The Influence of the Strength of Drug–Polymer Interactions on the Dissolution of Amorphous Solid Dispersions. Mol. Pharm. 2020, 18, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, C.; Chen, Z.; Su, C.; Hageman, M.; Hussain, M.; Haskell, R.; Stefanski, K.; Qian, F. Drug–Polymer–Water Interaction and Its Implication for the Dissolution Performance of Amorphous Solid Dispersions. Mol. Pharm. 2015, 12, 576–589. [Google Scholar] [CrossRef]
- Liu, L.; Chen, L.; Müllers, W.; Serno, P.; Qian, F. Water-Resistant Drug–Polymer Interaction Contributes to the Formation of Nano-Species during the Dissolution of Felodipine Amorphous Solid Dispersions. Mol. Pharm. 2022, 19, 2888–2899. [Google Scholar] [CrossRef]
- Ueda, K.; Hate, S.S.; Taylor, L.S. Impact of Hypromellose Acetate Succinate Grade on Drug Amorphous Solubility and In Vitro Membrane Transport. J. Pharm. Sci. 2020, 109, 2464–2473. [Google Scholar] [CrossRef]
- Sugihara, H.; Taylor, L.S. Evaluation of pazopanib phase behavior following pH-induced supersaturation. Mol. Pharm. 2018, 15, 1690–1699. [Google Scholar] [CrossRef]
- Schram, C.J.; Beaudoin, S.P.; Taylor, L.S. Impact of Polymer Conformation on the Crystal Growth Inhibition of a Poorly Water-Soluble Drug in Aqueous Solution. Langmuir 2015, 31, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liu, C.; Chen, Y.; Zhu, A.D.; Qian, F. Aggregation of Hydroxypropyl Methylcellulose Acetate Succinate under Its Dissolving pH and the Impact on Drug Supersaturation. Mol. Pharm. 2018, 15, 4643–4653. [Google Scholar] [CrossRef]
- Que, C.; Gao, Y.; Raina, S.A.; Zhang, G.G.Z.; Taylor, L.S. Paclitaxel Crystal Seeds with Different Intrinsic Properties and Their Impact on Dissolution of Paclitaxel-HPMCAS Amorphous Solid Dispersions. Cryst. Growth Des. 2018, 18, 1548–1559. [Google Scholar] [CrossRef]
- Qian, K.; Stella, L.; Jones, D.S.; Andrews, G.P.; Du, H.; Tian, Y. Drug-rich phases induced by amorphous solid dispersion: Arbitrary or intentional goal in oral drug delivery? Pharmaceutics 2021, 13, 889. [Google Scholar] [CrossRef] [PubMed]
- Saboo, S.; Mugheirbi, N.A.; Zemlyanov, D.Y.; Kestur, U.S.; Taylor, L.S. Congruent release of drug and polymer: A “sweet spot” in the dissolution of amorphous solid dispersions. J. Control. Release 2019, 298, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, S.; Wang, S.; Liu, C.; Su, C.; Hageman, M.; Hussain, M.; Haskell, R.; Stefanski, K.; Qian, F. Sodium Lauryl Sulfate Competitively Interacts with HPMC-AS and Consequently Reduces Oral Bioavailability of Posaconazole/HPMC-AS Amorphous Solid Dispersion. Mol. Pharm. 2016, 13, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Taylor, L.S. Partitioning of surfactant into drug-rich nanodroplets and its impact on drug thermodynamic activity and droplet size. J. Control. Release 2021, 330, 229–243. [Google Scholar] [CrossRef]
- Raina, S.A.; Zhang, G.G.Z.; Alonzo, D.E.; Wu, J.; Zhu, D.; Catron, N.D.; Gao, Y.; Taylor, L.S. Impact of Solubilizing Additives on Supersaturation and Membrane Transport of Drugs. Pharm. Res. 2015, 32, 3350–3364. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Bergstrom, C.A.S.; Taylor, L.S. Compromised in vitro dissolution and membrane transport of multidrug amorphous formulations. J. Control. Release 2016, 229, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Taylor, L.S. Tailoring supersaturation from amorphous solid dispersions. J. Control. Release 2018, 279, 114–125. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Q.; Chen, H.; Wang, Y.; Wang, R.; Xu, J.; Zhang, C. Amorphous Solid Dispersions: Role of the Polymer and Its Importance in Physical Stability and In Vitro Performance. Pharmaceutics 2022, 14, 1747. https://doi.org/10.3390/pharmaceutics14081747
Shi Q, Chen H, Wang Y, Wang R, Xu J, Zhang C. Amorphous Solid Dispersions: Role of the Polymer and Its Importance in Physical Stability and In Vitro Performance. Pharmaceutics. 2022; 14(8):1747. https://doi.org/10.3390/pharmaceutics14081747
Chicago/Turabian StyleShi, Qin, Haibiao Chen, Yanan Wang, Ruoxun Wang, Jia Xu, and Chen Zhang. 2022. "Amorphous Solid Dispersions: Role of the Polymer and Its Importance in Physical Stability and In Vitro Performance" Pharmaceutics 14, no. 8: 1747. https://doi.org/10.3390/pharmaceutics14081747
APA StyleShi, Q., Chen, H., Wang, Y., Wang, R., Xu, J., & Zhang, C. (2022). Amorphous Solid Dispersions: Role of the Polymer and Its Importance in Physical Stability and In Vitro Performance. Pharmaceutics, 14(8), 1747. https://doi.org/10.3390/pharmaceutics14081747