
Functional Duality of Chondrocyte Hypertrophy and Biomedical Application Trends in Osteoarthritis

 ,
, 
Abstract
1. Introduction
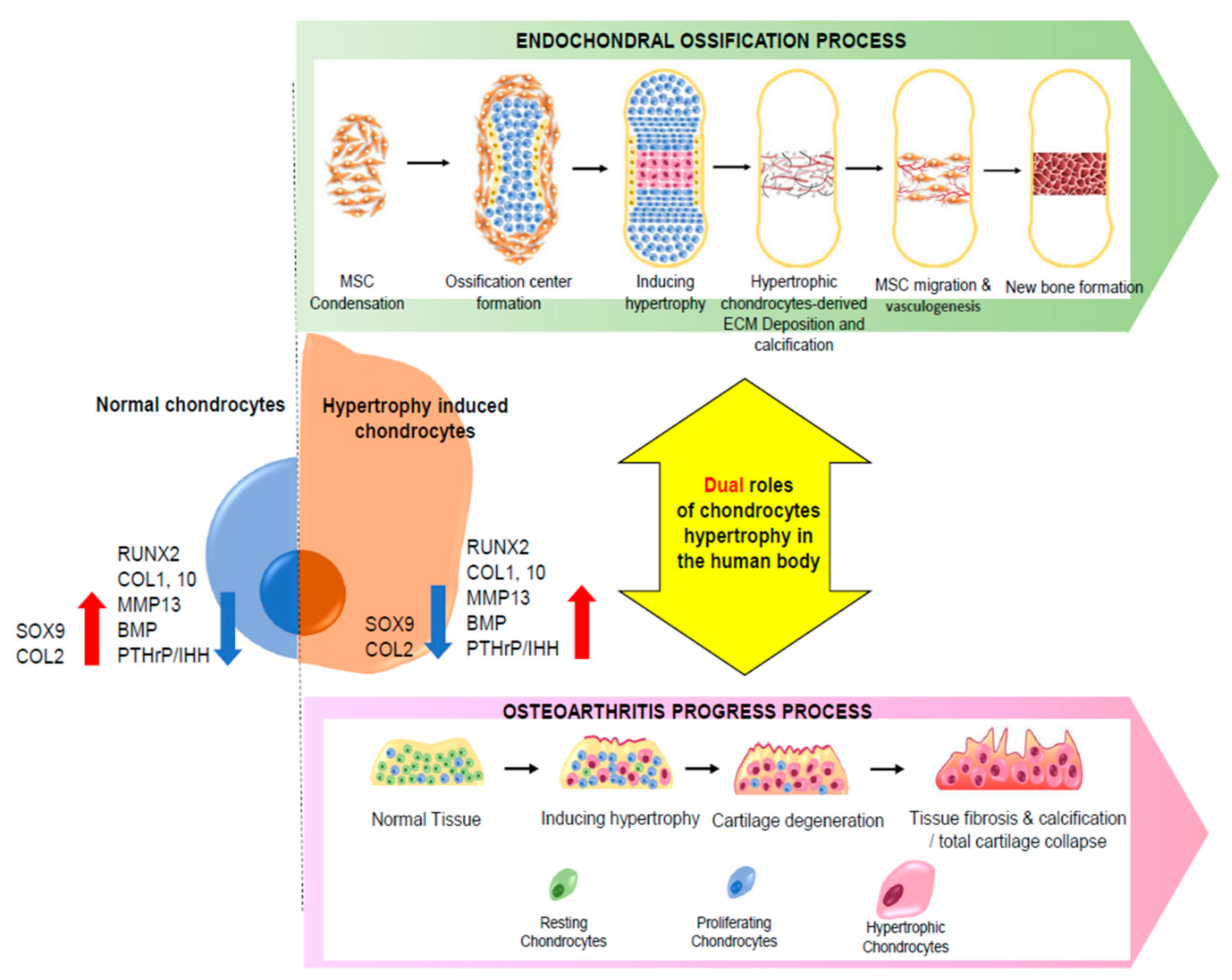
2. Dual Function of Chondrocyte Hypertrophy
2.1. Chondrocyte Hypertrophy in Endochondral Ossification
2.2. Hypertrophy in Cartilage Diseases
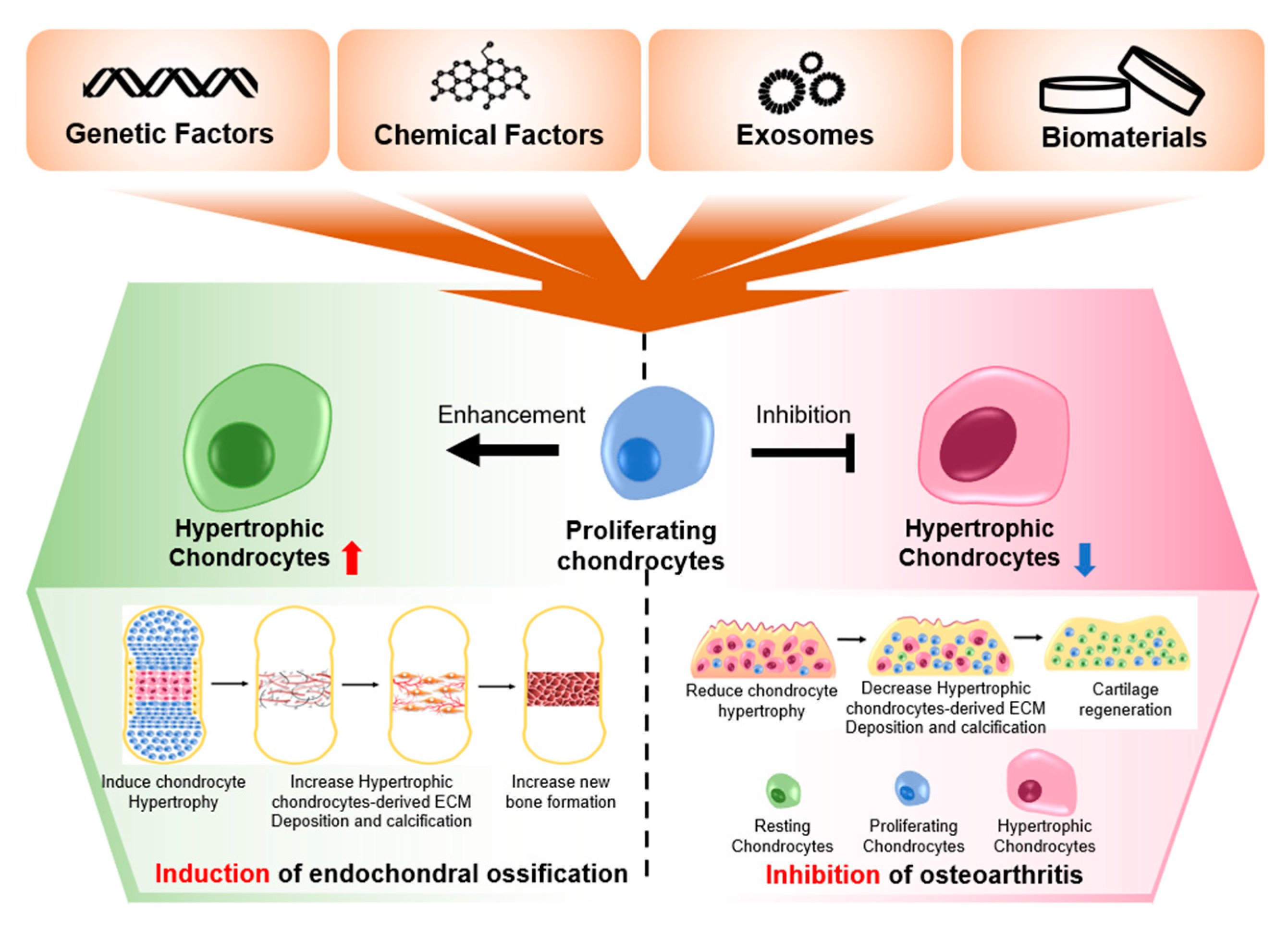
3. Current Trends of the Treatment of Chondrocyte Hypertrophy-Induced OA
3.1. Genetic Interventions against Chondrocyte Hypertrophy
SOX 9
3.2. Histone Deacetylase 4 (HDAC4)
NKX3.2
3.3. Protein Interventions for Chondrocyte Hypertrophy
3.3.1. TGF-β1
3.3.2. PTHrP
3.4. Inhibitors of Hypertrophic Markers
3.5. Small Molecules Preventing Chondrocyte Hypertrophy
Chemical Interventions Found in Nature
3.6. Three-Dimensional Environments Preventing Chondrocyte Hypertrophy
3.7. Exosomes
3.8. Stem Cell-Derived Exosomes
3.9. Exosomes Derived from Other Cell Types
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analy-sis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Peat, G. Knee pain and osteoarthritis in older adults: A review of community burden and current use of primary health care. Ann. Rheum. Dis. 2001, 60, 91–97. [Google Scholar] [CrossRef]
- Hunter, D.J.; McDougall, J.; Keefe, F.J. The Symptoms of Osteoarthritis and the Genesis of Pain. Med. Clin. N. Am. 2009, 93, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Schofield, D.; Callander, E. The individual and socioeconomic impact of osteoarthritis. Nat. Rev. Rheumatol. 2014, 10, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Archer, C.W.; Francis-West, P. The chondrocyte. Int. J. Biochem. Cell Biol. 2003, 35, 401–404. [Google Scholar] [CrossRef]
- van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef]
- Aghajanian, P.; Mohan, S. The art of building bone: Emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 2018, 6, 19. [Google Scholar] [CrossRef]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S.E. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef]
- Ortega, N.; Behonick, D.; Werb, Z. Matrix remodeling during endochondral ossification. Trends Cell Biol. 2004, 14, 86–93. [Google Scholar] [CrossRef]
- Brighton, C.T.; Sugioka, Y.; Hunt, R.M. Cytoplasmic structures of epiphyseal plate chondrocytes: Quantitative evalu-ation using electron micrographs of rat costochondral junctions with special reference to the fate of hypertrophic cells. J. Bone Joint Surg Am. 1973, 55, 771–784. [Google Scholar] [CrossRef]
- Mackie, E.; Tatarczuch, L.; Mirams, M. Thematic Review: The Skeleton: A Multi-Functional Complex Organ. The Growth Plate Chondrocyte and Endochondral Ossification. J. Endocrinol. 2011, 211, 109–121. [Google Scholar] [CrossRef]
- Usami, Y.; Gunawardena, A.T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt signaling in cartilage development and diseases: Lessons from animal studies. Lab. Investig. 2016, 96, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Hata, K.; Takahata, Y.; Murakami, T.; Nakamura, E.; Yagi, H. Regulation of Cartilage Development and Diseases by Transcription Factors. J. Bone Metab. 2017, 24, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Rolian, C. Endochondral ossification and the evolution of limb proportions. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e373. [Google Scholar] [CrossRef]
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Adams, C.S.; Srinivas, V.; Freeman, T.A. Chondrocyte hypertrophy and apoptosis at the cartilage-bone inter-face. In Bone and Osteoarthritis; Springer: London, UK, 2007; pp. 109–129. [Google Scholar]
- Ono, N.; Kronenberg, H.M. Developmental Biology of Musculoskeletal Tissues for Tissue Engineers. In Developmental Biology and Musculoskeletal Tissue Engineering; Elsevier BV: Amsterdam, The Netherlands, 2018; pp. 1–24. [Google Scholar]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.-S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Huang, X.; Karperien, M.; Post, J.N. The Regulatory Role of Signaling Crosstalk in Hypertrophy of MSCs and Human Articular Chondrocytes. Int. J. Mol. Sci. 2015, 16, 19225–19247. [Google Scholar] [CrossRef]
- Hollander, J.; Zeng, L. The Emerging Role of Glucose Metabolism in Cartilage Development. Curr. Osteoporos. Rep. 2019, 17, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Guntur, A.R.; Rosen, C.J. IGF-1 regulation of key signaling pathways in bone. BoneKEy Rep. 2013, 2, 437. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Cao, H.; Cu, F.; Xu, D.; Lei, Y.; Tan, Y.; Magdalou, J.; Wang, H.; Chen, L. Nicotine-induced retardation of chon-drogenesis through down-regulation of IGF-1 signaling pathway to inhibit matrix synthesis of growth plate chondrocytes in fetal rats. Toxicol. Appl. Pharmacol. 2013, 269, 25–33. [Google Scholar] [CrossRef]
- Leijten, J.; Emons, J.; Sticht, C.; Van Gool, S.; Decker, E.; Uitterlinden, A.; Rappold, G.; Hofman, A.; Rivadeneira, F.; Scherjon, S.; et al. Gremlin 1, Frizzled-related protein, and Dkk-1 are key regulators of human articular cartilage homeostasis. Arthritis Rheum. 2012, 64, 3302–3312. [Google Scholar] [CrossRef]
- Gawlitta, D.; Farrell, E.; Malda, J.; Creemers, L.; Alblas, J.; Dhert, W. Modulating Endochondral Ossification of Multipotent Stromal Cells for Bone Regeneration. Tissue Eng. Part B Rev. 2010, 16, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Sasano, Y.; Zhu, J.-X.; Tsubota, M.; Takahashi, I.; Onodera, K.; Mizoguchi, I.; Kagayama, M. Gene expression of MMP8 and MMP13 during embryonic development of bone and cartilage in the rat mandible and hind limb. J. Histochem. Cytochem. 2002, 50, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Wakabayashi, M.; Hata, K.; Matsubara, T.; Honma, S.; Wakisaka, S.; Kiyonari, H.; Shioi, G.; Yamaguchi, A.; Tsumaki, N.; et al. Osterix Regulates Calcification and Degradation of Chondrogenic Matrices through Matrix Metalloproteinase 13 (MMP13) Expression in Association with Transcription Factor Runx2 during Endochondral Ossification. J. Biol. Chem. 2012, 287, 33179–33190. [Google Scholar] [CrossRef] [PubMed]
- Pelttari, K.; Winter, A.; Steck, E.; Goetzke, K.; Hennig, T.; Ochs, B.G.; Aigner, T.; Richter, W. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells cor-relates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum 2006, 54, 3254–3266. [Google Scholar] [CrossRef]
- Nishimura, R.; Hata, K.; Takahata, Y.; Murakami, T.; Nakamura, E.; Ohkawa, M.; Ruengsinpinya, L. Role of Signal Trans-duction Pathways and Transcription Factors in Cartilage and Joint Diseases. Int. J. Mol. Sci. 2020, 21, 1340. [Google Scholar] [CrossRef]
- Komori, T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol. 2018, 149, 313–323. [Google Scholar] [CrossRef]
- Cleary, M.A.; van Osch, G.J.M.; Brama, P.A.; Hellingman, C.A.; Narcisi, R. FGF, TGFβ and Wnt crosstalk: Embryonic to in vitro cartilage development from mesenchymal stem cells. J. Tissue Eng. Regen. Med. 2015, 9, 332–342. [Google Scholar] [CrossRef]
- Riegger, J.; Brenner, R.E. Pathomechanisms of Posttraumatic Osteoarthritis: Chondrocyte Behavior and Fate in a Precarious Environment. Int. J. Mol. Sci. 2020, 21, 1560. [Google Scholar] [CrossRef] [PubMed]
- van der Kraan, P.; Matta, C.; Mobasheri, A. Age-related alterations in signaling pathways in articular chondrocytes: Implica-tions for the pathogenesis and progression of osteoarthritis-a mini-review. Gerontology 2017, 63, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Allas, L.; Rochoux, Q.; Leclercq, S.; Boumediene, K.; Baugé, C. Development of a simple osteoarthritis model useful to predict in vitro the anti-hypertrophic action of drugs. Lab. Investig. 2020, 100, 64–71. [Google Scholar] [CrossRef]
- Jing, J.; Hinton, R.J.; Feng, J.Q. Bmpr1a Signaling in Cartilage Development and Endochondral Bone Formation. Vitam. Horm. 2015, 99, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, J.; Zhang, S.; Ouyang, H.W. Inhibitory function of parathyroid hormone-related protein on chondrocyte hypertrophy: The implication for articular cartilage repair. Arthritis Res. Ther. 2012, 14, 221. [Google Scholar] [CrossRef]
- Wuelling, M.; Vortkamp, A. Transcriptional networks controlling chondrocyte proliferation and differentiation during endo-chondral ossification. Pediatric Nephrol. 2010, 25, 625–631. [Google Scholar] [CrossRef]
- Tsang, K.Y.; Cheah, K.S. The extended chondrocyte lineage: Implications for skeletal homeostasis and disorders. Curr. Opin. Cell Biol. 2019, 61, 132–140. [Google Scholar] [CrossRef]
- Ripmeester, E.G.J.; Timur, U.T.; Caron, M.M.J.; Welting, T.J.M. Recent Insights into the Contribution of the Changing Hypertrophic Chondrocyte Phenotype in the Development and Progression of Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- van der Kraan, P.M.; Blaney Davidson, E.N.; Blom, A.; van den Berg, W.B. TGF-beta signaling in chondrocyte terminal differentiation and osteoarthritis: Modulation and integration of signaling pathways through receptor-Smads. Osteoarthr. Cartil. 2009, 17, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhou, G.; Morello, R.; Chen, Y.; Garcia-Rojas, X.; Lee, B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte–specific expression in vivo. J. Cell Biol. 2003, 162, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Manner, P.A.; Horner, A.; Shum, L.; Tuan, R.S.; Nuckolls, G.H. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthr. Cartil. 2004, 12, 963–973. [Google Scholar] [CrossRef]
- Kamekura, S.; Hoshi, K.; Shimoaka, T.; Chung, U.; Chikuda, H.; Yamada, T.; Uchida, M.; Ogata, N.; Seichi, A.; Nakamura, K.; et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthr. Cartil. 2005, 13, 632–641. [Google Scholar] [CrossRef]
- Kamekura, S.; Kawasaki, Y.; Hoshi, K.; Shimoaka, T.; Chikuda, H.; Maruyama, Z.; Komori, T.; Sato, S.; Takeda, S.; Karsenty, G.; et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006, 54, 2462–2470. [Google Scholar] [CrossRef]
- Sandell, L.J.; Aigner, T. Articular cartilage and changes in Arthritis: Cell biology of osteoarthritis. Arthritis Res. 2001, 3, 107–113. [Google Scholar] [CrossRef]
- Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177–192. [Google Scholar] [CrossRef]
- Falah, M.; Nierenberg, G.; Soudry, M.; Hayden, M.; Volpin, G. Treatment of articular cartilage lesions of the knee. Int. Orthop. 2010, 34, 621–630. [Google Scholar] [CrossRef]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T. Tran-scriptional regulation of endochondral ossification by HIF-2α during skeletal growth and osteoarthritis development. Nat. Med. 2010, 16, 678. [Google Scholar] [CrossRef] [PubMed]
- Hirata, M.; Kugimiya, F.; Fukai, A.; Ohba, S.; Kawamura, N.; Ogasawara, T.; Kawasaki, Y.; Saito, T.; Yano, F.; Ikeda, T.; et al. C/EBPβ Promotes Transition from Proliferation to Hypertrophic Differentiation of Chondrocytes through Transactivation of p57Kip2. PLoS ONE 2009, 4, e4543. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.-H.; Chae, C.-S.; Kwak, J.-S.; Oh, H.; Shin, Y.; Huh, Y.H.; Lee, C.-G.; Park, Y.-W.; Chun, C.-H.; Kim, Y.-M. Hypoxia-inducible factor-2α is an essential catabolic regulator of inflammatory rheumatoid arthritis. PLoS Biol. 2014, 12, e1001881. [Google Scholar] [CrossRef]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.; Werb, Z.; Shah, M.H.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef]
- Chan, B.Y.; Little, C.B. The interaction of canonical bone morphogenetic protein- and Wnt-signaling pathways may play an important role in regulating cartilage degradation in osteoarthritis. Arthritis Res. Ther. 2012, 14, 119. [Google Scholar] [CrossRef] [PubMed]
- Mariani, E.; Pulsatelli, L.; Facchini, A. Signaling Pathways in Cartilage Repair. Int. J. Mol. Sci. 2014, 15, 8667–8698. [Google Scholar] [CrossRef]
- Hojo, H.; Yano, F.; Ohba, S.; Igawa, K.; Nakajima, K.; Komiyama, Y.; Kan, A.; Ikeda, T.; Yonezawa, T.; Woo, J.-T. Identifica-tion of oxytetracycline as a chondrogenic compound using a cell-based screening system. J. Bone Miner. Metab. Olism. 2010, 28, 627–633. [Google Scholar] [CrossRef]
- Huebner, J.L.; Johnson, K.; Kraus, V.B.; Terkeltaub, R.A. Transglutaminase 2 is a marker of chondrocyte hypertrophy and osteoarthritis severity in the Hartley guinea pig model of knee OA. Osteoarthr. Cartil. 2009, 17, 1056–1064. [Google Scholar] [CrossRef]
- Solomon, L.A.; Bérubé, N.G.; Beier, F. Transcriptional regulators of chondrocyte hypertrophy. Birth Defects Res. Part C Embryo Today Rev. 2008, 84, 123–130. [Google Scholar] [CrossRef]
- Buckwalter, J.A.; Anderson, D.D.; Brown, T.D.; Tochigi, Y.; Martin, J.A. The roles of mechanical stresses in the pathogenesis of osteoarthritis: Implications for treatment of joint injuries. Cartilage 2013, 4, 286–294. [Google Scholar] [CrossRef]
- Blain, E.J. Mechanical regulation of matrix metalloproteinases. Front. Biosci. 2007, 12, 507–527. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.-X. TGF-β/Smad3 signals repress chondrocyte hypertrophic differentia-tion and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef]
- Drissi, H.; Zuscik, M.; Rosier, R.; O’Keefe, R. Transcriptional regulation of chondrocyte maturation: Potential involvement of transcription factors in OA pathogenesis. Mol. Asp. Med. 2005, 26, 169–179. [Google Scholar] [CrossRef]
- Rutges, J.; Duit, R.; Kummer, J.; Oner, F.; van Rijen, M.; Verbout, A.; Castelein, R.; Dhert, W.; Creemers, L. Hypertrophic differentiation and calcification during intervertebral disc degeneration. Osteoarthr. Cartil. 2010, 18, 1487–1495. [Google Scholar] [CrossRef]
- Komuro, H.; Olee, T.; Kühn, K.; Quach, J.; Brinson, D.C.; Shikhman, A.; Valbracht, J.; Creighton-Achermann, L.; Lotz, M. The osteoprotegerin/receptor activator of nuclear factor κB/receptor activator of nuclear factor κB ligand system in cartilage. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2001, 44, 2768–2776. [Google Scholar] [CrossRef]
- Pilichou, A.; Papassotiriou, I.; Michalakakou, K.; Fessatou, S.; Fandridis, E.; Papachristou, G.; Terpos, E. High levels of synovial fluid osteoprotegerin (OPG) and increased serum ratio of receptor activator of nuclear factor-κB ligand (RANKL) to OPG correlate with disease severity in patients with primary knee osteoarthritis. Clin. Biochem. 2008, 41, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L. Osteo-protegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Simonet, W.; Lacey, D.; Dunstan, C.; Kelley, M.; Chang, M.-S.; Lüthy, R.; Nguyen, H.; Wooden, S.; Bennett, L.; Boone, T. Os-teoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Pfander, D.; Swoboda, B.; Kirsch, T. Expression of Early and Late Differentiation Markers (Proliferating Cell Nuclear Antigen, Syndecan-3, Annexin VI, and Alkaline Phosphatase) by Human Osteoarthritic Chondrocytes. Am. J. Pathol. 2001, 159, 1777–1783. [Google Scholar] [CrossRef]
- Roach, H.I. Association of Matrix Acid and Alkaline Phosphatases with Mineralization of Cartilage and Endochondral Bone. J. Mol. Histol. 1999, 31, 53–61. [Google Scholar] [CrossRef]
- Schmid, T.M.; Bonen, D.K.; Luchene, L.; Linsenmayer, T.F. Late events in chondrocyte differentiation: Hypertrophy, type X collagen synthesis and matrix calcification. In Vivo 1991, 5, 533–540. [Google Scholar]
- Moreno-Rubio, J.; Herrero-Beaumont, G.; Tardı, O.L.; álvarez-Soria, M.Á.; Largo, R. Nonsteroidal antiinflammatory drugs and prostaglandin E2 modulate the synthesis of osteoprotegerin and RANKL in the cartilage of patients with severe knee os-teoarthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2010, 62, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Antholz, C.R.; Cherian, J.J.; Elmallah, R.K.; Jauregui, J.J.; Pierce, T.P.; Mont, A.M. Selective Patellar Resurfacing: A Literature Review. Surg. Technol. Int. 2015, 26, 355–360. [Google Scholar]
- Boos, N.; Nerlich, A.G.; Wiest, I.; von der Mark, K.; Aebi, M. Immunolocalization of type X collagen in human lumbar inter-vertebral discs during ageing and degeneration. Histochem. Cell Biol. 1997, 108, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Nerlich, A.G.; Schleicher, E.D.; Boos, N. 1997 Volvo Award winner in basic science studies: Immunohistologic markers for age-related changes of human lumbar intervertebral discs. Spine 1997, 22, 2781–2795. [Google Scholar] [CrossRef]
- Roberts, S.; Bains, M.A.; Kwan, A.; Menage, J.; Eisenstein, S.M. Type X collagen in the human invertebral disc: An indication of repair or remodelling? J. Mol. Histol. 1998, 30, 89–95. [Google Scholar] [CrossRef]
- Akiyama, H. Control of chondrogenesis by the transcription factor Sox9. Mod. Rheumatol. 2008, 18, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Haseeb, A.; Kc, R.; Angelozzi, M.; de Charleroy, C.; Rux, D.; Tower, R.J.; Yao, L.; da Silva, R.P.; Pacifici, M.; Qin, L.; et al. SOX9 keeps growth plates and articular cartilage healthy by inhibiting chondrocyte dedifferentiation/osteoblastic redifferentiation. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Dvir-Ginzberg, M. SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect. Tissue Res. 2017, 58, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Jo, A.; Denduluri, S.; Zhang, B.; Wang, Z.; Yin, L.-J.; Yan, Z.; Kang, R.; Shi, L.L.; Mok, J.; Lee, M.J.; et al. The versatile functions of Sox9 in development, stem cells, and human diseases. Genes Dis. 2014, 1, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Deng, J.M.; Zhang, Z.; Behringer, R.R.; De Crombrugghe, B. Sox9 is required for cartilage formation. Nat. Genet. 1999, 22, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhang, K.; Liu, G.; Ding, W.; Zhao, C.; Xie, Y.; Yuan, J.; Sun, X.; Li, H.; Liu, C.; et al. Sox9 Gene Transfer Enhanced Regenerative Effect of Bone Marrow Mesenchymal Stem Cells on the Degenerated Intervertebral Disc in a Rabbit Model. PLoS ONE 2014, 9, e93570. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Kitoh, H.; Sugiura, F.; Ishiguro, N. Chondrogenesis enhanced by overexpression of sox9 gene in mouse bone marrow-derived mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2003, 301, 338–343. [Google Scholar] [CrossRef]
- Lefebvre, V.; Behringer, R.R.; De Crombrugghe, B. L-Sox5, Sox6 and Sox9 control essential steps of the chondrocyte differentiation pathway. Osteoarthr. Cartil. 2001, 9, S69–S75. [Google Scholar] [CrossRef]
- Liu, C.-F.; Lefebvre, V. The transcription factors SOX9 and SOX5/SOX6 cooperate genome-wide through super-enhancers to drive chondrogenesis. Nucleic Acids Res. 2015, 43, 8183–8203. [Google Scholar] [CrossRef]
- Wang, Z.; Qin, G.; Zhao, T.C. HDAC4: Mechanism of regulation and biological functions. Epigenomics 2014, 6, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Karamboulas, C.; Swedani, A.; Ward, C.; Al-Madhoun, A.S.; Wilton, S.; Boisvenue, S.; Ridgeway, A.G.; Skerjanc, I.S. HDAC activity regulates entry of mesoderm cells into the cardiac muscle lineage. J. Cell Sci. 2006, 119, 4305–4314. [Google Scholar] [CrossRef]
- Mielcarek, M.; Zielonka, D.; Carnemolla, A.; Marcinkowski, J.T.; Guidez, F. HDAC4 as a potential therapeutic target in neurodegenerative diseases: A summary of recent achievements. Front. Cell. Neurosci. 2015, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.; Zhao, Y.; Wang, J.; Dubielecka, P.M.; Zhuang, S.; Qin, G.; Chin, Y.E.; Kao, R.L.; Zhao, T.C. Myocyte-specific overexpressing HDAC4 promotes myocardial ischemia/reperfusion injury. Mol. Med. 2018, 24, 37. [Google Scholar] [CrossRef]
- Trazzi, S.; Fuchs, C.; Viggiano, R.; De Franceschi, M.; Valli, E.; Jedynak, P.; Hansen, F.K.; Perini, G.; Rimondini, R.; Kurz, T.; et al. HDAC4: A key factor underlying brain developmental alterations in CDKL5 disorder. Hum. Mol. Genet. 2016, 25, 3887–3907. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.-S.; Nasser, S.; Murray, L.B.; Ayyanar, K.; Arango, D.; Figueroa, M.E.; Melnick, A.; Kao, G.D.; Augenlicht, L.H.; et al. HDAC4 Promotes Growth of Colon Cancer Cells via Repression of p21. Mol. Biol. Cell 2008, 19, 4062–4075. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, Z.; Guo, L.; Wei, X.; Zhang, Y.; Wang, X.; Wei, L. The role of histone deacetylase 4 during chondrocyte hypertrophy and endochondral bone development. Bone Jt. Res. 2020, 9, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.-D.; Wei, L.; Li, P.-C.; Che, X.-D.; Zhao, R.-P.; Han, P.-F.; Lu, J.-G.; Wei, X.-C. Adenovirus-mediated transduction with Histone Deacetylase 4 ameliorates disease progression in an osteoarthritis rat model. Int. Immunopharmacol. 2019, 75, 105752. [Google Scholar] [CrossRef]
- Pei, M.; Chen, D.; Li, J.; Wei, L. Histone deacetylase 4 promotes TGF-β1-induced synovium-derived stem cell chondrogenesis but inhibits chondrogenically differentiated stem cell hypertrophy. Differentiation 2009, 78, 260–268. [Google Scholar] [CrossRef]
- Tribioli, C.; Frasch, M.; Lufkin, T. Bapxl: An evolutionary conserved homologue of the Drosophila bagpipe homeobox gene is expressed in splanchnic mesoderm and the embryonic skeleton. Mech. Dev. 1997, 65, 145–162. [Google Scholar] [CrossRef]
- Rainbow, R.S.; Won, H.K.; Zeng, L. The role of Nkx3.2 in chondrogenesis. Front. Biol. 2014, 9, 376–381. [Google Scholar] [CrossRef]
- Akazawa, H.; Komuro, I.; Sugitani, Y.; Yazaki, Y.; Nagai, R.; Noda, T. Targeted disruption of the homeobox transcription factorBapx1results in lethal skeletal dysplasia with asplenia and gastroduodenal malformation. Genes Cells 2000, 5, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Lettice, L.A.; Purdie, L.A.; Carlson, G.J.; Kilanowski, F.; Dorin, J.; Hill, R.E. The mouse bagpipe gene controls development of axial skeleton, skull, and spleen. Proc. Natl. Acad. Sci. USA 1999, 96, 9695–9700. [Google Scholar] [CrossRef] [PubMed]
- Provot, S.; Kempf, H.; Murtaugh, L.C.; Chung, U.-I.; Kim, D.-W.; Chyung, J.; Kronenberg, H.M.; Lassar, A.B. Nkx3.2/Bapx1 acts as a negative regulator of chondrocyte maturation. Development 2006, 133, 651–662. [Google Scholar] [CrossRef]
- Kawato, Y.; Hirao, M.; Ebina, K.; Shi, K.; Hashimoto, J.; Honjo, Y.; Yoshikawa, H.; Myoui, A. Nkx3.2 Promotes Primary Chondrogenic Differentiation by Upregulating Col2a1 Transcription. PLoS ONE 2012, 7, e34703. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, B.; Bronk, M.; van der Meer, T.; Bernards, R. Constitutive E2F1 Overexpression Delays Endochondral Bone Formation by Inhibiting Chondrocyte Differentiation. Mol. Cell. Biol. 2003, 23, 3656–3668. [Google Scholar] [CrossRef][Green Version]
- Liu, C.-F.; Samsa, W.E.; Zhou, G.; Lefebvre, V. Transcriptional control of chondrocyte specification and differentiation. Semin. Cell Dev. Biol. 2017, 62, 34–49. [Google Scholar] [CrossRef]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-β superfamily signaling pathways in human disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2008, 1782, 197–228. [Google Scholar] [CrossRef]
- Gilbert, R.W.; Vickaryous, M.K.; Viloria-Petit, A.M. Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration. J. Dev. Biol. 2016, 4, 21. [Google Scholar] [CrossRef]
- Thorp, B.; Anderson, I.; Jakowlew, S. Transforming growth factor-beta 1, -beta 2 and -beta 3 in cartilage and bone cells during endochondral ossification in the chick. Development 1992, 114, 907–911. [Google Scholar] [CrossRef]
- Ballock, R.; Heydemann, A.; Wakefield, L.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B. TGF-β1 Prevents Hypertrophy of Epiphyseal Chondrocytes: Regulation of Gene Expression for Cartilage Matrix Proteins and Metalloproteases. Dev. Biol. 1993, 158, 414–429. [Google Scholar] [CrossRef]
- Van Caam, A.; Madej, W.; Van Beuningen, H.; Davidson, E.B.; Van Der Kraan, P. TGF-beta blocks chondrocyte hypertrophy and maintains cell viability in cultured cartilage explants but does not protect against proteoglycan loss. Osteoarthr. Cartil. 2015, 23, A137–A138. [Google Scholar] [CrossRef][Green Version]
- Ongchai, S.; Somnoo, O.; Kongdang, P.; Peansukmanee, S.; Tangyuenyong, S. TGF-β1 upregulates the expression of hyalu-ronan synthase 2 and hyaluronan synthesis in culture models of equine articular chondrocytes. J. Vet Sci. 2018, 19, 735–743. [Google Scholar] [CrossRef]
- Aisenbrey, E.; Payne, K.; Bilousova, G.; Bryant, S.J. Chondrogenic Differentiation of Human Induced Pluripotent Stem Cells in a Photoclickable Biomimetic PEG Hydrogel. Available online: https://www.frontiersin.org/10.3389/conf.FBIOE.2016.01.02149/event_abstract (accessed on 14 May 2021).
- Zhu, S.; Chen, P.; Wu, Y.; Xiong, S.; Sun, H.; Xia, Q.; Shi, L.; Liu, H.; Ouyang, H.W. Programmed Application of Transforming Growth Factor β3 and Rac1 Inhibitor NSC23766 Committed Hyaline Cartilage Differentiation of Adipose-Derived Stem Cells for Osteochondral Defect Repair. Stem. Cells Transl. Med. 2014, 3, 1242–1251. [Google Scholar] [CrossRef]
- Suva, L.; Winslow, A.G.; Wettenhall, E.R.; Hammonds, G.; Moseley, J.M.; Diefenbach-Jagger, H.; Rodda, C.P.; E Kemp, B.; Rodriguez, H.; Chen, E.Y.; et al. A parathyroid hormone-related protein implicated in malignant hypercalcemia: Cloning and expression. Science 1987, 237, 893–896. [Google Scholar] [CrossRef]
- Richard, V.; Lairmore, M.D.; Green, P.L.; Feuer, G.; Erbe, R.S.; Albrecht, B.; D’Souza, C.; Keller, E.T.; Dai, J.; Rosol, T.J. Humoral Hypercalcemia of Malignancy: Severe Combined Immunodeficient/Beige Mouse Model of Adult T-Cell Lymphoma Independent of Human T-Cell Lymphotropic Virus Type-1 Tax Expression. Am. J. Pathol. 2001, 158, 2219–2228. [Google Scholar] [CrossRef]
- Bakre, M.M.; Zhu, Y.; Yin, H.; Burton, D.W.; Terkeltaub, R.; Deftos, L.J.; Varner, J. Parathyroid hormone–related peptide is a naturally occurring, protein kinase A–dependent angiogenesis inhibitor. Nat. Med. 2002, 8, 995–1003. [Google Scholar] [CrossRef]
- Karaplis, A.C.; Luz, A.; Glowacki, J.; Bronson, R.T.; Tybulewicz, V.L.; Kronenberg, H.M.; Mulligan, R.C. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994, 8, 277–289. [Google Scholar] [CrossRef]
- Guo, J.; Chung, U.-I.; Yang, D.; Karsenty, G.; Bringhurst, F.R.; Kronenberg, H.M. PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev. Biol. 2006, 292, 116–128. [Google Scholar] [CrossRef]
- Nishimori, S.; Lai, F.; Shiraishi, M.; Kobayashi, T.; Kozhemyakina, E.; Yao, T.-P.; Lassar, A.B.; Kronenberg, H.M. PTHrP targets HDAC4 and HDAC5 to repress chondrocyte hypertrophy. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef]
- Bo, N.; Peng, W.; Xinghong, P.; Ma, R. Early Cartilage Degeneration in a Rat Experimental Model of Developmental Dysplasia of the Hip. Connect. Tissue Res. 2012, 53, 513–520. [Google Scholar] [CrossRef]
- Wang, M.; Sampson, E.R.; Jin, H.; Li, J.; Ke, Q.H.; Im, H.-J.; Chen, D. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 2013, 15, R5. [Google Scholar] [CrossRef]
- Li, N.-G.; Shi, Z.-H.; Tang, Y.-P.; Wang, Z.-J.; Song, S.-L.; Qian, L.-H.; Qian, D.-W.; Duan, J.-A. New Hope for the Treatment of Osteoarthritis Through Selective Inhibition of MMP-13. Curr. Med. Chem. 2011, 18, 977–1001. [Google Scholar] [CrossRef]
- Ford, K.; Souberbielle, B.; Darling, D.; Farzaneh, F. Protein transduction: An alternative to genetic intervention? Gene Ther. 2001, 8, 1–4. [Google Scholar] [CrossRef]
- Dong, Y.-F.; Soung, D.Y.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor. J. Cell. Physiol. 2006, 208, 77–86. [Google Scholar] [CrossRef]
- Mak, K.K.-L.; Kronenberg, H.M.; Chuang, P.-T.; Mackem, S.; Yang, Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 2008, 135, 1947–1956. [Google Scholar] [CrossRef]
- Castro-Abril, H.; Guevara, J.; Moncayo, M.; Shefelbine, S.; Barrera, L.; Garzón-Alvarado, D. Cellular scale model of growth plate: An in silico model of chondrocyte hypertrophy. J. Theor. Biol. 2017, 428, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xie, R.; Hou, W.; Wang, B.; Shen, R.; Wang, X.; Wang, Q.; Zhu, T.; Jonason, J.H.; Chen, D. PTHrP prevents chondrocyte premature hypertrophy by inducing cyclin-D1-dependent Runx2 and Runx3 phosphorylation, ubiquitylation and proteasomal degradation. J. Cell Sci. 2009, 122, 1382–1389. [Google Scholar] [CrossRef]
- Zhen, X.; Wei, L.; Wu, Q.; Zhang, Y.; Chen, Q. Mitogen-activated Protein Kinase p38 Mediates Regulation of Chondrocyte Differentiation by Parathyroid Hormone. J. Biol. Chem. 2001, 276, 4879–4885. [Google Scholar] [CrossRef]
- Iwai, T.; Murai, J.; Yoshikawa, H.; Tsumaki, N. Smad7 Inhibits Chondrocyte Differentiation at Multiple Steps during Endochondral Bone Formation and Down-regulates p38 MAPK Pathways. J. Biol. Chem. 2008, 283, 27154–27164. [Google Scholar] [CrossRef]
- Lui, J.C.; Garrison, P.; Nguyen, Q.; Ad, M.; Keembiyehetty, C.; Chen, W.; Jee, Y.H.; Landman, E.; Nilsson, O.; Barnes, K.M.; et al. EZH1 and EZH2 promote skeletal growth by repressing inhibitors of chondrocyte proliferation and hypertrophy. Nat. Commun. 2016, 7, 13685. [Google Scholar] [CrossRef]
- Chen, L.; Wu, Y.; Wu, Y.; Wang, Y.; Sun, L.; Li, F. The inhibition of EZH2 ameliorates osteoarthritis development through the Wnt/β-catenin pathway. Sci. Rep. 2016, 6, 29176. [Google Scholar] [CrossRef]
- Deshmukh, V.; Hu, H.; Barroga, C.; Bossard, C.; Kc, S.; Dellamary, L.; Stewart, J.; Chiu, K.; Ibanez, M.; Pedraza, M.; et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr. Cartil. 2018, 26, 18–27. [Google Scholar] [CrossRef]
- Shkhyan, R.; Van Handel, B.; Bogdanov, J.; Lee, S.; Yu, Y.; Scheinberg, M.; Banks, N.W.; Limfat, S.; Chernostrik, A.; Franciozi, C.E.; et al. Drug-induced modulation of gp130 signalling prevents articular cartilage degeneration and promotes repair. Ann. Rheum. Dis. 2018, 77, 760–769. [Google Scholar] [CrossRef]
- Cao, Z.; Dou, C.; Dong, S. Curcumin Inhibits Chondrocyte Hypertrophy of Mesenchymal Stem Cells through IHH and Notch Signaling Pathways. Chem. Pharm. Bull. 2017, 65, 762–767. [Google Scholar] [CrossRef]
- Cao, Z.; Dou, C.; Li, J.; Tang, X.; Xiang, J.; Zhao, C.; Zhu, L.; Bai, Y.; Xiang, Q.; Dong, S. Cordycepin inhibits chondrocyte hypertrophy of mesenchymal stem cells through PI3K/Bapx1 and Notch signaling pathway. BMB Rep. 2016, 49, 548–553. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, Y.; Huang, Y. Icariin Reduces Cartilage Degeneration in a Mouse Model of Osteoarthritis and is Associated with the Changes in Expression of Indian Hedgehog and Parathyroid Hormone-Related Protein. Med. Sci. Monit. 2018, 24, 6695–6706. [Google Scholar] [CrossRef]
- Huang, X.; Xi, Y.; Mao, Z.; Chu, X.; Zhang, R.; Ma, X.; Ni, B.; Cheng, H.; You, H. Vanillic acid attenuates cartilage degeneration by regulating the MAPK and PI3K/AKT/NF-κB pathways. Eur. J. Pharmacol. 2019, 859, 172481. [Google Scholar] [CrossRef]
- Arai, Y.; Choi, B.; Kim, B.J.; Rim, W.; Park, S.; Park, H.; Ahn, J.; Lee, S.-H. Tauroursodeoxycholic acid (TUDCA) counters osteoarthritis by regulating intracellular cholesterol levels and membrane fluidity of degenerated chondrocytes. Biomater. Sci. 2019, 7, 3178–3189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, S.; Kuss, M.A.; Streubel, P.N.; Duan, B. Effects of Hydroxyapatite and Hypoxia on Chondrogenesis and Hypertrophy in 3D Bioprinted ADMSC Laden Constructs. ACS Biomater. Sci. Eng. 2016, 3, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fu, P.; Cong, R.; Wu, H.; Pei, M. Strategies to minimize hypertrophy in cartilage engineering and regeneration. Genes Dis. 2015, 2, 76–95. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, Z.; Yang, J.; Liu, J.; Wang, J.; Li, X.; Liu, Y. p38 MAPK mediated in compressive stress-induced chondrogenesis of rat bone marrow MSCs in 3D alginate scaffolds. J. Cell. Physiol. 2009, 221, 609–617. [Google Scholar] [CrossRef]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef]
- Geckil, H.; Xu, F.; Zhang, X.; Moon, S.; Demirci, U. Engineering hydrogels as extracellular matrix mimics. Nanomedicine 2010, 5, 469–484. [Google Scholar] [CrossRef]
- Schuh, E.; Hofmann, S.; Stok, K.; Notbohm, H.; Müller, R.; Rotter, N. Chondrocyte redifferentiation in 3D: The effect of adhesion site density and substrate elasticity. J. Biomed. Mater. Res. Part A 2011, 100, 38–47. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Singh, Y.P.; Mandal, B.B. Silk Fibroin Scaffold-Based 3D Co-Culture Model for Modulation of Chondrogenesis without Hypertrophy via Reciprocal Cross-talk and Paracrine Signaling. ACS Biomater. Sci. Eng. 2019, 5, 5240–5254. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The Collagen Family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Lu, Z.; Wu, H.; Liu, H.; Li, M.; Lei, D.; Zheng, L.; Zhao, J. Collagen, agarose, alginate, and Matrigel hydrogels as cell substrates for culture of chondrocytes in vitro: A comparative study. J. Cell. Biochem. 2018, 119, 7924–7933. [Google Scholar] [CrossRef] [PubMed]
- Hiramitsu, T.; Yasuda, T.; Ito, H.; Shimizu, M.; Julovi, S.; Kakinuma, T.; Akiyoshi, M.; Yoshida, M.; Nakamura, T. Intercellular adhesion molecule-1 mediates the inhibitory effects of hyaluronan on interleukin-1β-induced matrix metalloproteinase production in rheumatoid synovial fibroblasts via down-regulation of NF-κB and p38. Rheumatology 2006, 45, 824–832. [Google Scholar] [CrossRef]
- Comper, W.D. Extracellular Matrix: Molecular Components and Interactions; Harwood Academic Publishers: Reading, UK, 1996. [Google Scholar]
- Chen, W.Y.J.; Abatangelo, G. Functions of hyaluronan in wound repair. Wound Repair Regen. 1999, 7, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.B.; Lee, S.K.C.; Woods, E.C.; Acharya, A.P. Biomaterials-Based Modulation of the Immune System. BioMed Res. Int. 2013, 2013, 732182. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Feng, Q.; Sun, Y.; Li, G.; Bian, L. Effect of cartilaginous matrix components on the chondrogenesis and hypertrophy of mesenchymal stem cells in hyaluronic acid hydrogels. J. Biomed. Mater. Res. Part B Appl. Biomater. 2017, 105, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Hou, C.; Tous, E.; Rai, R.; Mauck, R.; Burdick, J.A. The influence of hyaluronic acid hydrogel crosslinking density and macromolecular diffusivity on human MSC chondrogenesis and hypertrophy. Biomaterials 2013, 34, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, S.; Takagi, M.; Wakitani, S.; Nihira, T.; Yoshida, T. Effect of chondroitin sulfate and hyaluronic acid on gene expression in a three-dimensional culture of chondrocytes. J. Biosci. Bioeng. 2005, 100, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, N.J.; Bryant, S.J. Chondroitin sulfate and dynamic loading alter chondrogenesis of human MSCs in PEG hydrogels. Biotechnol. Bioeng. 2012, 109, 2671–2682. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.; Hwang, N.S.; Canver, A.C.; Theprungsirikul, P.; Lin, D.W.; Elisseeff, J. Chondroitin sulfate based niches for chondrogenic differentiation of mesenchymal stem cells. Matrix Biol. 2008, 27, 12–21. [Google Scholar] [CrossRef]
- Müller, S.; Lindemann, S.; Gigout, A. Effects of Sprifermin, IGF1, IGF2, BMP7, or CNP on Bovine Chondrocytes in Monolayer and 3D Culture. J. Orthop. Res. 2020, 38, 653–662. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, J.; Chen, Y.; Zhang, Z.; Saunders, L.; Schipani, E.; Chen, Q.; Ma, P.X. Suppressing mesenchymal stem cell hypertrophy and endochondral ossification in 3D cartilage regeneration with nanofibrous poly(l-lactic acid) scaffold and matrilin-3. Acta Biomater. 2018, 76, 29–38. [Google Scholar] [CrossRef]
- Larson, C.M.; Kelley, S.S.; Blackwood, A.D.; Banes, A.J.; Lee, G.M. Retention of the native chondrocyte pericellular matrix results in significantly improved matrix production. Matrix Biol. 2002, 4, 349–359. [Google Scholar] [CrossRef]
- Cheng, N.; Estes, B.; Awad, H.; Guilak, F. Chondrogenic Differentiation of Adipose-Derived Adult Stem Cells by a Porous Scaffold Derived from Native Articular Cartilage Extracellular Matrix. Tissue Eng. Part A 2009, 15, 231–241. [Google Scholar] [CrossRef]
- Gigout, A.; Guehring, H.; Froemel, D.; Meurer, A.; Ladel, C.; Reker, D.; Bay-Jensen, A.; Karsdal, M.; Lindemann, S. Sprifermin (rhFGF18) enables proliferation of chondrocytes producing a hyaline cartilage matrix. Osteoarthr. Cartil. 2017, 25, 1858–1867. [Google Scholar] [CrossRef]
- Lai, Y.; Sun, Y.; Skinner, C.M.; Son, E.L.; Lu, Z.; Tuan, R.S.; Jilka, R.L.; Ling, J.; Chen, X.-D. Reconstitution of Marrow-Derived Extracellular Matrix Ex Vivo: A Robust Culture System for Expanding Large-Scale Highly Functional Human Mesenchymal Stem Cells. Stem Cells Dev. 2010, 19, 1095–1107. [Google Scholar] [CrossRef]
- Li, J.; Pei, M. Optimization of an In Vitro Three-Dimensional Microenvironment to Reprogram Synovium-Derived Stem Cells for Cartilage Tissue Engineering. Tissue Eng. Part A 2011, 17, 703–712. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and mi-croRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Javeed, N.; Mukhopadhyay, D. Exosomes and their role in the micro-/macro-environment: A comprehensive review. J. Biomed. Res. 2017, 31, 386. [Google Scholar]
- Toh, W.S.; Lai, R.C.; Hui, J.H.P.; Lim, S.K. MSC exosome as a cell-free MSC therapy for cartilage regeneration: Implications for osteoarthritis treatment. Semin. Cell Dev. Biol. 2017, 67, 56–64. [Google Scholar] [CrossRef]
- Murphy, M.; Fink, D.J.; Hunziker, E.B.; Barry, F.P. Stem cell therapy in a caprine model of osteoarthritis. Arthritis Rheum. 2003, 48, 3464–3474. [Google Scholar] [CrossRef]
- Uto, S.; Nishizawa, S.; Hikita, A.; Takato, T.; Hoshi, K. Application of induced pluripotent stem cells for cartilage regeneration in CLAWN miniature pig osteochondral replacement model. Regen. Ther. 2018, 9, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Pers, Y.-M.; Rackwitz, L.; Ferreira, R.; Pullig, O.; Delfour, C.; Barry, F.; Sensebe, L.; Casteilla, L.; Fleury, S.; Bourin, P.; et al. Adipose Mesenchymal Stromal Cell-Based Therapy for Severe Osteoarthritis of the Knee: A Phase I Dose-Escalation Trial. Stem. Cells Transl. Med. 2016, 5, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Al-Najar, M.; Khalil, H.; Al-Ajlouni, J.; Al-Antary, E.; Hamdan, M.; Rahmeh, R.; Alhattab, D.; Samara, O.; Yasin, M.; Al Abdullah, A.; et al. Intra-articular injection of expanded autologous bone marrow mesenchymal cells in moderate and severe knee osteoarthritis is safe: A phase I/II study. J. Orthop. Surg. Res. 2017, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Sierra, R.; Wyles, C.; Houdek, M.; Behfar, A. Mesenchymal stem cell therapy for osteoarthritis: Current perspectives. Stem Cells Cloning Adv. Appl. 2015, 8, 117–124. [Google Scholar] [CrossRef]
- Fu, Y.; Karbaat, L.; Wu, L.; Leijten, J.; Both, S.K.; Karperien, M. Trophic Effects of Mesenchymal Stem Cells in Tissue Regeneration. Tissue Eng. Part B Rev. 2017, 23, 515–528. [Google Scholar] [CrossRef]
- Cosenza, S.; Ruiz, M.; Toupet, K.; Jorgensen, C.; Noël, D. Mesenchymal stem cells derived exosomes and microparticles protect cartilage and bone from degradation in osteoarthritis. Sci. Rep. 2017, 7, 16214. [Google Scholar] [CrossRef]
- Zhang, S.; Chu, W.; Lai, R.C.; Lim, S.K.; Hui, J.H.P.; Toh, W. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthr. Cartil. 2016, 24, 2135–2140. [Google Scholar] [CrossRef]
- Zhang, S.; Teo, K.Y.W.; Chuah, S.J.; Lai, R.C.; Lim, S.K.; Toh, W.S. MSC exosomes alleviate temporomandibular joint osteoarthritis by attenuating inflammation and restoring matrix homeostasis. Biomaterials 2019, 200, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Lo Sicco, C.; Reverberi, D.; Balbi, C.; Ulivi, V.; Principi, E.; Pascucci, L.; Becherini, P.; Bosco, M.C.; Varesio, L.; Franzin, C.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Mediators of Anti-Inflammatory Effects: Endorsement of Macrophage Polarization. Stem Cells Transl. Med. 2017, 6, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, Y.; Zhao, B.; Niu, X.; Hu, B.; Li, Q.; Zhang, J.; Ding, J.; Chen, Y.; Wang, Y. Comparison of exosomes secreted by induced pluripotent stem cell-derived mesenchymal stem cells and synovial membrane-derived mesenchymal stem cells for the treatment of osteoarthritis. Stem Cell Res. Ther. 2017, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Domenis, R.; Zanutel, R.; Caponnetto, F.; Toffoletto, B.; Cifù, A.; Pistis, C.; Di Benedetto, P.; Causero, A.; Pozzi, M.; Bassini, F.; et al. Characterization of the Proinflammatory Profile of Synovial Fluid-Derived Exosomes of Patients with Osteoarthritis. Mediat. Inflamm. 2017, 2017, 4814987. [Google Scholar] [CrossRef]
- Mao, G.; Hu, S.; Zhang, Z.; Wu, P.; Zhao, X.; Lin, R.; Liao, W.; Kang, Y. Exosomal miR-95-5p regulates chondrogenesis and cartilage degradation via histone deacetylase 2/8. J. Cell. Mol. Med. 2018, 22, 5354–5366. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Zhang, Z.; Hu, S.; Zhang, Z.; Chang, Z.; Huang, Z.; Liao, W.; Kang, Y. Exosomes derived from miR-92a-3p-overexpressing human mesenchymal stem cells enhance chondrogenesis and suppress cartilage degradation via targeting WNT5A. Stem Cell Res. Ther. 2018, 9, 247. [Google Scholar] [CrossRef]
- Kato, T.; Miyaki, S.; Ishitobi, H.; Nakamura, Y.; Nakasa, T.; Lotz, M.K.; Ochi, M. Exosomes from IL-1β stimulated synovial fibroblasts induce osteoarthritic changes in articular chondrocytes. Arthritis Res. Ther. 2014, 16, R163-11. [Google Scholar] [CrossRef]
- Kolhe, R.; Hunter, M.; Liu, S.; Jadeja, R.N.; Pundkar, C.; Mondal, A.K.; Mendhe, B.; Drewry, M.; Rojiani, M.V.; Liu, Y.; et al. Gender-specific differential expression of exosomal miRNA in synovial fluid of patients with osteoarthritis. Sci. Rep. 2017, 7, 2029. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Choi, B.; Kim, B.J.; Park, S.; Park, H.; Moon, J.J.; Lee, S.-H. Cryptic ligand on collagen matrix unveiled by MMP13 accelerates bone tissue regeneration via MMP13/Integrin α3/RUNX2 feedback loop. Acta Biomater. 2021, 125, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Arai, Y.; Kim, B.J.; Bello, A.; Ashraf, S.; Park, H.; Park, K.-S.; Lee, S.-H. Suppression of SPRY4 Promotes Osteogenic Differentiation and Bone Formation of Mesenchymal Stem Cell. Tissue Eng. Part A 2019, 25, 1646–1657. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Type | Factors | Properties | In-Vitro Results | In-Vivo Results | Reference |
|---|---|---|---|---|---|
| Genetic Intervention | SOX9 | Master regulator of Chondrogenesis | Adenoviral transduction improves chondrogenesis, over-expression enhances BMMSC chondrogenesis | SOX9 promotes healthy growth plates and articular cartilage | [74] |
| Histone Deacetylase 4 (HDAC4) | Transcription factor, epigenetic regulator of histone proteins | Adenoviral transduction in rat chondrocyte prevents cartilage degeneration, interacts with and inhibits RUNX2 and MEF2C; Over-expression promotes TGF-B1 expression | Over-expression inhibits chondrocyte hypertrophy similar to RUNX2-loss of function phenotype | [89,90] | |
| NKX3.2 | Transcription factor, human homolog of the bagpipe gene (BAPX1) | Retroviral transduction prevents chondrocyte maturation and hypertrophy; over-expression enhanced COL2A1 and SOX9 expression | Over-expression causes skeletal dwarfism by delaying cartilage hypertrophy | [96] | |
| E2F1 | Transcription factor | E2F1 expression delays early and late phase differentiation of ATDC5 cells | over-expression prevents chondrocyte maturation indicated by reduced hypertrophic zone and disorganized growth plate | [97] | |
| Protein Intervention | TGF-Beta | Growth factor involved in chondrocyte and cartilage development | exogenous TGFB1 prevents terminal differentiation of chondrocytes; TGFB3 enhanced chondrogenesis and prevents hypertrophy in ASC | TGFB1 activation inhibits BMP signaling in cartilage | [103,106] |
| PTHrP | Homolog of the parathyroid hormone | Activates HDAC4/HDAC5 suppressing MEF2 and RUNX2 that promotes hypertrophy; over-expression reduces hypertrophic markers MMP13 and COL10 in MSC | Expression of PTHrP in IHH mutant mice prevents hypertrophy of chondrocyte | [6] |
| Type | Classification | Characteristics and Function | Reference | |
|---|---|---|---|---|
| Polymer-derived scaffold | Biomaterials (Natural polymer) | Silk fibroin |
| [138] |
| Collagen |
| [139,140] | ||
| Hyaluronic acid |
| [141,142,143,144,145,146] | ||
| Chondroitin sulfate |
| [147,148,149] | ||
| Bio-derived scaffold | Tissue | Articular cartilage |
| [152,153] |
| Cell | Chondrocyte |
| [154] | |
| Marrow- or synovium-derived stem cell |
| [155,156] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Bello, A.; Arai, Y.; Ahn, J.; Kim, D.; Cha, K.-Y.; Baek, I.; Park, H.; Lee, S.-H. Functional Duality of Chondrocyte Hypertrophy and Biomedical Application Trends in Osteoarthritis. Pharmaceutics 2021, 13, 1139. https://doi.org/10.3390/pharmaceutics13081139
Park S, Bello A, Arai Y, Ahn J, Kim D, Cha K-Y, Baek I, Park H, Lee S-H. Functional Duality of Chondrocyte Hypertrophy and Biomedical Application Trends in Osteoarthritis. Pharmaceutics. 2021; 13(8):1139. https://doi.org/10.3390/pharmaceutics13081139
Chicago/Turabian StylePark, Sunghyun, Alvin Bello, Yoshie Arai, Jinsung Ahn, Dohyun Kim, Kyung-Yup Cha, Inho Baek, Hansoo Park, and Soo-Hong Lee. 2021. "Functional Duality of Chondrocyte Hypertrophy and Biomedical Application Trends in Osteoarthritis" Pharmaceutics 13, no. 8: 1139. https://doi.org/10.3390/pharmaceutics13081139
APA StylePark, S., Bello, A., Arai, Y., Ahn, J., Kim, D., Cha, K.-Y., Baek, I., Park, H., & Lee, S.-H. (2021). Functional Duality of Chondrocyte Hypertrophy and Biomedical Application Trends in Osteoarthritis. Pharmaceutics, 13(8), 1139. https://doi.org/10.3390/pharmaceutics13081139