Fluorescent Dye Labeling Changes the Biodistribution of Tumor-Targeted Nanoparticles

 , ,
, ,  , , , ,
, , , ,  and
and 
Abstract
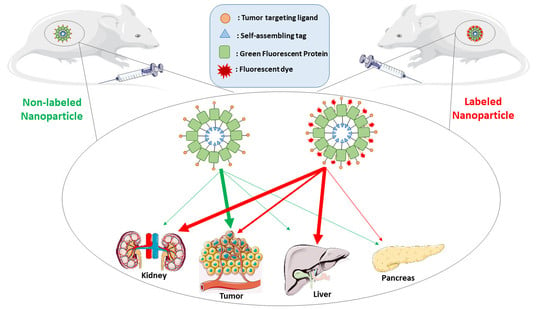
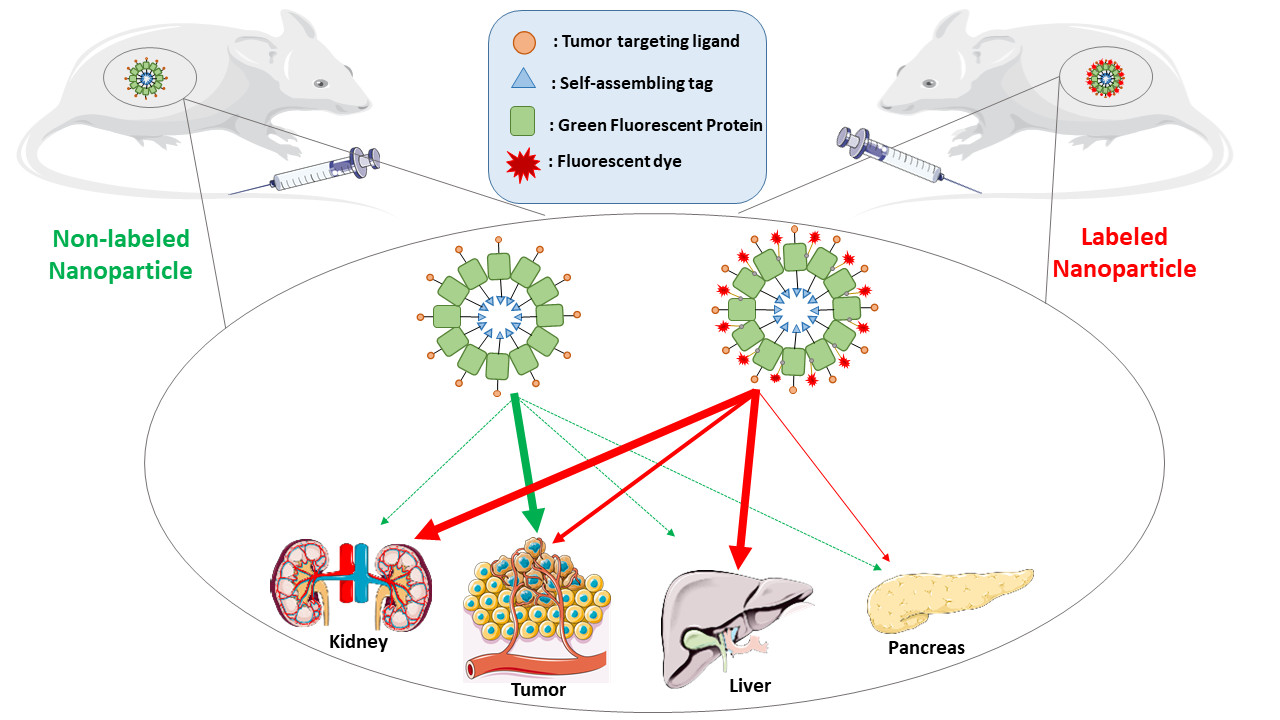{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Protein Production, Purification and Characterization
2.2. Fluorescent Dye Conjugation
2.3. Morphometric Characterization
2.4. Optical Properties of Nanoparticles and Free Dyes
2.5. Selection of Cell Lines for the Generation of In Vitro and In Vivo Models
2.6. Cell Culture, Protein Internalization and Competition Assays
2.7. CXCR4 Receptor Binding Affinity
2.8. Biodistribution in Subcutaneous DLBCL Mouse Model
2.9. Biodistribution in Subcutaneous Colorectal Cancer Mouse Model
2.10. Ex Vivo Biodistribution Fluorescent Image Analysis
2.11. Statistical Analysis
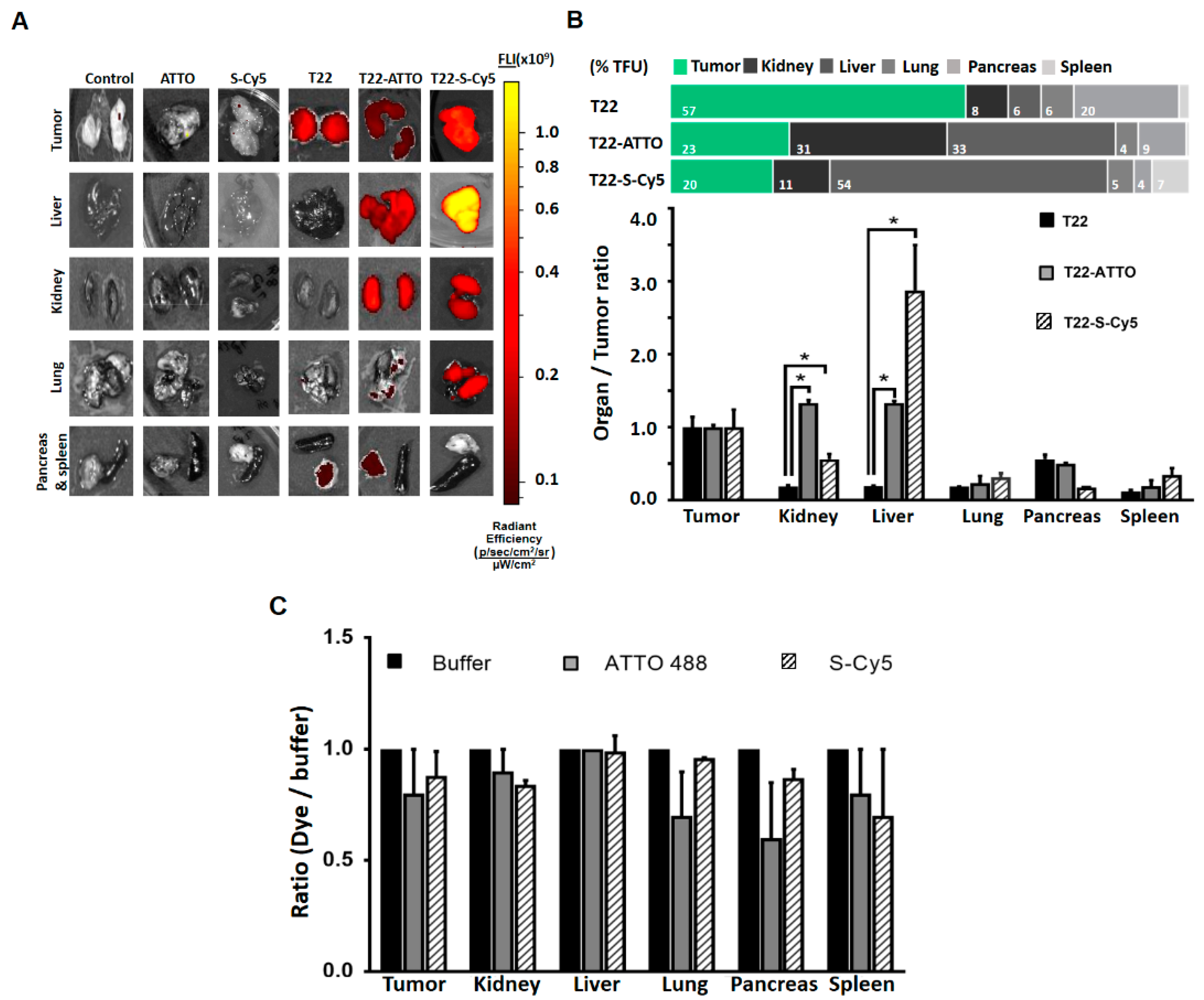
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mangues, R.; Vazquez, E.; Villaverde, A. Targeting in cancer therapies. Med. Sci. 2016, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Ozturk-Atar, K.; Eroglu, H.; Calis, S. Novel advances in targeted drug delivery. J. Drug Target. 2018, 26, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T. Macro-nanomedicine: Targeting the big picture. J. Control. Release Off. J. Control. Release Soc. 2019, 294, 372–375. [Google Scholar] [CrossRef]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef]
- Etheridge, M.L.; Campbell, S.A.; Erdman, A.G.; Haynes, C.L.; Wolf, S.M.; McCullough, J. The big picture on nanomedicine: The state of investigational and approved nanomedicine products. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 1–14. [Google Scholar] [CrossRef]
- Priem, B.; Tian, C.; Tang, J.; Zhao, Y.; Mulder, W.J. Fluorescent nanoparticles for the accurate detection of drug delivery. Expert Opin. Drug Deliv. 2015, 12, 1881–1894. [Google Scholar] [CrossRef]
- Perez-Medina, C.; Abdel-Atti, D.; Zhang, Y.; Longo, V.A.; Irwin, C.P.; Binderup, T.; Ruiz-Cabello, J.; Fayad, Z.A.; Lewis, J.S.; Mulder, W.J.; et al. A modular labeling strategy for in vivo PET and near-infrared fluorescence imaging of nanoparticle tumor targeting. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2014, 55, 1706–1711. [Google Scholar] [CrossRef]
- Merian, J.; Gravier, J.; Navarro, F.; Texier, I. Fluorescent nanoprobes dedicated to in vivo imaging: From preclinical validations to clinical translation. Molecules 2012, 17, 5564–5591. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, K.; Zuhorn, I.S.; Dierckx, R.A.; de Vries, E.F. Imaging of cells and nanoparticles: Implications for drug delivery to the brain. Pharm. Res. 2012, 29, 3213–3234. [Google Scholar] [CrossRef][Green Version]
- Terai, T.; Nagano, T. Small-molecule fluorophores and fluorescent probes for bioimaging. Pflug. Arch. Eur. J. Physiol. 2013, 465, 347–359. [Google Scholar] [CrossRef]
- Jones, C.G.; Stavila, V.; Conroy, M.A.; Feng, P.; Slaughter, B.V.; Ashley, C.E.; Allendorf, M.D. Versatile synthesis and fluorescent labeling of ZIF-90 nanoparticles for biomedical applications. ACS Appl. Mater. Interfaces 2016, 8, 7623–7630. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garcia, L.; Serna, N.; Alamo, P.; Sala, R.; Cespedes, M.V.; Roldan, M.; Sanchez-Chardi, A.; Unzueta, U.; Casanova, I.; Mangues, R.; et al. Self-assembling toxin-based nanoparticles as self-delivered antitumoral drugs. J. Control. Release 2018. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, W.G.; Mishra, M.K.; Jyoti, A.; Balakrishnan, B.; Zhang, F.; Nance, E.; Romero, R.; Kannan, S.; Kannan, R.M. Biodistribution of fluorescently labeled PAMAM dendrimers in neonatal rabbits: Effect of neuroinflammation. Mol. Pharm. 2013, 10, 4560–4571. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.K.; Tang, O.; Chen, X.M.; Kim, B.; Hartlieb, M.; Pollock, C.A.; Hawkett, B.S.; Perrier, S. Fluorescent labeling and biodistribution of latex nanoparticles formed by surfactant-Free RAFT emulsion polymerization. Macromol. Biosci. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bolinger, J.; Yu, Y.; Glass, Z.; Shi, N.; Yang, L.; Wang, M.; Xu, Q. Intracellular delivery and biodistribution study of CRISPR/Cas9 ribonucleoprotein loaded bioreducible lipidoid nanoparticles. Biomater. Sci. 2019, 7, 596–606. [Google Scholar] [CrossRef]
- Snipstad, S.; Hak, S.; Baghirov, H.; Sulheim, E.; Morch, Y.; Lelu, S.; von Haartman, E.; Back, M.; Nilsson, K.P.R.; Klymchenko, A.S.; et al. Labeling nanoparticles: Dye leakage and altered cellular uptake. Cytom. Part A J. Int. Soc. Anal. Cytol. 2017, 91, 760–766. [Google Scholar] [CrossRef]
- Debie, P.; Van Quathem, J.; Hansen, I.; Bala, G.; Massa, S.; Devoogdt, N.; Xavier, C.; Hernot, S. Effect of Dye and conjugation chemistry on the biodistribution profile of near-infrared-labeled nanobodies as tracers for image-guided surgery. Mol. Pharm. 2017, 14, 1145–1153. [Google Scholar] [CrossRef]
- Cilliers, C.; Nessler, I.; Christodolu, N.; Thurber, G.M. Tracking antibody distribution with near-infrared fluorescent dyes: Impact of dye structure and degree of labeling on plasma clearance. Mol. Pharm. 2017, 14, 1623–1633. [Google Scholar] [CrossRef]
- Adochite, R.C.; Moshnikova, A.; Golijanin, J.; Andreev, O.A.; Katenka, N.V.; Reshetnyak, Y.K. Comparative study of tumor targeting and biodistribution of pH (Low) insertion peptides (pHLIP((R)) Peptides) conjugated with different fluorescent dyes. Mol. Imaging Biol. 2016, 18, 686–696. [Google Scholar] [CrossRef]
- Zilkowski, I.; Theodorou, I.; Albrecht, K.; Duconge, F.; Groll, J. Subtle changes in network composition impact the biodistribution and tumor accumulation of nanogels. Chem. Commun. 2018, 54, 11777–11780. [Google Scholar] [CrossRef]
- Vasquez, K.O.; Casavant, C.; Peterson, J.D. Quantitative whole body biodistribution of fluorescent-labeled agents by non-invasive tomographic imaging. PLoS ONE 2011, 6, e20594. [Google Scholar] [CrossRef]
- Hensbergen, A.W.; van Willigen, D.M.; van Beurden, F.; van Leeuwen, P.J.; Buckle, T.; Schottelius, M.; Maurer, T.; Wester, H.J.; van Leeuwen, F.W.B. Image-guided surgery: Are we getting the most out of small-molecule prostate-specific-membrane-antigen-targeted Tracers? Bioconjugate Chem. 2020, 31, 375–395. [Google Scholar] [CrossRef]
- Hensbergen, A.W.; Buckle, T.; van Willigen, D.M.; Schottelius, M.; Welling, M.M.; van der Wijk, F.A.; Maurer, T.; van der Poel, H.G.; van der Pluijm, G.; van Weerden, W.M.; et al. Hybrid tracers based on cyanine backbones targeting prostate-specific membrane antigen: Tuning pharmacokinetic properties and exploring dye-protein interaction. J. Nucl. Med. 2020, 61, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Buckle, T.; van Willigen, D.M.; Spa, S.J.; Hensbergen, A.W.; van der Wal, S.; de Korne, C.M.; Welling, M.M.; van der Poel, H.G.; Hardwick, J.C.H.; van Leeuwen, F.W.B. Tracers for fluorescence-guided surgery: How elongation of the polymethine chain in cyanine dyes alters the pharmacokinetics of a dual-modality c[RGDyK] tracer. J. Nucl. Med. 2018, 59, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Bunschoten, A.; van Willigen, D.M.; Buckle, T.; van den Berg, N.S.; Welling, M.M.; Spa, S.J.; Wester, H.J.; van Leeuwen, F.W. Tailoring fluorescent dyes to optimize a hybrid RGD-tracer. Bioconjugate Chem. 2016, 27, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Liang, X. CXCR4, inhibitors and mechanisms of action. Chem. Biol. Drug Des. 2008, 72, 97–110. [Google Scholar] [CrossRef]
- Murakami, T.; Zhang, T.Y.; Koyanagi, Y.; Tanaka, Y.; Kim, J.; Suzuki, Y.; Minoguchi, S.; Tamamura, H.; Waki, M.; Matsumoto, A.; et al. Inhibitory mechanism of the CXCR4 antagonist T22 against human immunodeficiency virus type 1 infection. J. Virol. 1999, 73, 7489–7496. [Google Scholar] [CrossRef]
- Balkwill, F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin. Cancer Biol. 2004, 14, 171–179. [Google Scholar] [CrossRef]
- Kim, J.; Mori, T.; Chen, S.L.; Amersi, F.F.; Martinez, S.R.; Kuo, C.; Turner, R.R.; Ye, X.; Bilchik, A.J.; Morton, D.L.; et al. Chemokine receptor CXCR4 expression in patients with melanoma and colorectal cancer liver metastases and the association with disease outcome. Ann. Surg. 2006, 244, 113–120. [Google Scholar] [CrossRef]
- Unzueta, U.; Ferrer-Miralles, N.; Cedano, J.; Zikung, X.; Pesarrodona, M.; Saccardo, P.; Garcia-Fruitos, E.; Domingo-Espin, J.; Kumar, P.; Gupta, K.C.; et al. Non-amyloidogenic peptide tags for the regulatable self-assembling of protein-only nanoparticles. Biomaterials 2012, 33, 8714–8722. [Google Scholar] [CrossRef]
- Unzueta, U.; Roldan, M.; Pesarrodona, M.; Benitez, R.; Sanchez-Chardi, A.; Conchillo-Sole, O.; Mangues, R.; Villaverde, A.; Vazquez, E. Self-assembling as regular nanoparticles dramatically minimizes photobleaching of tumour-targeted GFP. Acta Biomater. 2020, 103, 272–280. [Google Scholar] [CrossRef]
- Cespedes, M.V.; Unzueta, U.; Tatkiewicz, W.; Sanchez-Chardi, A.; Conchillo-Sole, O.; Alamo, P.; Xu, Z.; Casanova, I.; Corchero, J.L.; Pesarrodona, M.; et al. In vivo architectonic stability of fully de novo designed protein-only nanoparticles. ACS Nano 2014, 8, 4166–4176. [Google Scholar] [CrossRef]
- Unzueta, U.; Cespedes, M.V.; Ferrer-Miralles, N.; Casanova, I.; Cedano, J.; Corchero, J.L.; Domingo-Espin, J.; Villaverde, A.; Mangues, R.; Vazquez, E. Intracellular CXCR4(+) cell targeting with T22-empowered protein-only nanoparticles. Int. J. Nanomed. 2012, 7, 4533–4544. [Google Scholar] [CrossRef]
- Cespedes, M.V.; Unzueta, U.; Alamo, P.; Gallardo, A.; Sala, R.; Casanova, I.; Pavon, M.A.; Mangues, M.A.; Trias, M.; Lopez-Pousa, A.; et al. Cancer-specific uptake of a liganded protein nanocarrier targeting aggressive CXCR4+ colorectal cancer models. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1987–1996. [Google Scholar] [CrossRef]
- Cespedes, M.V.; Unzueta, U.; Avino, A.; Gallardo, A.; Alamo, P.; Sala, R.; Sanchez-Chardi, A.; Casanova, I.; Mangues, M.A.; Lopez-Pousa, A.; et al. Selective depletion of metastatic stem cells as therapy for human colorectal cancer. EMBO Mol. Med. 2018. [Google Scholar] [CrossRef]
- Serna, N.C.M.; Sánchez-García, L.; Unzueta, U.; Sala, R.; Sánchez-Chardi, A.; Cortés, F.; Ferrer-Miralles, N.; Mangues, R.; Vázquez, E.; Villaverde, A. Peptide-based nanostructured materials with intrinsic proapoptotic activities in CXCR4+ solid tumors. Adv. Funct. Mater. 2017, 27, 1700919. [Google Scholar] [CrossRef]
- Diaz, R.; Pallares, V.; Cano-Garrido, O.; Serna, N.; Sanchez-Garcia, L.; Falgas, A.; Pesarrodona, M.; Unzueta, U.; Sanchez-Chardi, A.; Sanchez, J.M.; et al. Selective CXCR4(+) cancer cell targeting and potent antineoplastic effect by a nanostructured version of recombinant ricin. Small 2018, e1800665. [Google Scholar] [CrossRef]
- Pallares, V.; Unzueta, U.; Falgas, A.; Sanchez-Garcia, L.; Serna, N.; Gallardo, A.; Morris, G.A.; Alba-Castellon, L.; Alamo, P.; Sierra, J.; et al. An Auristatin nanoconjugate targeting CXCR4+ leukemic cells blocks acute myeloid leukemia dissemination. J. Hematol. Oncol. 2020, 13, 36. [Google Scholar] [CrossRef]
- Falgas, A.; Pallares, V.; Serna, N.; Sanchez-Garcia, L.; Sierra, J.; Gallardo, A.; Alba-Castellon, L.; Alamo, P.; Unzueta, U.; Villaverde, A.; et al. Selective delivery of T22-PE24-H6 to CXCR4(+) diffuse large B-cell lymphoma cells leads to wide therapeutic index in a disseminated mouse model. Theranostics 2020, 10, 5169–5180. [Google Scholar] [CrossRef]
- Lopez-Laguna, H.; Unzueta, U.; Conchillo-Sole, O.; Sanchez-Chardi, A.; Pesarrodona, M.; Cano-Garrido, O.; Volta, E.; Sanchez-Garcia, L.; Serna, N.; Saccardo, P.; et al. Assembly of histidine-rich protein materials controlled through divalent cations. Acta Biomater. 2019, 83, 257–264. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Falgas, A.; Pallares, V.; Unzueta, U.; Cespedes, M.V.; Arroyo-Solera, I.; Moreno, M.J.; Sierra, J.; Gallardo, A.; Mangues, M.A.; Vazquez, E.; et al. A CXCR4-targeted nanocarrier achieves highly selective tumor uptake in diffuse large B-cell lymphoma mouse models. Haematologica 2020, 105, 741–753. [Google Scholar] [CrossRef]
- Rueda, F.; Cespedes, M.V.; Conchillo-Sole, O.; Sanchez-Chardi, A.; Seras-Franzoso, J.; Cubarsi, R.; Gallardo, A.; Pesarrodona, M.; Ferrer-Miralles, N.; Daura, X.; et al. Bottom-up instructive quality control in the biofabrication of smart protein materials. Adv. Mater. 2015, 27, 7816–7822. [Google Scholar] [CrossRef]
- Hughes, L.D.; Rawle, R.J.; Boxer, S.G. Choose your label wisely: Water-soluble fluorophores often interact with lipid bilayers. PLoS ONE 2014, 9, e87649. [Google Scholar] [CrossRef]
- Kim, M.T.; Chen, Y.; Marhoul, J.; Jacobson, F. Statistical modeling of the drug load distribution on trastuzumab emtansine (Kadcyla), a lysine-linked antibody drug conjugate. Bioconjugate Chem. 2014, 25, 1223–1232. [Google Scholar] [CrossRef]
- Pawig, L.; Klasen, C.; Weber, C.; Bernhagen, J.; Noels, H. Diversity and inter-connections in the CXCR4 chemokine receptor/ligand family: Molecular perspectives. Front. Immunol. 2015, 6, 429. [Google Scholar] [CrossRef]
- Meng, L.; Zhang, X.; Lu, Q.; Fei, Z.; Dyson, P.J. Single walled carbon nanotubes as drug delivery vehicles: Targeting doxorubicin to tumors. Biomaterials 2012, 33, 1689–1698. [Google Scholar] [CrossRef]
- Bhat, I.A.; Nazir, M.I.; Ahmad, I.; Pathakota, G.B.; Chanu, T.I.; Goswami, M.; Sundaray, J.K.; Sharma, R. Fabrication and characterization of chitosan conjugated eurycomanone nanoparticles: In vivo evaluation of the biodistribution and toxicity in fish. Int. J. Biol. Macromol. 2018, 112, 1093–1103. [Google Scholar] [CrossRef]
- Vincenzi, B.; Armento, G.; Spalato Ceruso, M.; Catania, G.; Leakos, M.; Santini, D.; Minotti, G.; Tonini, G. Drug-induced hepatotoxicity in cancer patients—implication for treatment. Expert Opin. Drug Saf. 2016, 15, 1219–1238. [Google Scholar] [CrossRef]
- Sarges, P.; Steinberg, J.M.; Lewis, J.H. Drug-induced liver injury: Highlights from a review of the 2015 literature. Drug Saf. 2016, 39, 801–821. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Y.; Dong, M.; He, X.; Wang, Z.; Li, J.; Wang, Y. Observation of hepatotoxicity during long-term gefitinib administration in patients with non-small-cell lung cancer. Anticancer Drugs 2016, 27, 245–250. [Google Scholar] [CrossRef]
- Damodar, G.; Smitha, T.; Gopinath, S.; Vijayakumar, S.; Rao, Y. An evaluation of hepatotoxicity in breast cancer patients receiving injection Doxorubicin. Ann. Med. Health Sci. Res. 2014, 4, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of drug-antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody-maytansinoid conjugates. Bioconjugate Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Orcutt, K.D.; Rhoden, J.J.; Ruiz-Yi, B.; Frangioni, J.V.; Wittrup, K.D. Effect of small-molecule-binding affinity on tumor uptake in vivo: A systematic study using a pretargeted bispecific antibody. Mol. Cancer Ther. 2012, 11, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.P.; Schier, R.; McCall, A.M.; Simmons, H.H.; Horak, E.M.; Alpaugh, R.K.; Marks, J.D.; Weiner, L.M. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001, 61, 4750–4755. [Google Scholar]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef]
- Liu, M.; Apriceno, A.; Sipin, M.; Scarpa, E.; Rodriguez-Arco, L.; Poma, A.; Marchello, G.; Battaglia, G.; Angioletti-Uberti, S. Combinatorial entropy behaviour leads to range selective binding in ligand-receptor interactions. Nat. Commun. 2020, 11, 4836. [Google Scholar] [CrossRef]
- Carlson, C.B.; Mowery, P.; Owen, R.M.; Dykhuizen, E.C.; Kiessling, L.L. Selective tumor cell targeting using low-affinity, multivalent interactions. ACS Chem. Biol. 2007, 2, 119–127. [Google Scholar] [CrossRef]
- Martinez-Veracoechea, F.J.; Frenkel, D. Designing super selectivity in multivalent nano-particle binding. Proc. Natl. Acad. Sci. USA 2011, 108, 10963–10968. [Google Scholar] [CrossRef]
- Chen, L.; Wang, L.; Shion, H.; Yu, C.; Yu, Y.Q.; Zhu, L.; Li, M.; Chen, W.; Gao, K. In-depth structural characterization of Kadcyla(R) (ado-trastuzumab emtansine) and its biosimilar candidate. mAbs 2016, 8, 1210–1223. [Google Scholar] [CrossRef]
- Zanetti-Domingues, L.C.; Tynan, C.J.; Rolfe, D.J.; Clarke, D.T.; Martin-Fernandez, M. Hydrophobic fluorescent probes introduce artifacts into single molecule tracking experiments due to non-specific binding. PLoS ONE 2013, 8, e74200. [Google Scholar] [CrossRef]
- Okamoto, H.; Oitate, M.; Hagihara, K.; Shiozawa, H.; Furuta, Y.; Ogitani, Y.; Kuga, H. Pharmacokinetics of trastuzumab deruxtecan (T-DXd), a novel anti-HER2 antibody-drug conjugate, in HER2-positive tumour-bearing mice. Xenobiotica Fate Foreign Compd. Biol. Syst. 2020, 50, 1242–1250. [Google Scholar] [CrossRef]
- Walles, M.; Rudolph, B.; Wolf, T.; Bourgailh, J.; Suetterlin, M.; Moenius, T.; Peraus, G.; Heudi, O.; Elbast, W.; Lanshoeft, C.; et al. New insights in tissue distribution, metabolism, and excretion of [3H]-labeled antibody maytansinoid conjugates in female tumor-bearing nude rats. Drug Metab. Dispos. Biol. Fate Chem. 2016, 44, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Azhdarinia, A. Application of immuno-PET in antibody-drug conjugate development. Mol. Imaging 2018, 17, 1536012118801223. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2016. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álamo, P.; Pallarès, V.; Céspedes, M.V.; Falgàs, A.; Sanchez, J.M.; Serna, N.; Sánchez-García, L.; Voltà-Duràn, E.; Morris, G.A.; Sánchez-Chardi, A.; et al. Fluorescent Dye Labeling Changes the Biodistribution of Tumor-Targeted Nanoparticles. Pharmaceutics 2020, 12, 1004. https://doi.org/10.3390/pharmaceutics12111004
Álamo P, Pallarès V, Céspedes MV, Falgàs A, Sanchez JM, Serna N, Sánchez-García L, Voltà-Duràn E, Morris GA, Sánchez-Chardi A, et al. Fluorescent Dye Labeling Changes the Biodistribution of Tumor-Targeted Nanoparticles. Pharmaceutics. 2020; 12(11):1004. https://doi.org/10.3390/pharmaceutics12111004
Chicago/Turabian StyleÁlamo, Patricia, Victor Pallarès, María Virtudes Céspedes, Aïda Falgàs, Julieta M. Sanchez, Naroa Serna, Laura Sánchez-García, Eric Voltà-Duràn, Gordon A. Morris, Alejandro Sánchez-Chardi, and et al. 2020. "Fluorescent Dye Labeling Changes the Biodistribution of Tumor-Targeted Nanoparticles" Pharmaceutics 12, no. 11: 1004. https://doi.org/10.3390/pharmaceutics12111004
APA StyleÁlamo, P., Pallarès, V., Céspedes, M. V., Falgàs, A., Sanchez, J. M., Serna, N., Sánchez-García, L., Voltà-Duràn, E., Morris, G. A., Sánchez-Chardi, A., Casanova, I., Mangues, R., Vazquez, E., Villaverde, A., & Unzueta, U. (2020). Fluorescent Dye Labeling Changes the Biodistribution of Tumor-Targeted Nanoparticles. Pharmaceutics, 12(11), 1004. https://doi.org/10.3390/pharmaceutics12111004