Erythrocyte Membrane Cloaked Curcumin-Loaded Nanoparticles for Enhanced Chemotherapy


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Preparation and Characterization of p-PLGA NPs
2.3. Loading of Cur
2.4. Isolation of RBCM
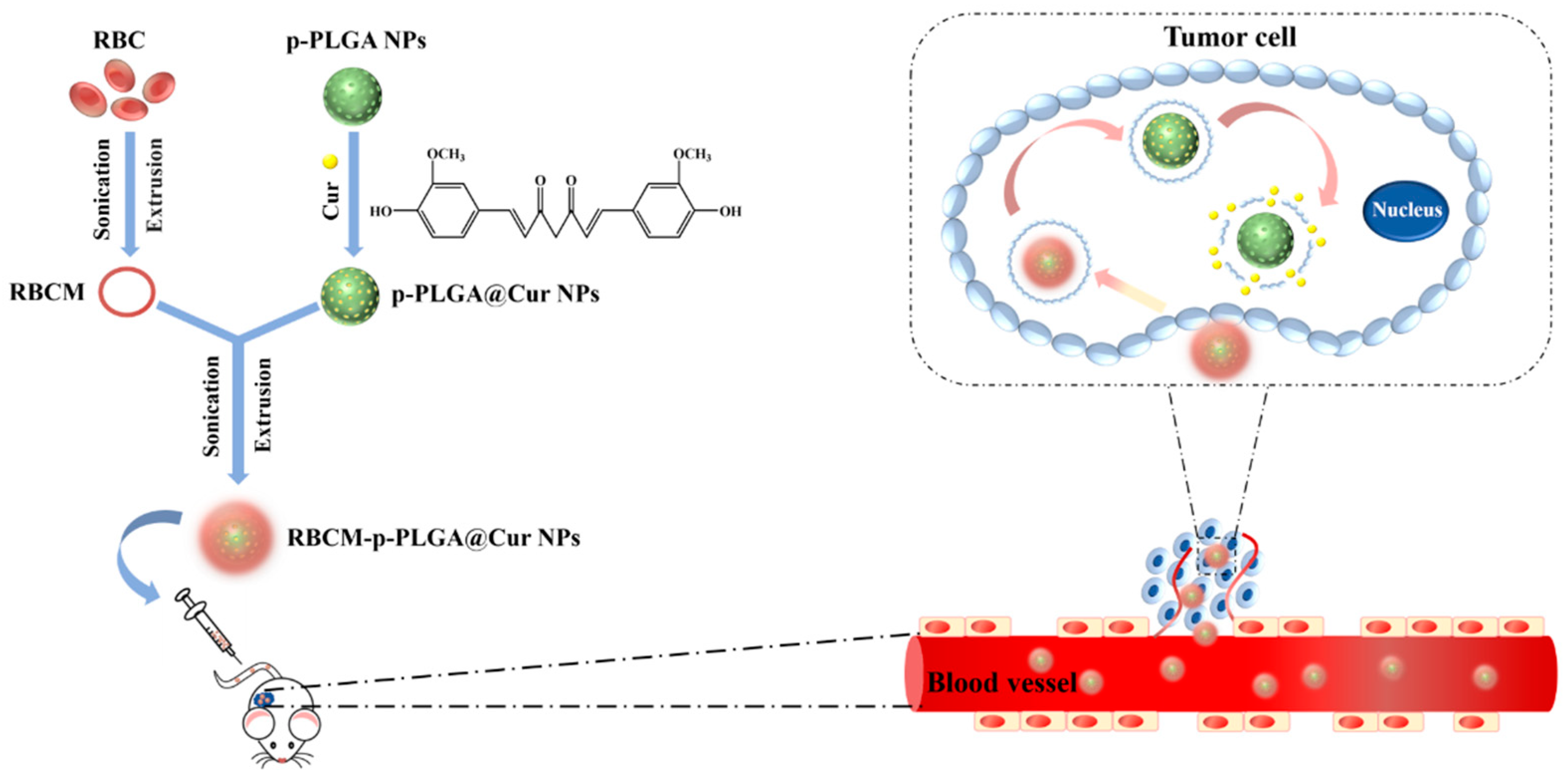
2.5. Preparation and Characterization of RBCM-p-PLGA@Cur NPs
2.6. Characterization Techniques
2.7. Drug Release
2.8. Cytotoxicity Measurements
2.9. Cellular Uptake
2.10. In Vivo Murine Tumor Model
2.11. In Vivo Antitumor Efficacy
2.12. Statistical Analysis
3. Results and Discussion
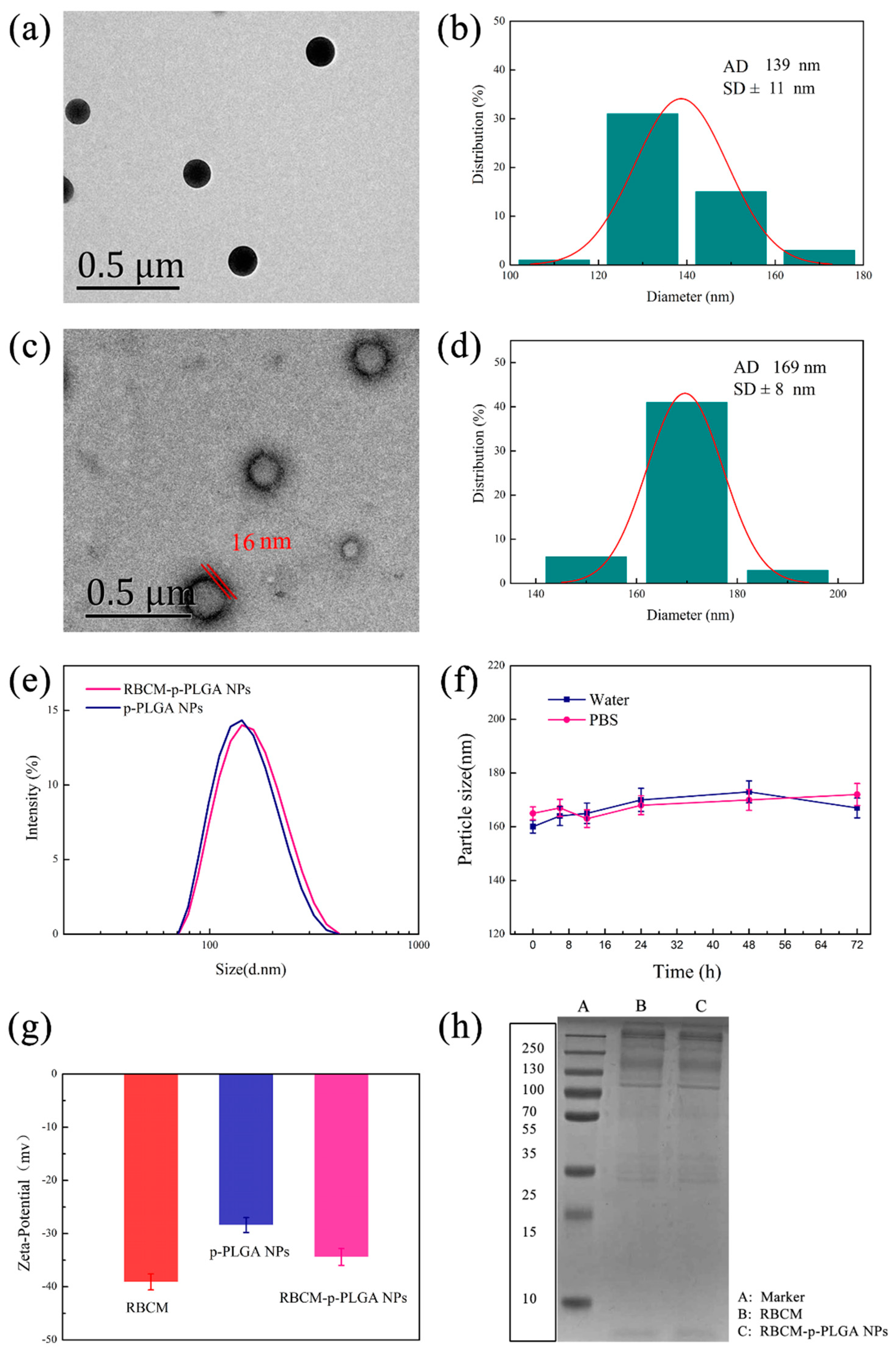
3.1. Characterization of NPs
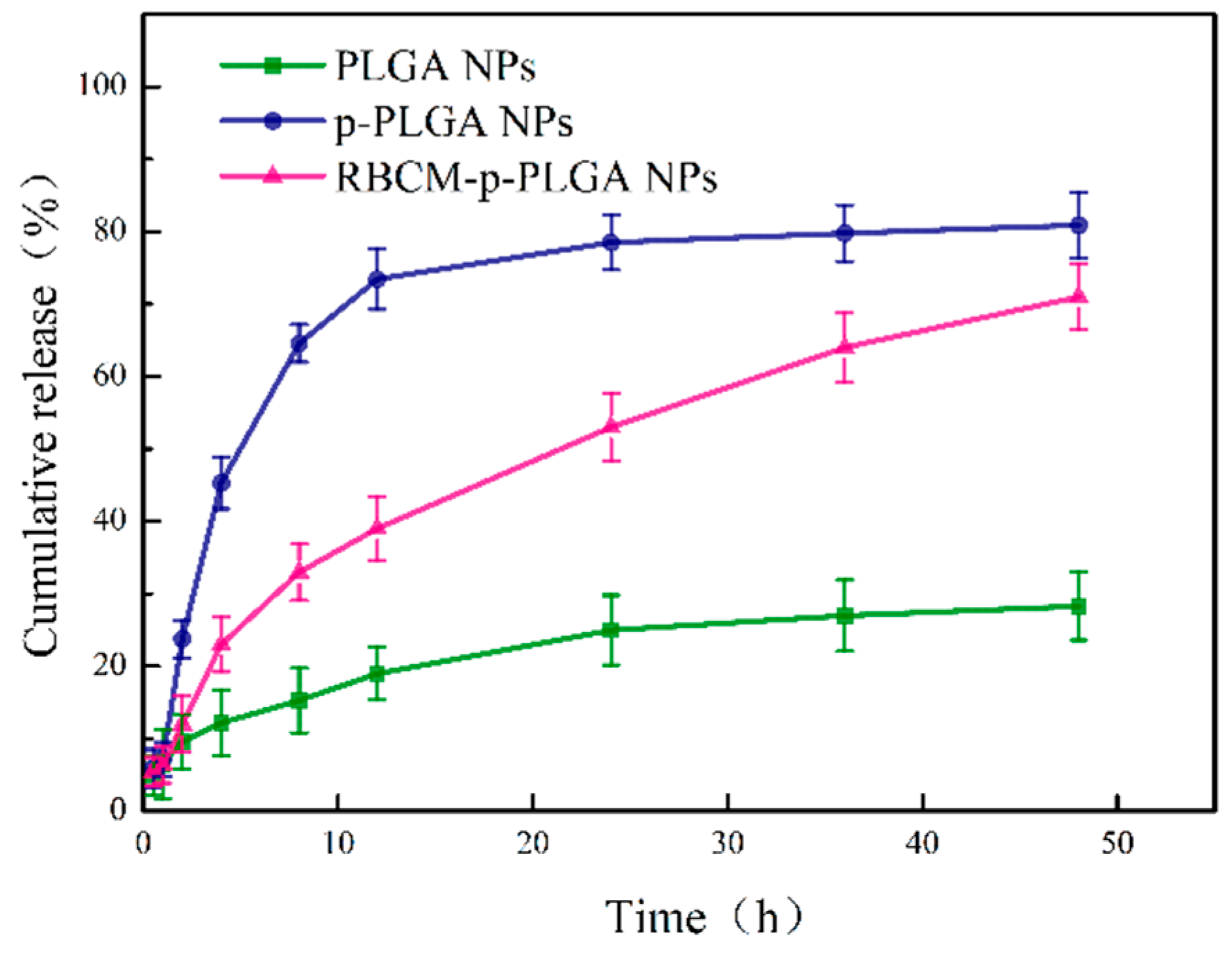
3.2. Drug Loading and Release Behavior
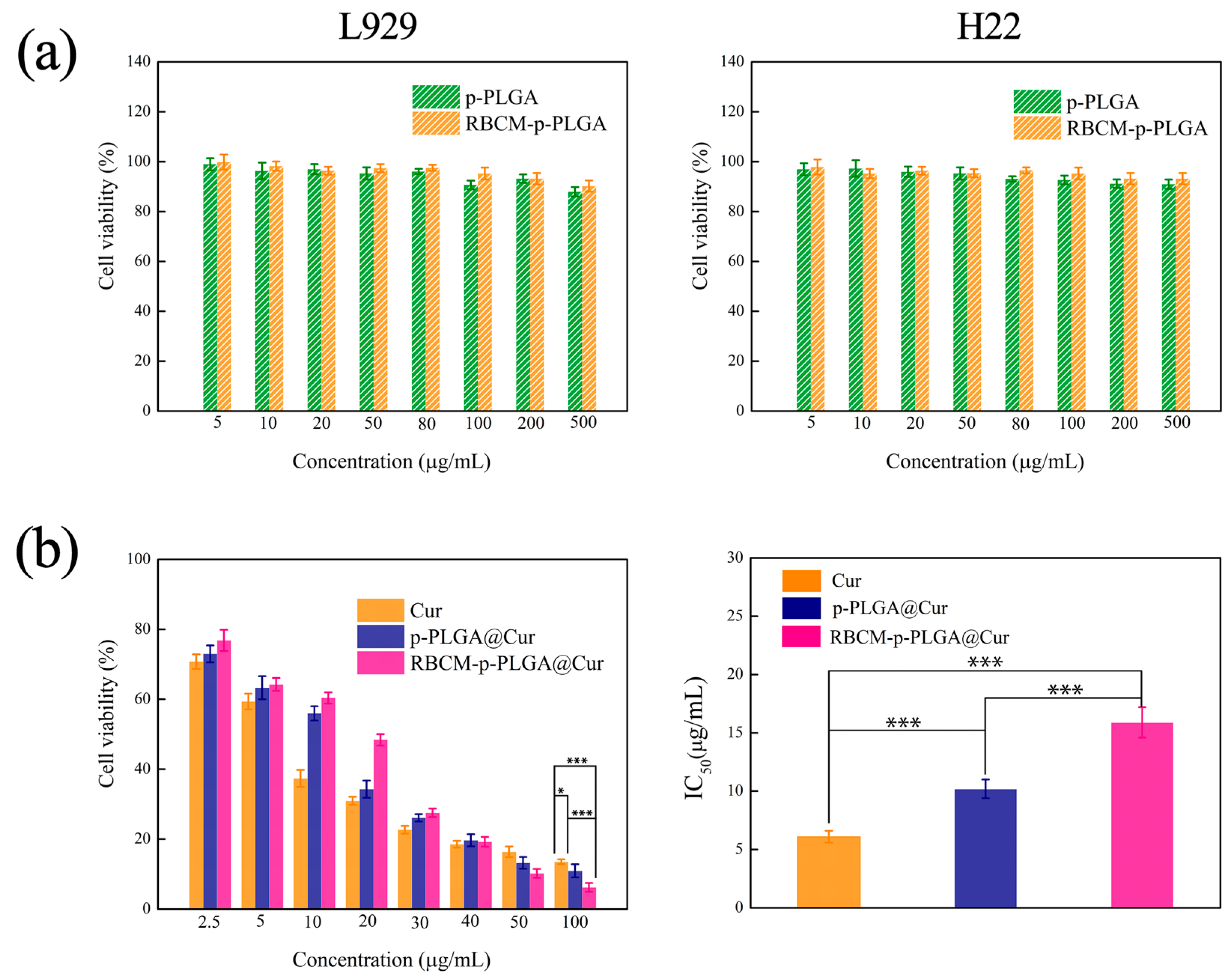
3.3. In Vitro Cytotoxicity
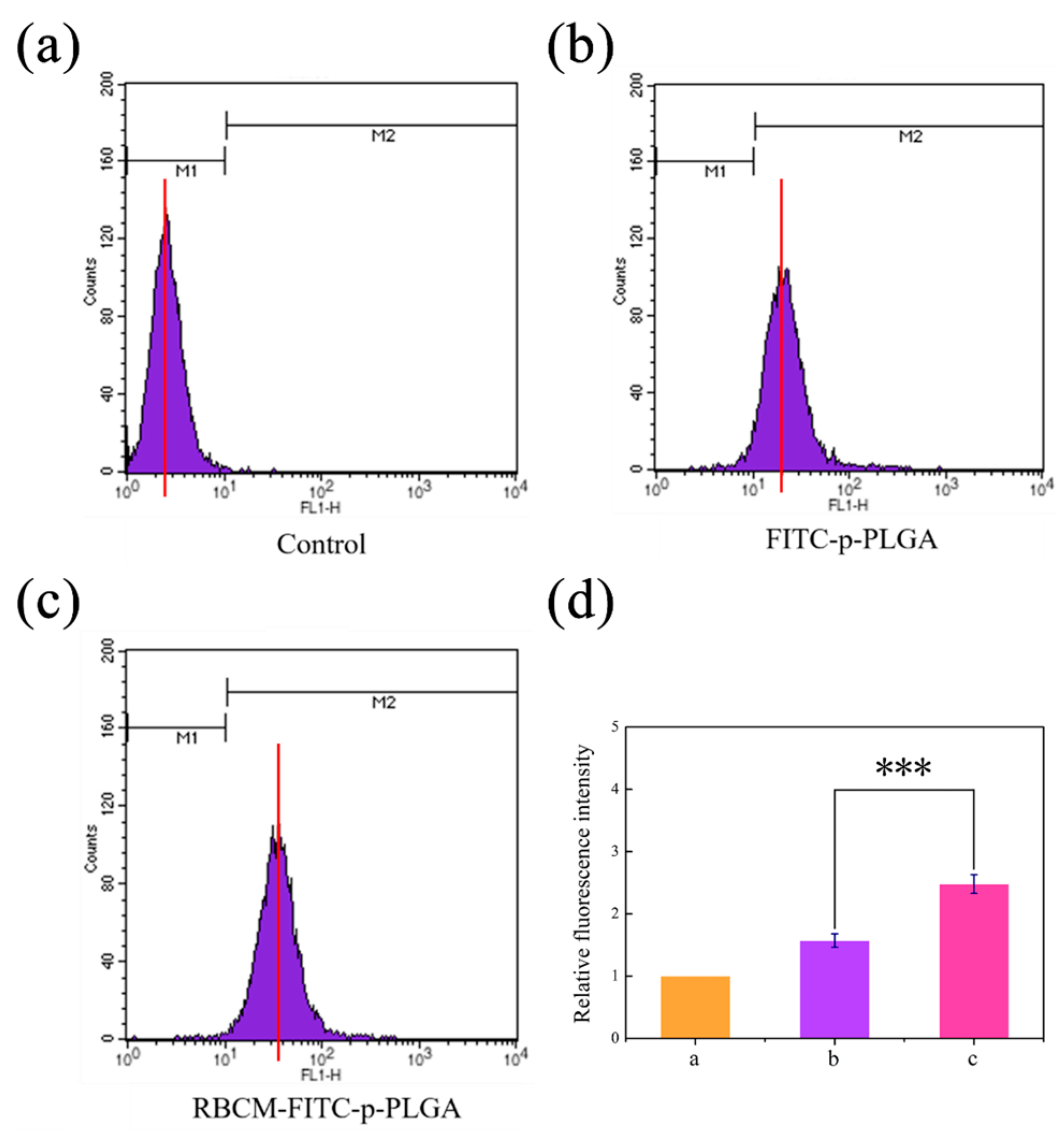
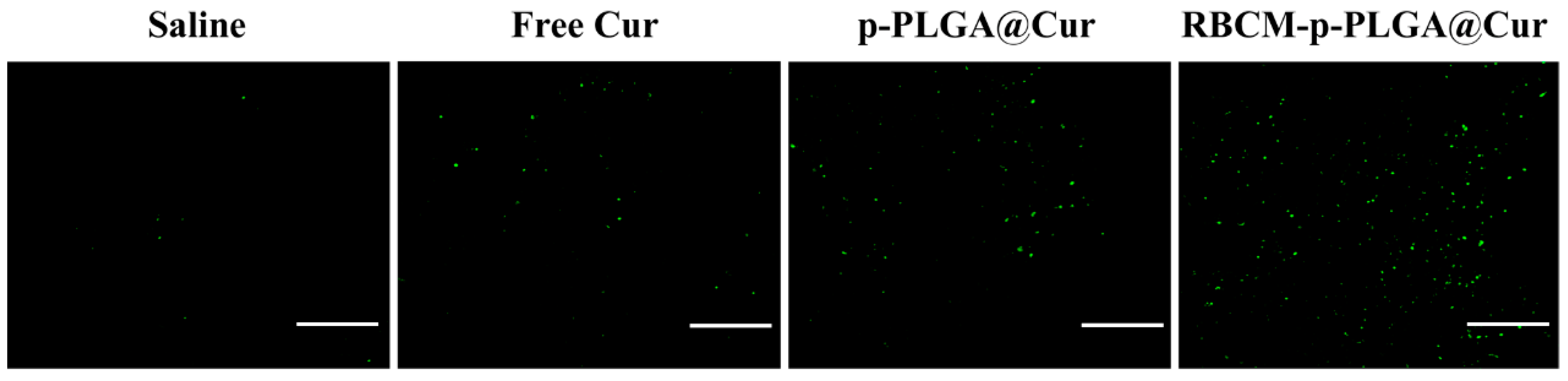
3.4. Cellular Uptake In Vitro
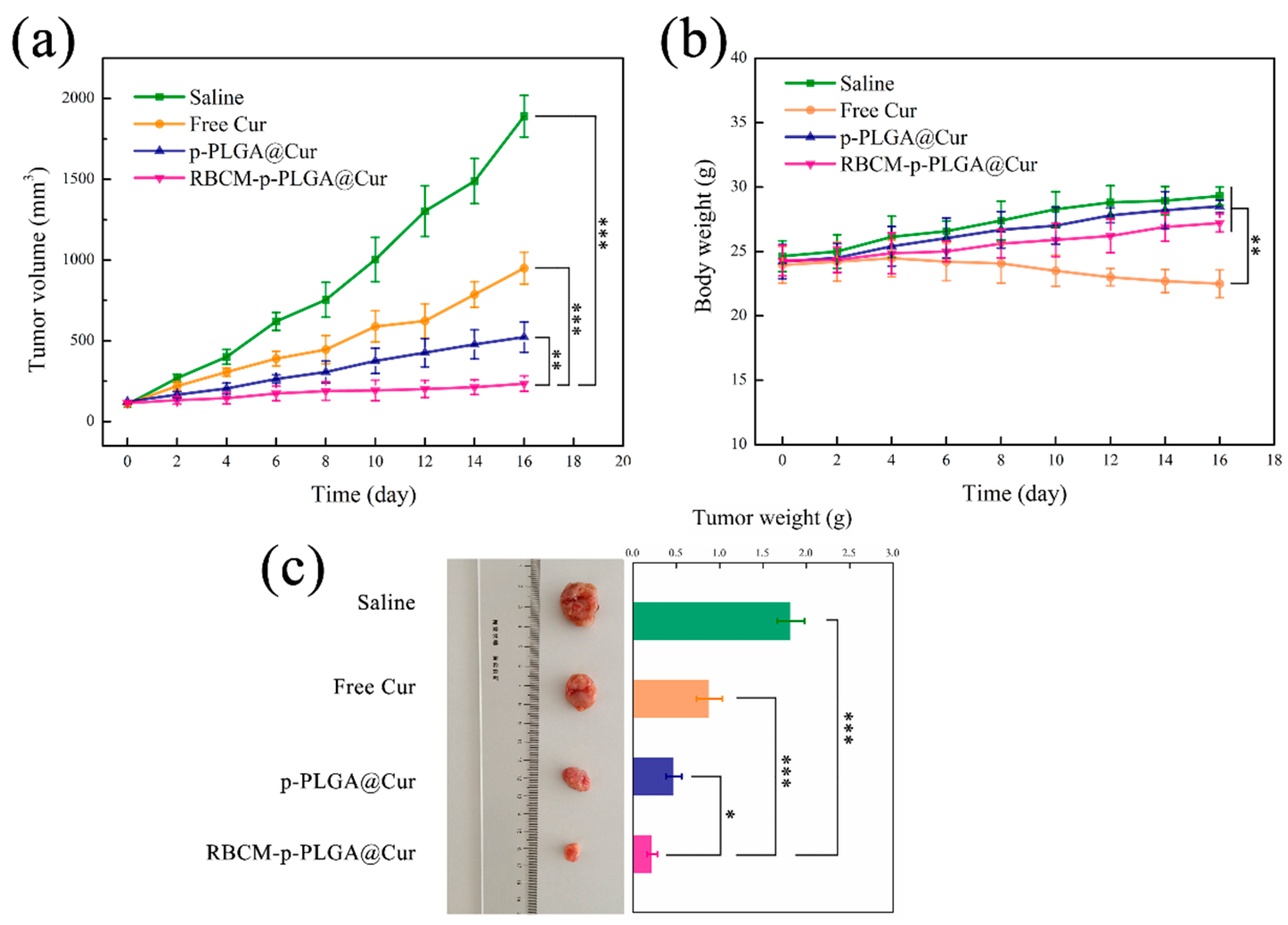
3.5. In Vivo Antitumor Efficacy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shi, J.J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.L.; Gupta, P.; Chen, Y.L.; Wang, E.J.; Ji, L.N.; Chao, H.; Chen, Z.S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Nie, J.P.; Xu, L.; Liang, C.Y.; Peng, Y.; Liu, G.; Wang, T.; Mei, L.; Huang, L.Q.; Zeng, X.W. pH-Sensitive Delivery Vehicle Based on Folic Acid-Conjugated Polydopamine-Modified Mesoporous Silica Nanoparticles for Targeted Cancer Therapy. ACS Appl. Mater. Interfaces 2017, 9, 18462–18473. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.M.; Lee, Y.S.; Gillies, R.J.; Hruby, V.J. Synthesis and evaluation of bivalent ligands for binding to the human melanocortin-4 receptor. Bioorgan. Med. Chem. 2014, 22, 6360–6365. [Google Scholar] [CrossRef] [PubMed]
- Low, P.S.; Henne, W.A.; Doorneweerd, D.D. Discovery and development of folic-acid-based receptor targeting for Imaging and therapy of cancer and inflammatory diseases. Accounts Chem. Res. 2008, 41, 120–129. [Google Scholar] [CrossRef] [PubMed]
- van Waarde, A.; Rybczynska, A.A.; Ramakrishnan, N.K.; Ishiwata, K.; Elsinga, P.H.; Dierckx, R. Potential applications for sigma receptor ligands in cancer diagnosis and therapy. BBA Biomembr. 2015, 1848, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.; Quattrocchi, N.; van de Ven, A.L.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.O.; Brown, B.S.; Khaled, S.Z.; Yazdi, I.K.; Vittoria Enzo, M.; et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2013, 8, 61–68. [Google Scholar] [CrossRef]
- Wang, H.J.; Williams, G.R.; Wu, J.R.; Wu, J.Z.; Niu, S.W.; Xie, X.T.; Li, S.D.; Zhu, L.M. Pluronic F127-based micelles for tumor-targeted bufalin delivery. Int. J. Pharm. 2019, 559, 289–298. [Google Scholar] [CrossRef]
- Rapido, F.; Brittenham, G.M.; Bandyopadhyay, S.; La Carpia, F.; L’Acqua, C.; McMahon, D.J.; Rebbaa, A.; Wojczyk, B.S.; Netterwald, J.; Wang, H.L.; et al. Prolonged red cell storage before transfusion increases extravascular hemolysis. J. Clin. Investig. 2017, 127, 375–382. [Google Scholar] [CrossRef]
- Salvagno, G.L.; Sanchis-Gomar, F.; Picanza, A.; Lippi, G. Red blood cell distribution width: A simple parameter with multiple clinical applications. Crit. Rev. Clin. Lab. Sci. 2015, 52, 86–105. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Yamada, S.; Sakamoto, W.; Usugi, E.; Watanabe, M.; Yogo, T. Red Blood Cell-Shaped Microparticles with a Red Blood Cell Membrane Demonstrate Prolonged Circulation Time in Blood. ACS Biomater. Sci. Eng. 2018, 4, 2729–2732. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, W.; Wang, L.Y.; Zhu, B.Y.; Qu, M.K.; Yang, L.Q.; Sun, X.; Gong, T.; Zhang, Z.R.; Lin, Q.; et al. Erythrocyte-Membrane-Camouflaged Nanoplatform for Intravenous Glucose-Responsive Insulin Delivery. Adv. Funct. Mater. 2018, 28, 1802250. [Google Scholar] [CrossRef]
- Gao, M.; Liang, C.; Song, X.J.; Chen, Q.; Jin, Q.T.; Wang, C.; Liu, Z. Erythrocyte-Membrane-Enveloped Perfluorocarbon as Nanoscale Artificial Red Blood Cells to Relieve Tumor Hypoxia and Enhance Cancer Radiotherapy. Adv. Mater. 2017, 29, 201701429. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed]
- Jutkova, A.; Chorvat, D.; Miskovsky, P.; Jancura, D.; Datta, S. Encapsulation of anticancer drug curcumin and co-loading with photosensitizer hypericin into lipoproteins investigated by fluorescence resonance energy transfer. Int. J. Pharm. 2019, 564, 369–378. [Google Scholar] [CrossRef]
- Tsuda, T. Curcumin as a functional food-derived factor: Degradation products, metabolites, bioactivity, and future perspectives. Food Funct. 2018, 9, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Bondi, M.L.; Emma, M.R.; Botto, C.; Augello, G.; Azzolina, A.; Di Gaudio, F.; Craparo, E.F.; Cavallaro, G.; Bachvarov, D.; Cervello, M. Biocompatible Lipid Nanoparticles as Carriers to Improve Curcumin Efficacy in Ovarian Cancer Treatment. J. Agric. Food. Chem. 2017, 65, 1342–1352. [Google Scholar] [CrossRef]
- Song, W.X.; Muthana, M.; Mukherjee, J.; Falconer, R.J.; Biggs, C.A.; Zhao, X.B. Magnetic-Silk Core-Shell Nanoparticles as Potential Carriers for Targeted Delivery of Curcumin into Human Breast Cancer Cells. ACS Biomater. Sci. Eng. 2017, 3, 1027–1038. [Google Scholar] [CrossRef]
- Zou, J.; Zhang, S.S.; Li, P.Y.; Zheng, X.M.; Feng, D. Supplementation with curcumin inhibits intestinal cholesterol absorption and prevents atherosclerosis in high-fat diet-fed apolipoprotein E knockout mice. Nutr. Res. 2018, 56, 32–40. [Google Scholar] [CrossRef]
- Akbar, M.U.; Zia, K.M.; Akash, M.S.H.; Nazir, A.; Zuber, M.; Ibrahim, M. In-vivo anti-diabetic and wound healing potential of chitosan/alginate/maltodextrin/pluronic-based mixed polymeric micelles: Curcumin therapeutic potential. Int. J. Biol. Macromol. 2018, 120, 2418–2430. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, M.; Corsini, E.; Rosini, M.; Racchi, M.; Lanni, C. Immunomodulators Inspired by Nature: A Review on Curcumin and Echinacea. Molecules 2018, 23, 2778. [Google Scholar] [CrossRef] [PubMed]
- Shrivash, M.K.; Mishra, S.; Pandey, J.; Misra, K. In-silico designing, chemical synthesis, characterization and in-vitro assessment of antibacterial properties of some analogues of curcumin. Microb. Pathog. 2018, 123, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Lelli, D.; Sahebkar, A.; Johnston, T.P.; Pedone, C. Curcumin use in pulmonary diseases: State of the art and future perspectives. Pharmacol. Res. 2017, 115, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Allijn, I.E.; Schiffelers, R.M.; Storm, G. Comparison of pharmaceutical nanoformulations for curcumin: Enhancement of aqueous solubility and carrier retention. Int. J. Pharm. 2016, 506, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Pereverzeva, E.; Treschalin, I.; Treschalin, M.; Arantseva, D.; Ermolenko, Y.; Kumskova, N.; Maksimenko, O.; Balabanyan, V.; Kreuter, J.; Gelperina, S. Toxicological study of doxorubicin-loaded PLGA nanoparticles for the treatment of glioblastoma. Int. J. Pharm. 2019, 554, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Kamel, R.; Abbas, H. PLGA-based monolithic filaments prepared by hot-melt extrusion: In-vitro comparative study. Ann. Pharm. Fr. 2018, 76, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, N.B.; Patil, S.S.; Pandit, J.K.; Lele, A.K.; Kulkarni, M.G.; Mashelkar, R.A. Morphological changes in degrading PLGA and P(DL)LA microspheres: Implications for the design of controlled release systems. J. Microencapsul. 2001, 18, 783–800. [Google Scholar]
- Liu, D.D.; Cheng, B.C.; Li, D.Y.; Li, J.Y.; Wu, Q.Y.; Pan, H. Investigations on the interactions between curcumin loaded vitamin E TPGS coated nanodiamond and Caco-2 cell monolayer. Int. J. Pharm. 2018, 551, 177–183. [Google Scholar] [CrossRef]
- Sun, M.; Wang, X.; Cheng, X.; He, L.; Yan, G.Q.; Tang, R.P. TPGS-functionalized and ortho ester-crosslinked dextran nanogels for enhanced cytotoxicity on multidrug resistant tumor cells. Carbohyd. Polym. 2018, 198, 142–154. [Google Scholar] [CrossRef]
- Zhu, H.J.; Chen, H.B.; Zeng, X.W.; Wang, Z.Y.; Zhang, X.D.; Wu, Y.P.; Gao, Y.F.; Zhang, J.X.; Liu, K.W.; Liu, R.Y.; et al. Co-delivery of chemotherapeutic drugs with vitamin E TPGS by porous PLGA nanoparticles for enhanced chemotherapy against multi-drug resistance. Biomaterials 2014, 35, 2391–2400. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Wu, J.Z.; Williams, G.R.; Fan, Q.; Niu, S.W.; Wu, J.R.; Xie, X.T.; Zhu, L.M. Platelet-membrane-biomimetic nanoparticles for targeted antitumor drug delivery. J. Nanobiotechnol. 2019, 17. [Google Scholar] [CrossRef] [PubMed]
- Dehaini, D.; Wei, X.L.; Fang, R.H.; Masson, S.; Angsantikul, P.; Luk, B.T.; Zhang, Y.; Ying, M.; Jiang, Y.; Kroll, A.V.; et al. Erythrocyte-Platelet Hybrid Membrane Coating for Enhanced Nanoparticle Functionalization. Adv. Mater. 2017, 29, 1606209. [Google Scholar] [CrossRef] [PubMed]
- Kroll, A.V.; Fang, R.H.; Zhang, L.F. Biointerfacing and Applications of Cell Membrane-Coated Nanoparticles. Bioconjug. Chem. 2017, 28, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.W.; Fang, R.H.; Thamphiwatana, S.; Luk, B.T.; Li, J.M.; Angsantikul, P.; Zhang, Q.Z.; Hu, C.M.J.; Zhang, L.F. Modulating Antibacterial Immunity via Bacterial Membrane-Coated Nanoparticles. Nano Lett. 2015, 15, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.J.; Fang, R.H.; Wang, K.C.; Luk, B.T.; Thamphiwatana, S.; Dehaini, D.; Nguyen, P.; Angsantikul, P.; Wen, C.H.; Kroll, A.V.; et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature 2015, 526, 118. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Luo, Z.M.; Men, Y.Z.; Yang, P.; Peng, H.B.; Guo, R.R.; Tian, Y.; Pang, Z.Q.; Yang, W.L. Red blood cell membrane-camouflaged melanin nanoparticles for enhanced photothermal therapy. Biomaterials 2017, 143, 29–45. [Google Scholar] [CrossRef]
- Xu, P.P.; Zuo, H.Q.; Chen, B.; Wang, R.J.; Ahmed, A.; Hu, Y.; Ouyang, J. Doxorubicin-loaded platelets as a smart drug delivery system: An improved therapy for lymphoma. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Jiang, T.; Zhang, B.; Zhang, L.; Wu, X.; Li, H.; Shen, S.; Luo, Z.; Liu, X.; Hu, Y.; Pang, Z.; et al. Biomimetic nanoparticles delivered hedgehog pathway inhibitor to modify tumour microenvironment and improved chemotherapy for pancreatic carcinoma. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1088–1101. [Google Scholar] [CrossRef]
- Rao, L.; Bu, L.L.; Meng, Q.F.; Cai, B.; Deng, W.W.; Li, A.; Li, K.Y.; Guo, S.S.; Zhang, W.F.; Liu, W.; et al. Antitumor Platelet-Mimicking Magnetic Nanoparticles. Adv. Funct. Mater. 2017, 27. [Google Scholar] [CrossRef]
- Barzegar-Jalali, M.; Adibkia, K.; Valizadeh, H.; Shadbad, M.R.S.; Hasan, M. Kinetic analysis of drug release from nanoparticles. J. Pharm. Pharm. Sci. 2008, 11, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Xuan, M.J.; Shao, J.X.; Zhao, J.; Li, Q.; Dai, L.R.; Li, J.B. Magnetic Mesoporous Silica Nanoparticles Cloaked by Red Blood Cell Membranes: Applications in Cancer Therapy. Angew. Chem. Int. Ed. 2018, 57, 6049–6053. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.; Mitragotri, S. Long circulating nanoparticles via adhesion on red blood cells: Mechanism and extended circulation. Exp. Biol. Med. 2007, 232, 958–966. [Google Scholar]
- Hu, C.M.J.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L.F. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.Q.; Qian, K.Y.; Cai, J.; Yang, Y.; Zhu, L.J.; Liu, B.R. Therapy for Gastric Cancer with Peritoneal Metastasis Using Injectable Albumin Hydrogel Hybridized with Paclitaxel-Loaded Red Blood Cell Membrane Nanoparticles. ACS Biomater. Sci. Eng. 2019, 5, 1100–1112. [Google Scholar] [CrossRef]
- Peng, J.R.; Yang, Q.; Li, W.T.; Tan, L.W.; Xiao, Y.; Chen, L.J.; Hao, Y.; Qian, Z.Y. Erythrocyte-Membrane-Coated Prussian Blue/Manganese Dioxide Nanoparticles as H2O2-Responsive Oxygen Generators to Enhance Cancer Chemotherapy/Photothermal Therapy. AC. Appl. Mater. Inter. 2017, 9, 44410–44422. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.F.; Luo, Z.Y.; Zheng, M.B.; Tian, H.; Gong, P.; Gao, G.H.; Pan, H.; Liu, L.L.; Ma, A.Q.; et al. Cancer Cell Membrane-Biomimetic Nanoparticles for Homologous-Targeting Dual-Modal Imaging and Photothermal Therapy. ACS Nano 2016, 10, 10049–10057. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wu, S.; Zhou, X.Q. Bioinspired nanocarriers for an effective chemotherapy of hepatocellular carcinoma. J. Biomater. Appl. 2018, 33, 72–81. [Google Scholar] [CrossRef]
- Dwivedi, P.; Yuan, S.; Han, S.; Mangrio, F.A.; Zhu, Z.; Lei, F.; Ming, Z.; Cheng, L.; Liu, Z.; Si, T.; et al. Core-shell microencapsulation of curcumin in PLGA microparticles: Programmed for application in ovarian cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, S481–S491. [Google Scholar] [CrossRef]
- Jamali, Z.; Khoobi, M.; Hejazi, S.M.; Eivazi, N.; Abdolahpour, S.; Imanparast, F.; Moradi-Sardareh, H.; Paknejad, M. Evaluation of targeted curcumin (CUR) loaded PLGA nanoparticles for in vitro photodynamic therapy on human glioblastoma cell line. Photodiagn. Photodyn. Ther. 2018, 23, 190–201. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, X.; Wang, H.; Williams, G.R.; Yang, Y.; Zheng, Y.; Wu, J.; Zhu, L.-M. Erythrocyte Membrane Cloaked Curcumin-Loaded Nanoparticles for Enhanced Chemotherapy. Pharmaceutics 2019, 11, 429. https://doi.org/10.3390/pharmaceutics11090429
Xie X, Wang H, Williams GR, Yang Y, Zheng Y, Wu J, Zhu L-M. Erythrocyte Membrane Cloaked Curcumin-Loaded Nanoparticles for Enhanced Chemotherapy. Pharmaceutics. 2019; 11(9):429. https://doi.org/10.3390/pharmaceutics11090429
Chicago/Turabian StyleXie, Xiaotian, Haijun Wang, Gareth R. Williams, Yanbo Yang, Yongli Zheng, Junzi Wu, and Li-Min Zhu. 2019. "Erythrocyte Membrane Cloaked Curcumin-Loaded Nanoparticles for Enhanced Chemotherapy" Pharmaceutics 11, no. 9: 429. https://doi.org/10.3390/pharmaceutics11090429
APA StyleXie, X., Wang, H., Williams, G. R., Yang, Y., Zheng, Y., Wu, J., & Zhu, L.-M. (2019). Erythrocyte Membrane Cloaked Curcumin-Loaded Nanoparticles for Enhanced Chemotherapy. Pharmaceutics, 11(9), 429. https://doi.org/10.3390/pharmaceutics11090429