
Chronic Kidney Disease-Induced Arterial Media Calcification in Rats Prevented by Tissue Non-Specific Alkaline Phosphatase Substrate Supplementation Rather Than Inhibition of the Enzyme

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiment
2.2. Analysis of Biochemical Parameters
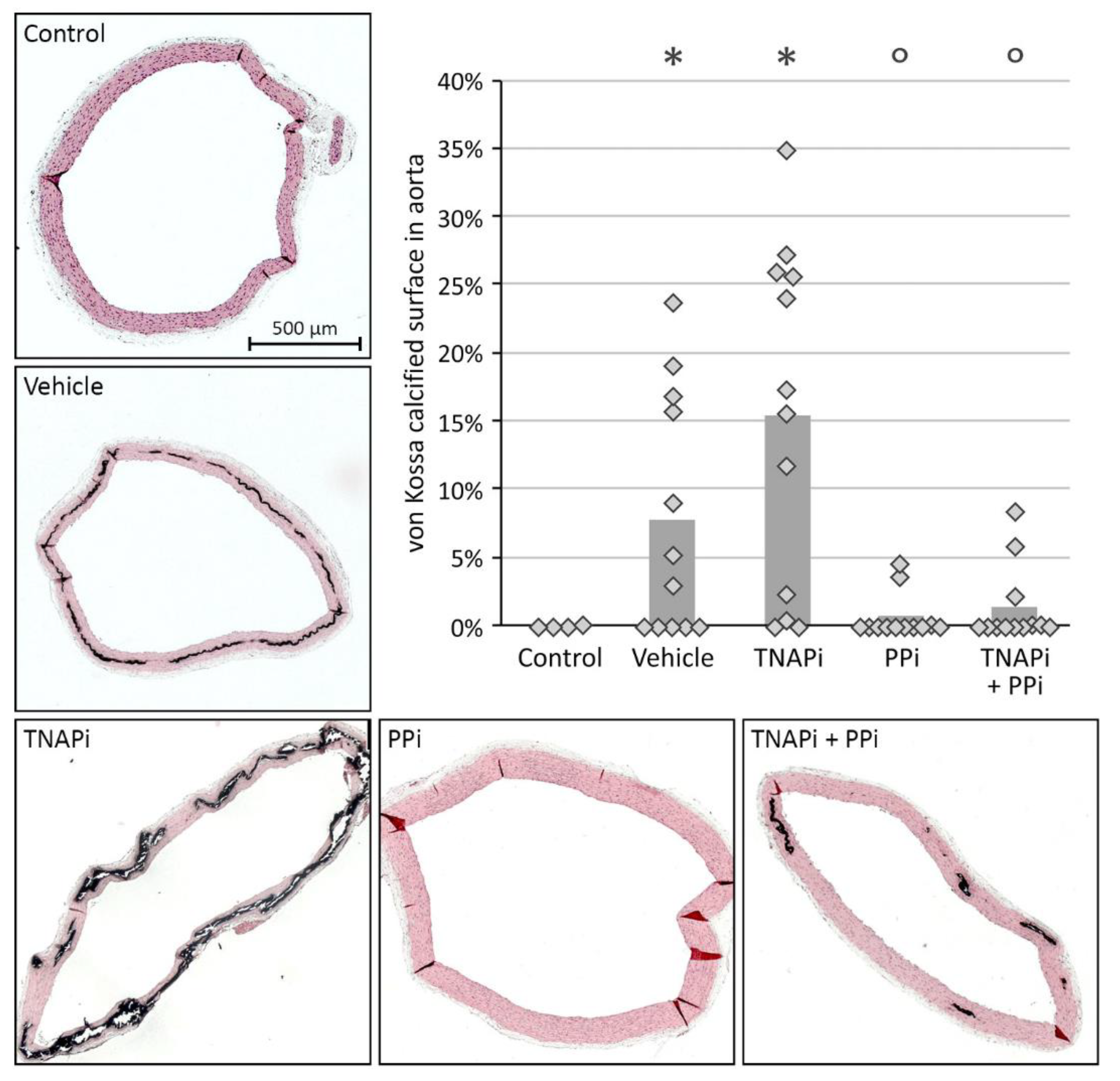
2.3. Quantification of Arterial Calcification
2.4. Quantitative Real Time PCR
2.5. Bone Histomorphometry
2.6. Statistical Analysis
3. Results
3.1. Adenine-Fed Rats Developed a Stable Chronic Renal Failure Based on Serum Creatinine, Phosphorus and Calcium Levels
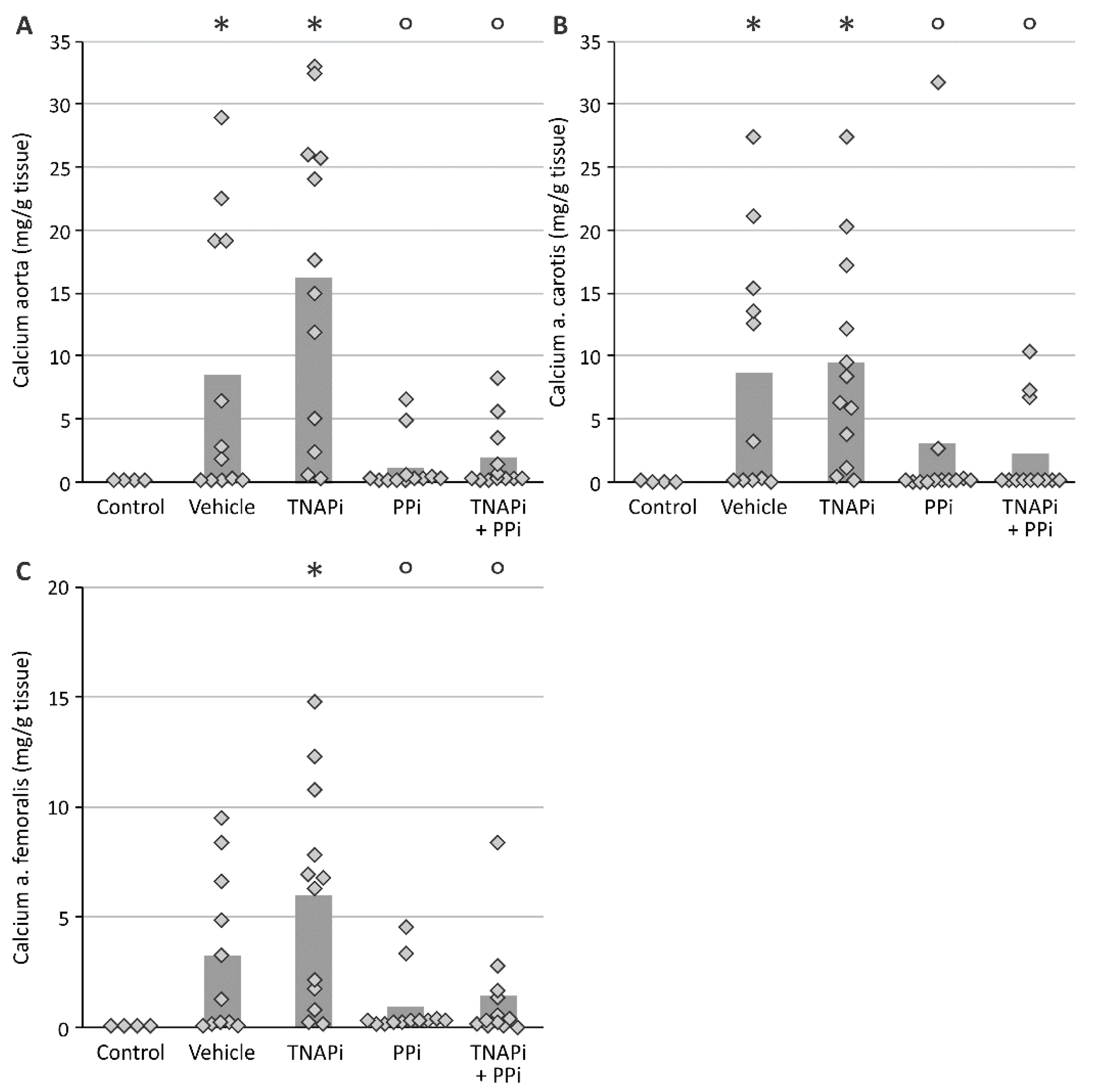
3.2. Exposure to PPi, but Not TNAP Inhibition, Prevented the Development of Arterial Media Calcification
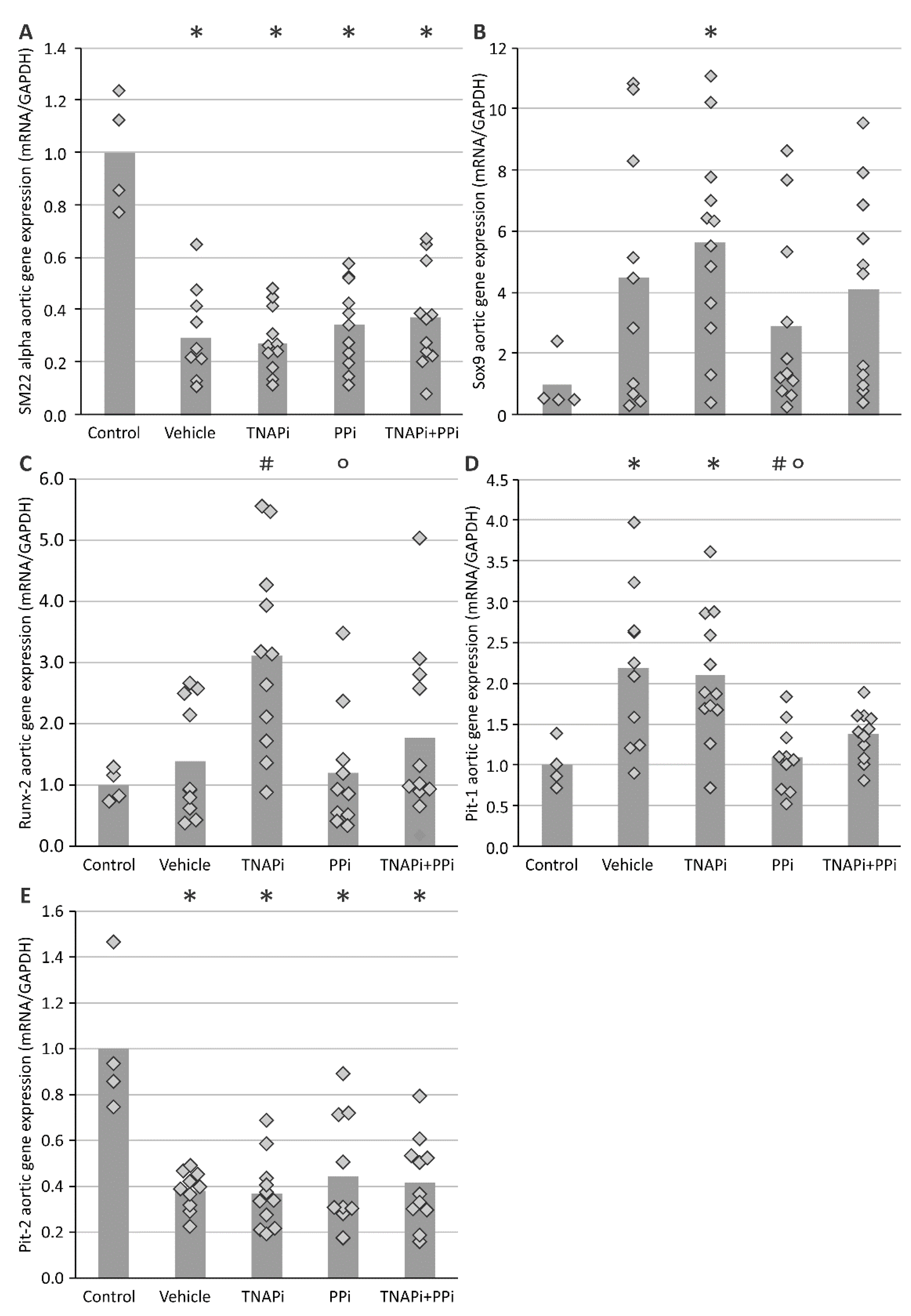
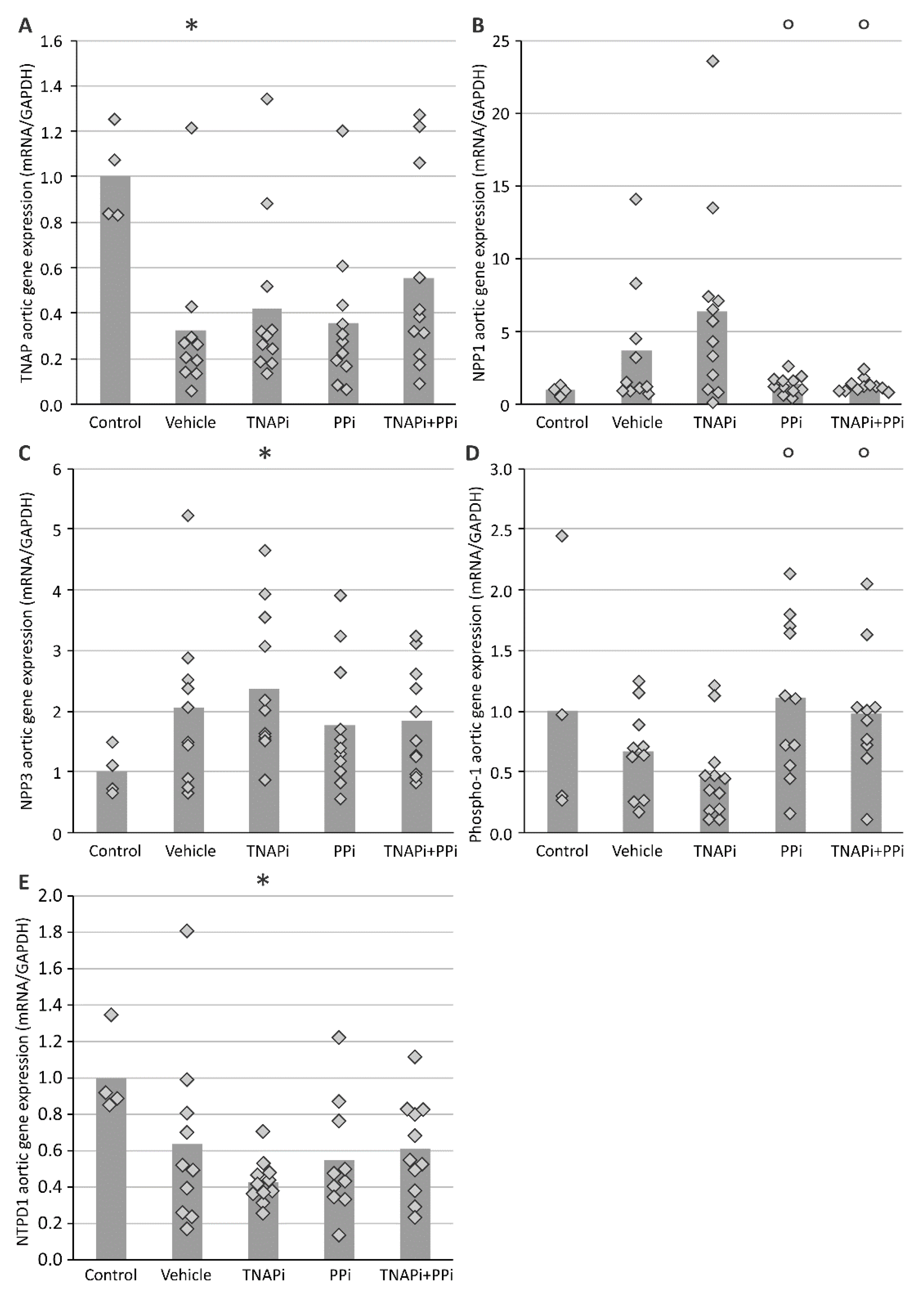
3.3. mRNA Expression of Genes Involved in CKD-Related Arterial Media Calcification
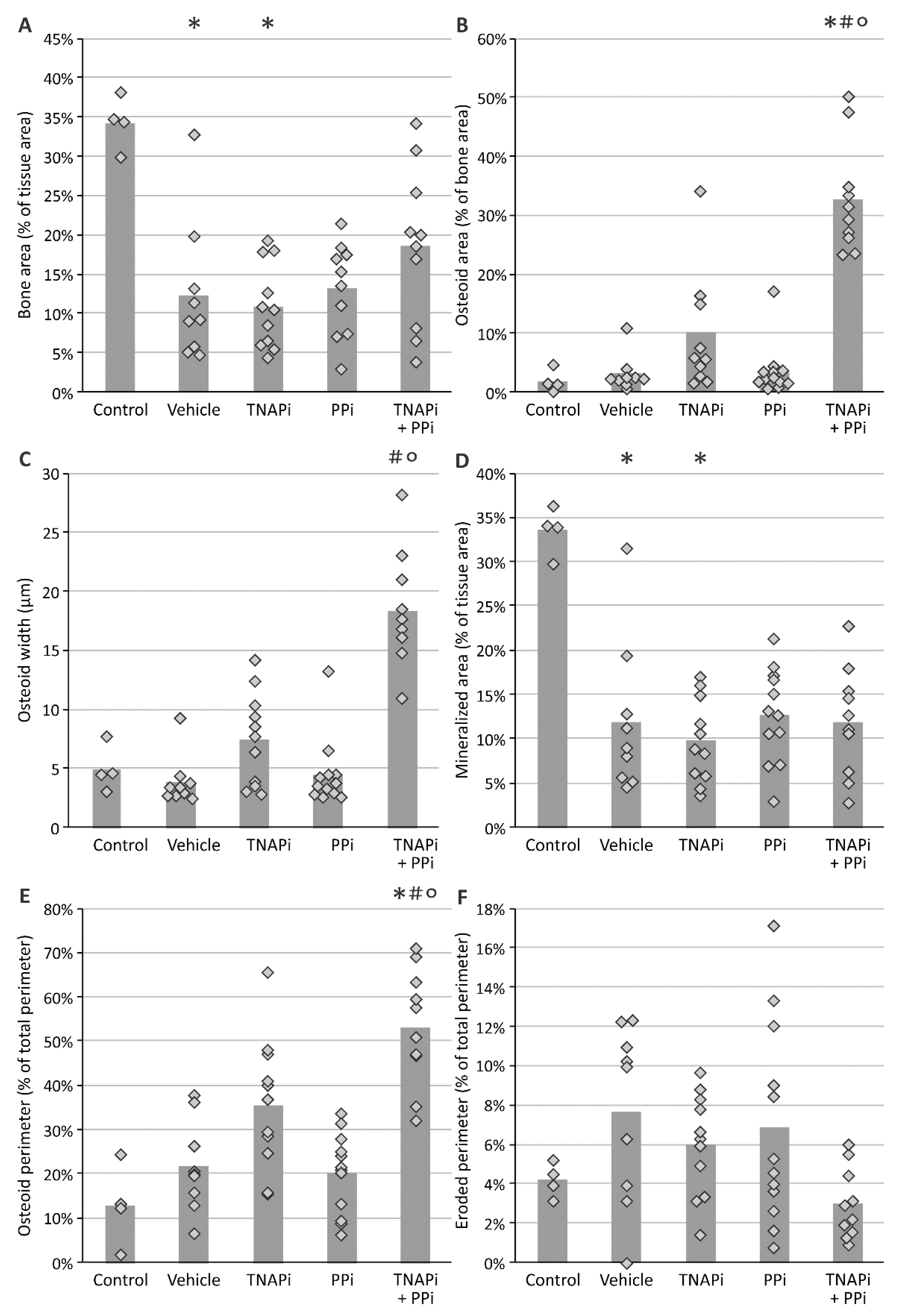
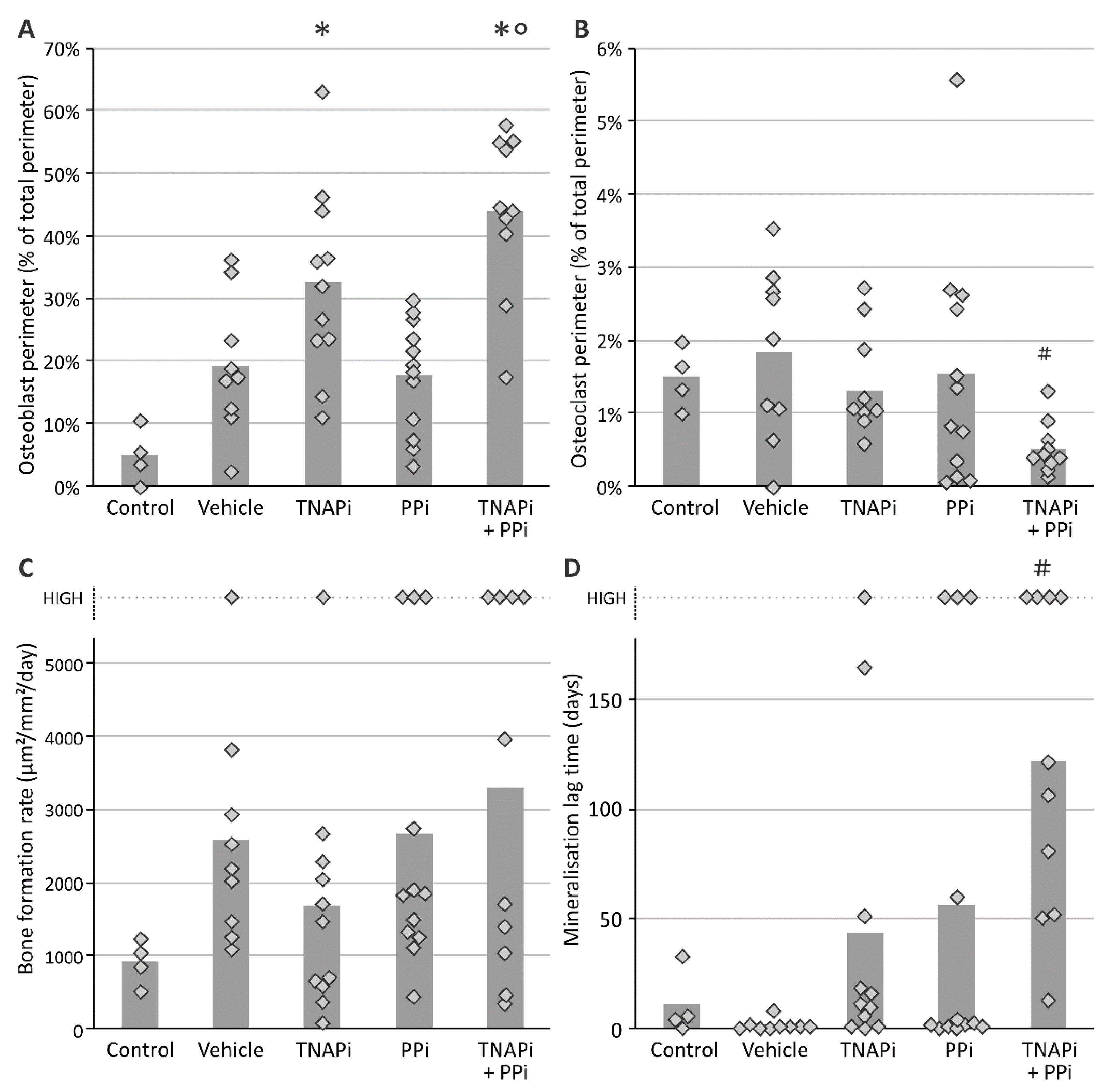
3.4. Treatment with SBI-425 with or without PPi Administration Worsens Bone Metabolism in a CKD Context in the Rat
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sigrist, M.K.; Taal, M.W.; Bungay, P.; McIntyre, C.W. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin. J. Am. Soc. Nephrol. CJASN 2007, 2, 1241–1248. [Google Scholar] [CrossRef]
- Neven, E.; De Schutter, T.M.; Dams, G.; Gundlach, K.; Steppan, S.; Buchel, J.; Passlick-Deetjen, J.; D’Haese, P.C.; Behets, G.J. A magnesium based phosphate binder reduces vascular calcification without affecting bone in chronic renal failure rats. PLoS ONE 2014, 9, e107067. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.H.; Hayward, R.; Rajan, R.; Whitehead, M.; Cobb, A.M.; Ahmad, S.; Sun, M.; Goldberga, I.; Li, R.; Bashtanova, U.; et al. Poly(ADP-Ribose) Links the DNA Damage Response and Biomineralization. Cell Rep. 2019, 27, 3124–3138.e13. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef]
- Van den Bergh, G.; Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. The Vicious Cycle of Arterial Stiffness and Arterial Media Calcification. Trends Mol. Med. 2019, 25, 1133–1146. [Google Scholar] [CrossRef]
- Townsend, R.R. Arterial Stiffness in CKD: A Review. Am. J. Kidney Dis. 2019, 73, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Düsing, P.; Zietzer, A.; Goody, P.R.; Hosen, M.R.; Kurts, C.; Nickenig, G.; Jansen, F. Vascular pathologies in chronic kidney disease: Pathophysiological mechanisms and novel therapeutic approaches. J. Mol. Med. 2021, 99, 335–348. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Mallamaci, F.; Zoccali, C. Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update. J. Clin. Med. 2020, 9, 2359. [Google Scholar] [CrossRef] [PubMed]
- Neven, E.; De Schutter, T.M.; De Broe, M.E.; D’Haese, P.C. Cell biological and physicochemical aspects of arterial calcification. Kidney Int. 2011, 79, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Roumeliotis, A.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Biomarkers of vascular calcification in serum. Adv. Clin. Chem. 2020, 98, 91–147. [Google Scholar] [CrossRef]
- Zimmermann, H.; Zebisch, M.; Strater, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef]
- Fleisch, H.; Russell, R.G.; Straumann, F. Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature 1966, 212, 901–903. [Google Scholar] [CrossRef]
- Nitschke, Y.; Rutsch, F. Inherited Arterial Calcification Syndromes: Etiologies and Treatment Concepts. Curr. Osteoporos. Rep. 2017, 15, 255–270. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Khawandi, W.; O’Neill, W.C. Reduced plasma pyrophosphate levels in hemodialysis patients. J. Am. Soc. Nephrol. JASN 2005, 16, 2495–2500. [Google Scholar] [CrossRef]
- O’Neill, W.C.; Sigrist, M.K.; McIntyre, C.W. Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, A.B.; Sergienko, E.; Bravo, Y.; Dahl, R.; Ma, C.T.; Sun, Q.; Jackson, M.R.; Cosford, N.D.P.; Millan, J.L. Discovery of 5-((5-chloro-2-methoxyphenyl)sulfonamido)nicotinamide (SBI-425), a potent and orally bioavailable tissue-nonspecific alkaline phosphatase (TNAP) inhibitor. Bioorgan. Med. Chem. Lett. 2018, 28, 31–34. [Google Scholar] [CrossRef]
- Li, Q.; Huang, J.; Pinkerton, A.B.; Millan, J.L.; van Zelst, B.D.; Levine, M.A.; Sundberg, J.P.; Uitto, J. Inhibition of Tissue-Nonspecific Alkaline Phosphatase Attenuates Ectopic Mineralization in the Abcc6(-/-) Mouse Model of PXE but Not in the Enpp1 Mutant Mouse Models of GACI. J. Investig. Dermatol. 2019, 139, 360–368. [Google Scholar] [CrossRef]
- Romanelli, F.; Corbo, A.; Salehi, M.; Yadav, M.C.; Salman, S.; Petrosian, D.; Rashidbaigi, O.J.; Chait, J.; Kuruvilla, J.; Plummer, M.; et al. Overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in endothelial cells accelerates coronary artery disease in a mouse model of familial hypercholesterolemia. PLoS ONE 2017, 12, e0186426. [Google Scholar] [CrossRef] [PubMed]
- Sheen, C.R.; Kuss, P.; Narisawa, S.; Yadav, M.C.; Nigro, J.; Wang, W.; Chhea, T.N.; Sergienko, E.A.; Kapoor, K.; Jackson, M.R.; et al. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J. Bone Miner. Res. 2015, 30, 824–836. [Google Scholar] [CrossRef]
- Tani, T.; Fujiwara, M.; Orimo, H.; Shimizu, A.; Narisawa, S.; Pinkerton, A.B.; Millan, J.L.; Tsuruoka, S. Inhibition of tissue-nonspecific alkaline phosphatase protects against medial arterial calcification and improved survival probability in the CKD-MBD mouse model. J. Pathol. 2019. [Google Scholar] [CrossRef]
- Ziegler, S.G.; Ferreira, C.R.; MacFarlane, E.G.; Riddle, R.C.; Tomlinson, R.E.; Chew, E.Y.; Martin, L.; Ma, C.T.; Sergienko, E.; Pinkerton, A.B.; et al. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Neven, E.; Millán, J.L.; Pinkerton, A.B.; D’Haese, P.C.; Verhulst, A. Pharmacological TNAP inhibition efficiently inhibits arterial media calcification in a warfarin rat model but deserves careful consideration of potential physiological bone formation/mineralization impairment. Bone 2020, 137, 115392. [Google Scholar] [CrossRef]
- Evenepoel, P.; Opdebeeck, B.; David, K.; D’Haese, P.C. Bone-Vascular Axis in Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2019, 26, 472–483. [Google Scholar] [CrossRef]
- Evenepoel, P.; Behets, G.J.S.; Laurent, M.R.; D’Haese, P.C. Update on the role of bone biopsy in the management of patients with CKD-MBD. J. Nephrol. 2017, 30, 645–652. [Google Scholar] [CrossRef]
- Bowers, G.N., Jr.; McComb, R.B. A continuous spectrophotometric method for measuring the activity of serum alkaline phosphatase. Clin. Chem. 1966, 12, 70–89. [Google Scholar] [CrossRef]
- Dempster, D.W.; Compston, J.E.; Drezner, M.K.; Glorieux, F.H.; Kanis, J.A.; Malluche, H.; Meunier, P.J.; Ott, S.M.; Recker, R.R.; Parfitt, A.M. Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2013, 28, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Renda, R. Can salivary creatinine and urea levels be used to diagnose chronic kidney disease in children as accurately as serum creatinine and urea levels? A case-control study. Ren. Fail. 2017, 39, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Swapna, L.A.; Koppolu, P.; Prince, J. Oral health in diabetic and nondiabetic patients with chronic kidney disease. Saudi J. Kidney Dis. Transplant. 2017, 28, 1099–1105. [Google Scholar] [CrossRef]
- Kramer, H.; Toto, R.; Peshock, R.; Cooper, R.; Victor, R. Association between chronic kidney disease and coronary artery calcification: The Dallas Heart Study. J. Am. Soc. Nephrol. JASN 2005, 16, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Qunibi, W.Y.; Abouzahr, F.; Mizani, M.R.; Nolan, C.R.; Arya, R.; Hunt, K.J. Cardiovascular calcification in Hispanic Americans (HA) with chronic kidney disease (CKD) due to type 2 diabetes. Kidney Int. 2005, 68, 271–277. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Russo, D.; Palmiero, G.; De Blasio, A.P.; Balletta, M.M.; Andreucci, V.E. Coronary artery calcification in patients with CRF not undergoing dialysis. Am. J. Kidney Dis. 2004, 44, 1024–1030. [Google Scholar] [CrossRef]
- Dedinszki, D.; Szeri, F.; Kozák, E.; Pomozi, V.; Tőkési, N.; Mezei, T.R.; Merczel, K.; Letavernier, E.; Tang, E.; Le Saux, O.; et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol. Med. 2017, 9, 1463–1470. [Google Scholar] [CrossRef]
- Pomozi, V.; Julian, C.B.; Zoll, J.; Pham, K.; Kuo, S.; Tokesi, N.; Martin, L.; Varadi, A.; Le Saux, O. Dietary Pyrophosphate Modulates Calcification in a Mouse Model of Pseudoxanthoma Elasticum: Implication for Treatment of Patients. J. Investig. Dermatol. 2019, 139, 1082–1088. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.Y.; Giachelli, C.M. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef]
- Couasnay, G.; Bon, N.; Devignes, C.S.; Sourice, S.; Bianchi, A.; Véziers, J.; Weiss, P.; Elefteriou, F.; Provot, S.; Guicheux, J.; et al. PiT1/Slc20a1 Is Required for Endoplasmic Reticulum Homeostasis, Chondrocyte Survival, and Skeletal Development. J. Bone Miner. Res. 2019, 34, 387–398. [Google Scholar] [CrossRef]
- Salaün, C.; Leroy, C.; Rousseau, A.; Boitez, V.; Beck, L.; Friedlander, G. Identification of a novel transport-independent function of PiT1/SLC20A1 in the regulation of TNF-induced apoptosis. J. Biol. Chem. 2010, 285, 34408–34418. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Levi, M.; Sorribas, V. Vascular smooth muscle cell calcification and SLC20 inorganic phosphate transporters: Effects of PDGF, TNF-alpha, and Pi. Pflug. Arch. Eur. J. Physiol. 2009, 458, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Bogaert, Y.E.; Levi, M.; Sorribas, V. Characterization of phosphate transport in rat vascular smooth muscle cells: Implications for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1030–1036. [Google Scholar] [CrossRef]
- Crouthamel, M.H.; Lau, W.L.; Leaf, E.M.; Chavkin, N.W.; Wallingford, M.C.; Peterson, D.F.; Li, X.; Liu, Y.; Chin, M.T.; Levi, M.; et al. Sodium-dependent phosphate cotransporters and phosphate-induced calcification of vascular smooth muscle cells: Redundant roles for PiT-1 and PiT-2. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Leaf, E.M.; Chia, J.J.; Cox, T.C.; Speer, M.Y.; Giachelli, C.M. PiT-2, a type III sodium-dependent phosphate transporter, protects against vascular calcification in mice with chronic kidney disease fed a high-phosphate diet. Kidney Int. 2018, 94, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Wang, X.; Millan, J.L.; Dubyak, G.R.; O’Neill, W.C. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H61–H68. [Google Scholar] [CrossRef] [PubMed]
- Harmey, D.; Hessle, L.; Narisawa, S.; Johnson, K.A.; Terkeltaub, R.; Millan, J.L. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: An integrated model of the pathogenesis of mineralization disorders. Am. J. Pathol. 2004, 164, 1199–1209. [Google Scholar] [CrossRef]
- Orriss, I.R.; Utting, J.C.; Brandao-Burch, A.; Colston, K.; Grubb, B.R.; Burnstock, G.; Arnett, T.R. Extracellular nucleotides block bone mineralization in vitro: Evidence for dual inhibitory mechanisms involving both P2Y2 receptors and pyrophosphate. Endocrinology 2007, 148, 4208–4216. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Synthesis of Extracellular Pyrophosphate Increases in Vascular Smooth Muscle Cells during Phosphate-Induced Calcification. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2137–2147. [Google Scholar] [CrossRef]
- Ciancaglini, P.; Yadav, M.C.; Simão, A.M.; Narisawa, S.; Pizauro, J.M.; Farquharson, C.; Hoylaerts, M.F.; Millán, J.L. Kinetic analysis of substrate utilization by native and TNAP-, NPP1-, or PHOSPHO1-deficient matrix vesicles. J. Bone Miner. Res. 2010, 25, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Kiffer-Moreira, T.; Yadav, M.C.; Zhu, D.; Narisawa, S.; Sheen, C.; Stec, B.; Cosford, N.D.; Dahl, R.; Farquharson, C.; Hoylaerts, M.F.; et al. Pharmacological inhibition of PHOSPHO1 suppresses vascular smooth muscle cell calcification. J. Bone Miner. Res. 2013, 28, 81–91. [Google Scholar] [CrossRef]
- Roberts, S.; Narisawa, S.; Harmey, D.; Millan, J.L.; Farquharson, C. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J. Bone Miner. Res. 2007, 22, 617–627. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Impact of magnesium:calcium ratio on calcification of the aortic wall. PLoS ONE 2017, 12, e0178872. [Google Scholar] [CrossRef]
- Narisawa, S.; Yadav, M.C.; Millán, J.L. In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J. Bone Miner. Res. 2013, 28, 1587–1598. [Google Scholar] [CrossRef]
- Babler, A.; Schmitz, C.; Buescher, A.; Herrmann, M.; Gremse, F.; Gorgels, T.; Floege, J.; Jahnen-Dechent, W. Microvasculopathy and soft tissue calcification in mice are governed by fetuin-A, magnesium and pyrophosphate. PLoS ONE 2020, 15, e0228938. [Google Scholar] [CrossRef]
- Millán, J.L.; Narisawa, S.; Lemire, I.; Loisel, T.P.; Boileau, G.; Leonard, P.; Gramatikova, S.; Terkeltaub, R.; Camacho, N.P.; McKee, M.D.; et al. Enzyme replacement therapy for murine hypophosphatasia. J. Bone Miner. Res. 2008, 23, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Buchet, R.; Millan, J.L.; Magne, D. Multisystemic functions of alkaline phosphatases. Methods Mol. Biol. 2013, 1053, 27–51. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.S.; Walter, G.L.; Walker, R.M. Clinical Pathology in Non-Clinical Toxicology Testing. In Haschek and Rousseaux’s Handbook of Toxicologic Pathology, 3rd ed.; Haschek, W.M., Rousseaux, C.G., Wallig, M.A., Eds.; Academic Press: Boston, MA, USA, 2013; Chapter 18; pp. 565–594. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Control | Vehicle | TNAPi | PPi | TNAPi + PPi |
|---|---|---|---|---|---|
| Number of rats | 4 | 11 | 12 | 12 | 12 |
| Creatinine (mg/dl) | 0.70 ± 0.09 | 6.38 ± 0.43 a | 6.27 ± 0.54 a | 6.28 ± 0.37 a | 6.40 ± 0.36 a |
| Creatinine clearance (mL/min) | 2.42 ± 0.45 | 0.15 ± 0.02 a | 0.11 ± 0.01 a | 0.14 ± 0.01 a | 0.12 ± 0.01 a |
| Phosphorus (mg/dl) | 8.85 ± 1.64 | 23.36 ± 0.80 a | 22.86 ± 0.69 a | 24.72 ± 1.28 a | 23.87 ± 1.32 a |
| Calcium (mg/dl) | 13.74 ± 0.34 | 11.29 ± 0.25 a | 11.63 ± 0.26 | 11.28 ± 0.24 a | 11.41 ± 0.54 a |
| FGF23 (pg/µL) | 0.22 ± 0.02 | 83 ± 26 | 130 ± 22 a | 55 ± 11 | 116 ± 23 a |
| ALP (U/L) | 182 ± 12 | 127 ± 9 | 170 ± 20 | 122 ± 12 | 159 ± 11 |
| PPi (µM) | 6.2 ± 1.56 | 8.2 ± 1.30 | 7.5 ± 1.39 | 6.5 ± 1.06 | 10.4 ± 1.20 |
| AST (U/L) | 78.5 ± 2.2 | 72.6 ± 5.6 | 76.0 ± 6.9 | 79.2 ± 6.4 | 82.2 ± 5.8 |
| ALT (U/L) | 37.5 ± 2.8 | 18.6 ± 2.1 | 17.8 ± 2.5 a | 23.5 ± 5.5 | 19.0 ± 2.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Opdebeeck, B.; Neven, E.; Millán, J.L.; Pinkerton, A.B.; D’Haese, P.C.; Verhulst, A. Chronic Kidney Disease-Induced Arterial Media Calcification in Rats Prevented by Tissue Non-Specific Alkaline Phosphatase Substrate Supplementation Rather Than Inhibition of the Enzyme. Pharmaceutics 2021, 13, 1138. https://doi.org/10.3390/pharmaceutics13081138
Opdebeeck B, Neven E, Millán JL, Pinkerton AB, D’Haese PC, Verhulst A. Chronic Kidney Disease-Induced Arterial Media Calcification in Rats Prevented by Tissue Non-Specific Alkaline Phosphatase Substrate Supplementation Rather Than Inhibition of the Enzyme. Pharmaceutics. 2021; 13(8):1138. https://doi.org/10.3390/pharmaceutics13081138
Chicago/Turabian StyleOpdebeeck, Britt, Ellen Neven, José Luis Millán, Anthony B. Pinkerton, Patrick C. D’Haese, and Anja Verhulst. 2021. "Chronic Kidney Disease-Induced Arterial Media Calcification in Rats Prevented by Tissue Non-Specific Alkaline Phosphatase Substrate Supplementation Rather Than Inhibition of the Enzyme" Pharmaceutics 13, no. 8: 1138. https://doi.org/10.3390/pharmaceutics13081138
APA StyleOpdebeeck, B., Neven, E., Millán, J. L., Pinkerton, A. B., D’Haese, P. C., & Verhulst, A. (2021). Chronic Kidney Disease-Induced Arterial Media Calcification in Rats Prevented by Tissue Non-Specific Alkaline Phosphatase Substrate Supplementation Rather Than Inhibition of the Enzyme. Pharmaceutics, 13(8), 1138. https://doi.org/10.3390/pharmaceutics13081138