Quantitative Prediction of Human Pharmacokinetics and Pharmacodynamics of CKD519, a Potent Inhibitor of Cholesteryl Ester Transfer Protein (CETP)


Abstract
1. Introduction
2. Materials and Methods
2.1. Overall Strategy
- Perform full PK samplings in three animal species to build-up animal PK datasets.
- Develop exploratory PK models by species from the datasets built in Step 1.
- Establish an overall (interspecies) PK model with allometric relationships for PK parameters in consideration of the model structure in Step 2.
- Human PK parameter prediction using the overall PK model.
- Human PK/PD modeling and simulation incorporating the PD information of the comparator drug.
2.2. Animal Full PK Study
2.3. Animal PK Model Development
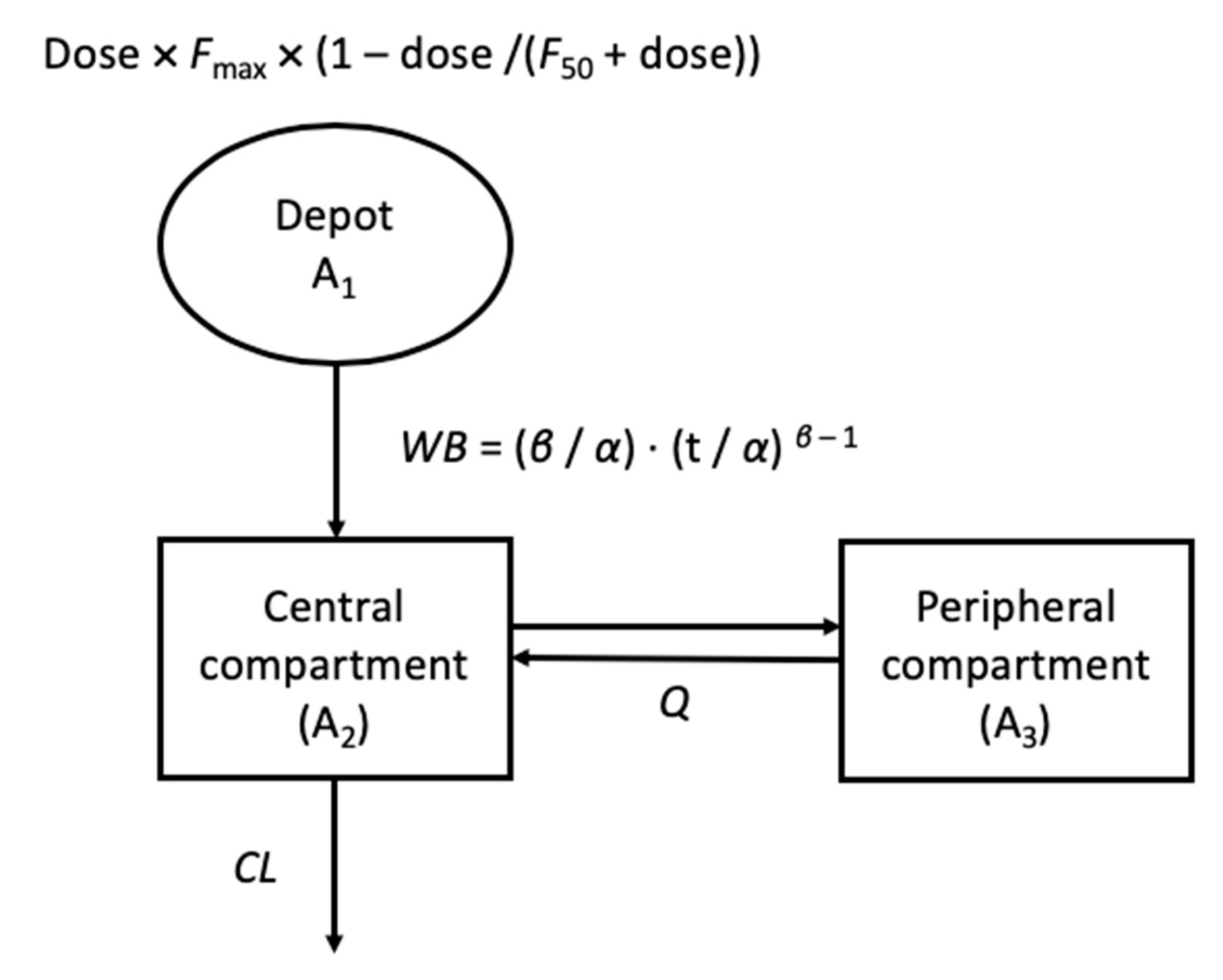
2.4. Incorporation of Allometry and Human PK Parameter Prediction
2.5. Human PK/PD Modeling and Simulation
3. Results
3.1. Animal PK Dataset and Exploratory Data Analysis
3.2. Animal PK Model by Species
3.3. Overall (Interspecies) PK Model and Allometric Relationship
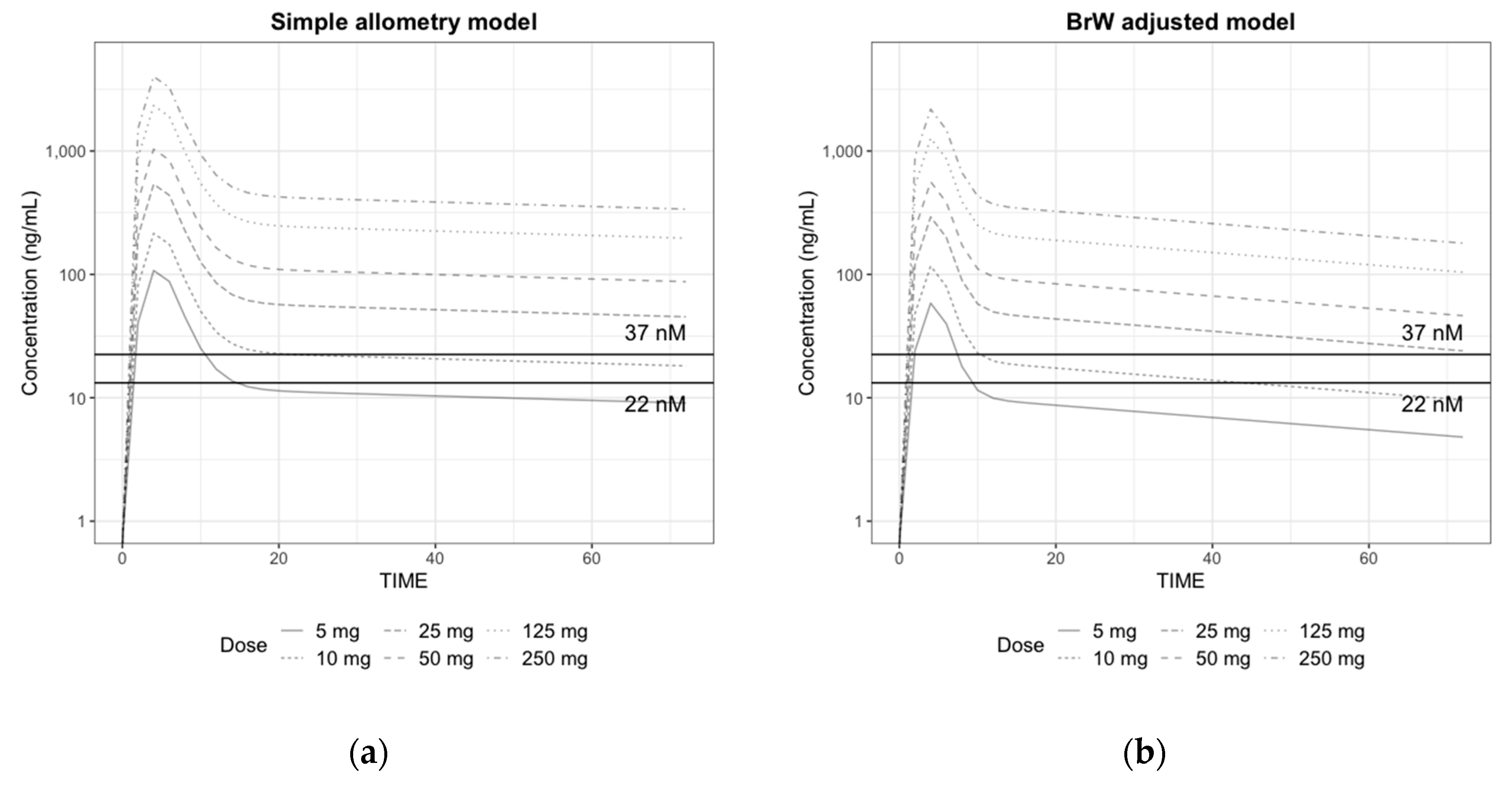
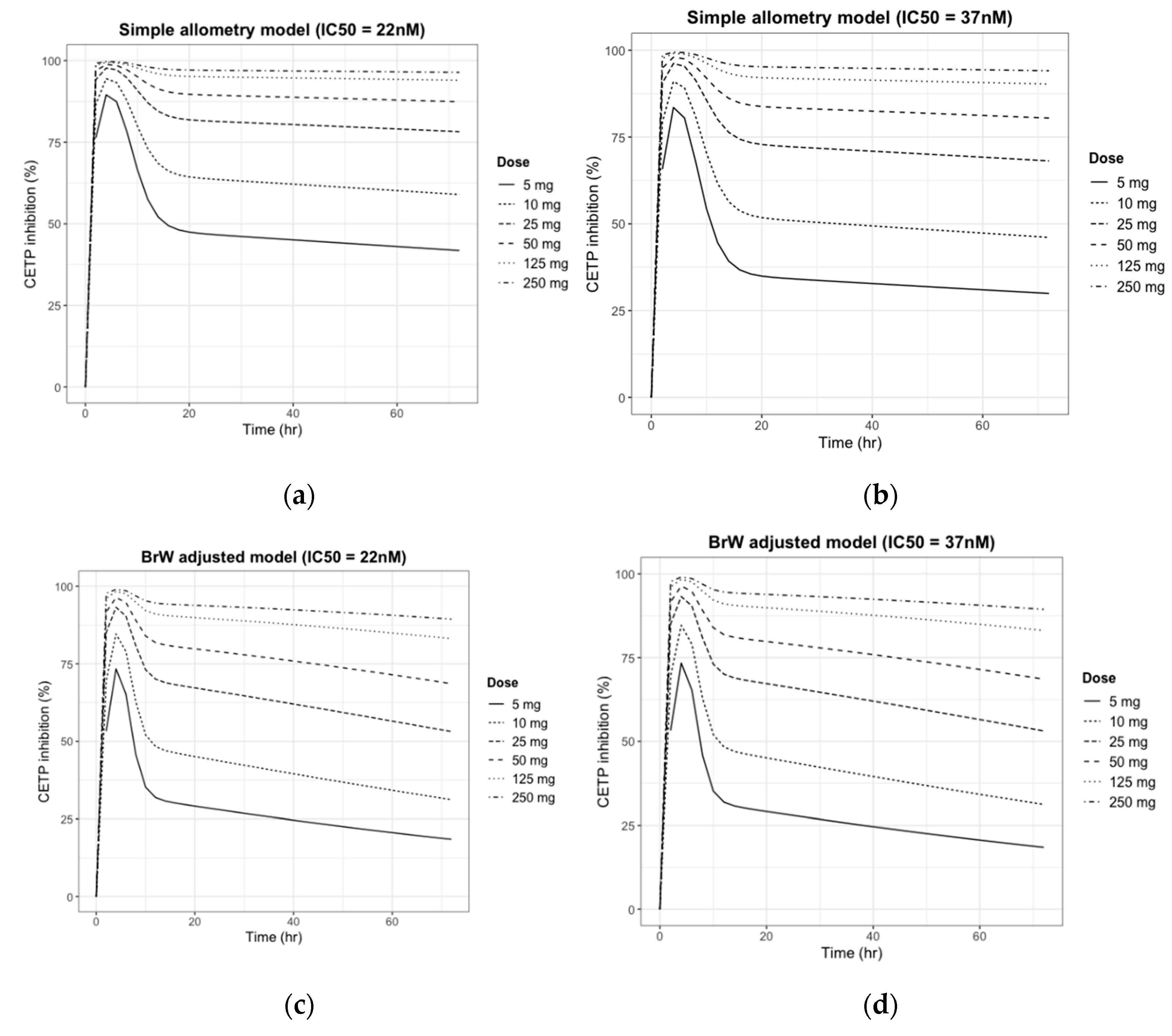
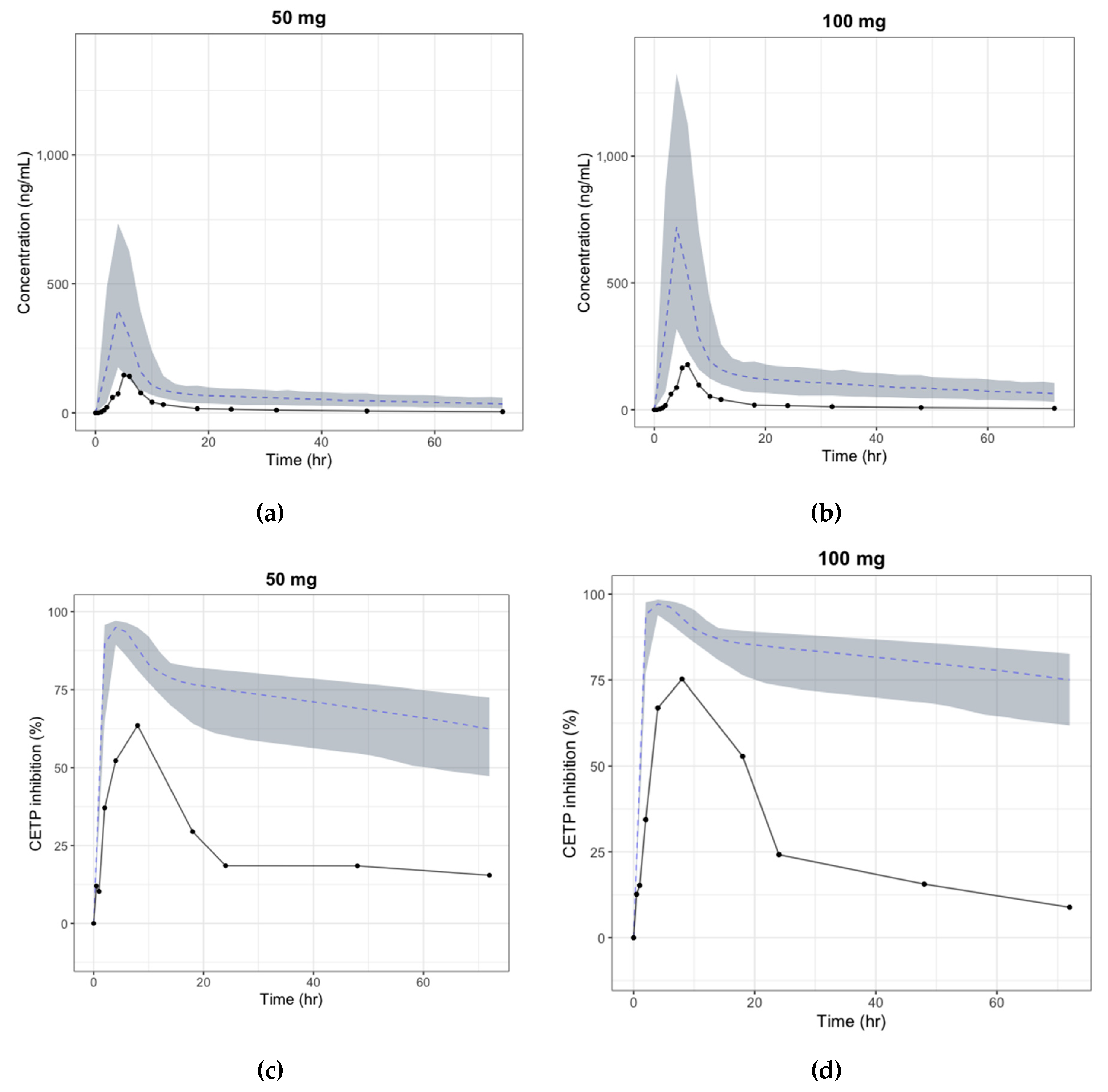
3.4. Human PK/PD Simulation
3.5. Prediction of the Human Efficacious Dose
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kopin, L.; Lowenstein, C. Dyslipidemia. Ann. Intern. Med. 2017, 167, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.; Wentworth, D.; Neaton, J.D. Is relationship between serum cholesterol and risk of premature death from coronary heart disease continuous and graded? Finding in 356,222 primary screens of the Multiple Risk Factor Intervention Trial (MRFIT). JAMA 1986, 256, 2823–2828. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.Y.; Riverra, J.J.; Blumenthal, J.S. Residual risk in statin-treated patients: Future therapeutic options. Curr. Cardiol. Rep. 2007, 9, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R. Plasma cholesteryl ester transfer protein. J. Lipid Res. 1993, 34, 1255–1274. [Google Scholar] [PubMed]
- Rhoads, G.G.; Gulbrandsen, C.L.; Kagan, A. Serum lipoproteins and coronary heart disease in a population study of Hawaii Japanese men. N. Engl. J. Med. 1976, 294, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Brewer, H.B.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl Ester Transfer Protein. Arter. Thromb. Vasc. Boil. 2003, 23, 160–167. [Google Scholar] [CrossRef]
- Sikorski, J.A. Oral cholesteryl ester transfer protein (CETP) inhibitors: A potential new approach for treating coronary artery disease. J. Med. Chem. 2006, 49, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Joy, T.; Hegele, R.A. The end of the road for CETP inhibitors after torcetrapib? Curr. Opin. Cardiol. 2009, 24, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; López-Sendón, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of Torcetrapib in Patients at High Risk for Coronary Events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Neeli, H.; Rader, D.J. Cholesteryl ester transfer protein (CETP) inhibitors: Is there life after torcetrapib? Cardiol. Clin. 2008, 26, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.P.; Van-Leuven, S.I.; Burgess, L.; Evans, G.W.; Kuivenhoven, J.A.; Barter, P.J.; Revkin, J.H.; Grobbee, D.E.; Riley, W.A.; Shear, C.L.; et al. Effect of torcetrapib on carotid atherosclerosis in familiar hypercholesterolemia. N. Engl. J. Med 2007, 356, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.Y.; Hartmann, G.; Chen, Q.; Pereira, A.; Bradley, S.; Doss, G.; Zhang, A.S.; Ho, J.Z.; Braun, M.P.; Dean, D.C.; et al. Pharmacokinetics, metabolism, and excretion of anacetrapib, a novel inhibitor of the cholesteryl ester transfer protein, in rats and rhesus monkeys. Drug Metab. Dispos. 2010, 38, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Bergman, A.J.; Green, M.; Dockendorf, M.F.; Wagner, J.A.; Dykstra, K. Model-based development of anacetrapib, a novel cholesteryl ester transfer protein inhibitor. AAPS J. 2011, 13, 179–190. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krishna, R.; Bergman, A.J.; Jin, B.; Fallon, M.; Cote, J.; Van Hoydonck, P.; Laethem, T.; Gendrano, I.N., 3rd; Van Dyck, K.; Hilliard, D.; et al. Multiple-dose pharmacodynamics and pharmacokinetics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Clin. Pharmacol. Ther. 2008, 84, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Garg, A.; Panebianco, D.; Cote, J.; Bergman, A.J.; Van Hoydonck, P.; Laethem, T.; Van Dyck, K.; Chen, J.; Chavez-Eng, C.; et al. Single-dose pharmacokinetics and pharmacodynamics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Br. J. Clin. Pharmacol. 2009, 68, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tan, E.Y.; Hartmann, G.; Biddle, Z.; Bergman, A.J.; Dru, J.; Ho, J.Z.; Jones, A.N.; Staskiewicz, S.J.; Braun, M.P.; et al. Metabolism and excretion of anacetrapib, a novel inhibitor of the cholesteryl ester transfer protein, in humans. Drug Metab. Dispos. 2010, 38, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Shah, S.; Sapre, A.; Ashraf, T.B.; Tobias, S.C.; Sahin, T.; Ye, P.; Dong, Y.; Sheu, W.H.; Kang, D.H.; et al. A multiregional, randomized evaluation of the lipid-modifying efficacy and tolerability of anacetrapib added to ongoing statin therapy in patients with hypercholesterolemia or low high-density lipoprotein cholesterol. Am. J. Cardiol. 2017, 120, 569–576. [Google Scholar] [CrossRef]
- Schmeck, C.; Gielen-Haertwig, H.; Vakalopoulos, A.; Bischoff, H.; Li, V.; Wirtz, G.; Weber, O. Novel tetrahydrochinoline derived CETP-inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1740–1743. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.F.; Heinig, R.; Schmeck, C.; Kohlsdorfer, C.; Ludwig, M.; Schaefer, A.; Gelfert-Peukert, S.; Wensing, G.; Weber, O. Single dose pharmacokinetics, pharmacodynamics, tolerability and safety of BAY 60-5521, a potent inhibitor of cholesteryl ester transfer protein. Br. J. Clin. Pharmacol. 2012, 73, 210–218. [Google Scholar] [CrossRef]
- Weber, O.; Willmann, S.; Bischoff, H.; Li, V.; Vakalopoulos, A.; Lustig, K.; Hafner, F.T.; Heinig, R.; Schmeck, C.; Buehner, K. Prediction of a potentially effective dose in humans for BAY 60-5521, a potent inhibitor of cholesteryl ester transfer protein (CETP) by allometric species scaling and combined pharmacodynamic and physiologically-based pharmacokinetic modelling. Br. J. Clin. Pharmacol. 2012, 73, 219–231. [Google Scholar] [CrossRef]
- Wang, X.; Li, W.; Hao, L.; Xie, H.; Hao, C.; Liu, C.; Li, W.; Xiong, X.; Zhao, D. The therapeutic potential of CETP inhibitors: A patent review. Expert Opin. Ther. Pat. 2018, 28, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ma, X.; Liu, Y.; Zhou, H.; Shi, C.; Wu, F.; Jiang, J.; Hu, P. Quantitative prediction of human pharmacokinetics and pharmacodynamics of imigliptin, a novel DPP-4 inhibitor, using allometric scaling, IVIVE and PK/PD modeling methods. Eur. J. Pharm. Sci. 2016, 89, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Morsy, M.A.; Jacob, S. Dose translation between laboratory animals and human in preclinical and clinical phases of drug development. Drug Dev. Res. 2018, 79, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I. Application of allometric principles for the prediction of pharmacokinetics in human and veterinary drug development. Adv. Drug Deliv. Rev. 2007, 59, 1177–1192. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Yu, Y.; Zheng, N.; Yang, Y.; Paholak, H.J.; Yu, L.X.; Sun, D. Applications of human pharmacokinetic prediction in first-in-human dose estimation. AAPS J. 2012, 14, 262–281. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Riviere, J.E. The application of allometric scaling principles to predict pharmacokinetic parameters across species. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1241–1253. [Google Scholar] [CrossRef]
- Huh, Y.; Smith, D.E.; Feng, M.R. Interspecies scaling and prediction of human clearance: Comparison of small- and macro-molecule drugs. Xenobiotica 2011, 41, 972–987. [Google Scholar] [CrossRef]
- Rajman, I. Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front. Pharmacol. 2014, 5, 1–16. [Google Scholar] [CrossRef]
- Boxenbaum, H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 1982, 10, 201–227. [Google Scholar] [CrossRef]
- Gabrielsson, J.; Weiner, D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications, 4th ed.; Swedish Pharmaceutical Press: Stockholm, Sweden, 2007; p. 192. [Google Scholar]
- Clark, R.W.; Sutfin, T.A.; Ruggeri, R.B.; Willauer, A.T.; Sugarman, E.D.; Magnus-Aryitey, G.; Cosgrove, P.G.; Sand, T.M.; Wester, R.T.; Williams, J.A.; et al. Raising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein: An initial multidose study of torcetrapib. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 490–497. [Google Scholar] [CrossRef]
- Brousseau, M.E.; Schaefer, E.J.; Wolfe, M.L.; Bloedon, L.T.; Digenio, A.G.; Clark, R.W.; Mancuso, J.P.; Rader, D.J. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N. Engl. J. Med. 2004, 350, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H. Pharmacokinetic strategies in deciphering atypical drug absorption profiles. J. Clin. Pharm. 2003, 43, 211–227. [Google Scholar] [CrossRef]
- Faeste, C.K.; Ivanova, L.; Sayyari, A.; Hansen, U.; Sivertsen, T.; Uhlig, S. Prediction of deoxynivalenol toxicokinetics in humans by in vitro-to-in vivo extrapolation and allometric scaling of in vivo animal data. Arch. Toxicol. 2018, 92, 2195–2216. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Léger, F.; Le Meur, Y.; Saint-Marcoux, F.; Paintaud, G.; Buchler, M.; Marquet, P. Population pharmacokinetic modeling of oral cyclosporin using NONMEM: Comparison of absorption pharmacokinetic models and design of a Bayesian estimator. Ther. Drug Monit. 2004, 26, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Park, G.J.; Park, W.S.; Bae, S.; Park, S.M.; Han, S.; Yim, D.S. Population pharmacokinetics of imatinib mesylate in healthy Korean subjects. Transl. Clin. Pharmacol. 2016, 24, 96–104. [Google Scholar] [CrossRef]
- Padoin, C.; Tod, M.; Brion, N.; Louchahi, K.; Le Gros, V.; Petitjean, O. Pharmacokinetics of amoxicillin coadministered with a saline-polyethylene glycol solution in healthy volunteers. Biopharm. Drug Dispos. 1995, 16, 169–176. [Google Scholar] [CrossRef]
- Desai, A.; Kovanda, L.; Kowalski, D.; Lu, Q.; Townsend, R.; Bonate, P.L. Population pharmacokinetics of isavuconazole from phase 1 and phase 3 (SECURE) trials in adults and target attainment in patients with invasive infections due to Aspergillus and other filamentous fungi. Antimicrob. Agents Chemother. 2016, 60, 5483–5491. [Google Scholar] [CrossRef]
- Choi, H.Y.; Choi, S.; Kim, Y.H.; Lim, H.S. Population pharmacokinetic and pharmacodynamic modeling analysis of GCC-4401C, a novel direct factor Xa inhibitor, in healthy volunteers. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 532–543. [Google Scholar] [CrossRef]
- Svensson, R.J.; Aarnoutse, R.E.; Diacon, A.H.; Dawson, R.; Gillespie, S.H.; Boeree, M.J.; Simonsson, U.S.H. A Population Pharmacokinetic Model Incorporating Saturable Pharmacokinetics and Autoinduction for High Rifampicin Doses. Clin. Pharmacol. Ther. 2018, 103, 674–683. [Google Scholar] [CrossRef]
- Musther, H.; Olivares-Morales, A.; Hatley, O.J.; Liu, B.; Rostami-Hodjugan, A. Animal versus human oral drug bioavailability: Do they correlate? Eur. J. Pharm. Sci. 2014, 57, 280–291. [Google Scholar] [CrossRef]
- Bonate, P.L.; Howard, D.R. Pharmacokinetics in Drug Development: Clinical Study Design and Analysis, 1st ed; American Association of Pharmaceutical Scientists: Arlington, VA, USA, 2004; pp. 423–432. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Weight (kg) | Brain Weight (g) | Maximum Life Span (years) |
|---|---|---|---|
| Hamster | 0.15 | 1.4 | 2.7 |
| Rat | 0.25 | 1.8 | 4.7 |
| Cynomolgus monkey | 3 | 74 | 22.3 |
| Human | 60 | 1500 | 93.4 |
| Species | Administration Route | Sample Size | Sex | Dose (mg/kg) | Cmax (ng/mL) | Tmax (h) | AUClast (ng∙h/mL) | CL/F * (mL/h) | V/F * (mL) | t1/2 (h) | ke (h−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Hamster | Intravenous | 3 | F | 0.5 | 0.083 (0.083–0.083) | 8760 ± 635 | 6.36 ± 1.20 | 138 ± 89.9 | 16.9 ± 14.1 | 0.0616 ± 0.0446 | |
| Oral | 4 | F | 3 | 659 ± 89.3 | 3.5 (3–4) | 6140 ± 471 | 34.7 ± 3.08 | 1570 ± 163 | 31.5 ± 4.41 | 0.0223 ± 0.00294 | |
| 4 | F | 15 | 3250 ± 465 | 4 (3–4) | 18800 ± 4310 | 66.6 ± 20.1 | 2460 ± 162 | 26.8 ± 5.23 | 0.0268 ± 0.00613 | ||
| 4 | F | 45 | 3160 ± 614 | 4 (3–6) | 26700 ± 2330 | 145 ± 9.52 | 4330 ± 572 | 20.7 ± 1.76 | 0.0338 ± 0.00311 | ||
| Rat | Intravenous | 5 | F | 0.5 | 0.083 (0.083–0.083) | 3940 ± 585 | 28.9 ± 6.24 | 494 ± 126 | 12.0 ± 2.98 | 0.0677 ± 0.0214 | |
| Oral | 5 | F | 5 | 893 ± 231 | 3 (3–4) | 6640 ± 932 | 188 ± 28.6 | 2860 ± 1930 | 10.0 ± 4.78 | 0.0784 ± 0.0245 | |
| 5 | F | 15 | 2040 ± 448 | 2 (2–4) | 11600 ± 3240 | 323 ± 82.8 | 4770 ± 1740 | 10.1 ± 1.82 | 0.0701 ± 0.0107 | ||
| 5 | F | 45 | 184 ± 235 | 3 (2–5) | 12700 ± 1900 | 887 ± 140 | 10100 ± 1380 | 7.97 ± 0.782 | 0.0876 ± 0.00791 | ||
| Monkey | Intravenous | 4 | F | 0.1 | 0.1665 (0.083–0.25) | 5150 ± 1530 | 33.5 ± 20.3 | 3500 ± 1380 | 104 ± 80.0 | 0.0105 ± 0.00836 | |
| Oral | 4 | F | 1 | 752 ± 148 | 5.5 (5–6) | 8940 ± 954 | 230 ± 64.6 | 17600 ± 4000 | 59.9 ± 35.5 | 0.0140 ± 0.00570 | |
| 4 | F | 5 | 4700 ± 1100 | 7 (6–8) | 29500 ± 4840 | 309 ± 77.3 | 50500 ± 16900 | 125 ± 64.6 | 0.00707 ± 0.00412 | ||
| 4 | F | 30 | 10200 ± 2390 | 8 (8–8) | 79100 ± 15600 | 999 ± 123 | 67800 ± 44900 | 46.3 ± 26.8 | 0.0194 ± 0.0104 |
| Species | CL (L/h) | Vc (L) | Q (L/h) | Vp (L) | α | β | Fmax | F50 |
|---|---|---|---|---|---|---|---|---|
| Hamster | 0.00377 (7.40%) | 0.00472 (9.43%) | 0.00617 (10.3%) | 0.0924 (9.78%) | 4.20 (4.14%) | 2.62 (4.47%) | 0.129 (11.3%) | 12.8 (16.7%) |
| Rat | 0.0326 (5.58%) | 0.0287 (6.41%) | 0.0404 (12.0%) | 0.285 (8.21%) | 3.86 (3.94%) | 2.59 (4.59%) | 0.43 (18.5%) | 5.09 (23.2%) |
| Monkey | 0.0344 (24.0%) | 0.383 (74.7%) | 0.0895 (49.9%) | 3.40 (51.5%) | 5.44 (11.8%) | 3.54 (6.50%) | 0.18 (27.4%) | 12.3 (35.4%) |
| Structures | Objective Function Value(OFV) |
|---|---|
| CL(Q) = a·BWb | 4670.56 |
| CL(Q) = a·BWb·BrWc | 5226.38 |
| CL(Q) = a·BrWb/MLP | 5263.84 |
| CL(Q) = a·BrWb/BW | 5419.25 |
| CL(Q) = a·BWb/BrW | 4608.67 |
| CL(Q) = a·BWb/MLP | 4902.96 |
| Parameter | Description | Simple Allometry | Brain Weight-Corrected | ||
|---|---|---|---|---|---|
| Estimates | Bootstrap Median (90% Confidence Interval(CI)) | Estimates | Bootstrap Median (90% Confidence Interval(CI)) | ||
| CL * = θ1 × WT θ2 (/BrW) * | |||||
| θ1 | Coefficient for CL | 0.0308 | 0.0308 (0.0259–0.0379) | 0.488 | 0.490 (0.420–0.588) |
| θ2 | Exponent for CL | 0.525 | 0.526 (0.469–0.586) | 1.90 | 1.90 (1.83–1.95) |
| Vc = θ3 × WTθ4 | |||||
| θ3 | Coefficient for Vc | 0.0755 | 0.0763 (0.0628–0.0943) | 0.0735 | 0.0756 (0.0626–0.0954) |
| θ4 | Exponent of Vc | 1 (fix) | 1 (fix) | ||
| Q = θ5 × WTθ6 (/BrW) * | |||||
| θ5 | Coefficient for Q | 0.0549 | 0.0553 (0.0392–0.0760) | 0.846 | 0.837 (0.595–1.16) |
| θ6 | Exponent of Q | 0.670 | 0.663 (0.504–0.833) | 2.04 | 2.03 (1.85–2.21) |
| Vp = θ7 × WTθ8 | |||||
| θ7 | Coefficient for Vp | 0.829 | 0.832 (0.724–0.999) | 0.813 | 0.813 (0.719–0.960) |
| θ8 | Exponent of Vp | 1 (fix) | 1 (fix) | ||
| WB = (β/α) × (t/α) β − 1 | |||||
| α | Scale parameter of WB | 4.50 | 4.50 (4.21–4.80) | 4.51 | 4.49 (4.21–4.80) |
| β | Shape parameter of WB | 3.01 | 3.10 (2.89–3.32) | 3.11 | 3.10 (2.79–3.38) |
| F = Fmax × (1 − dose/(F50 + dose)) | |||||
| Fmax | Maximum bioavailability | 0.183 | 0.182 (0.151–0.219) | 0.192 | 0.192 (0.159–0.229) |
| F50 | Amount of dose reaching 50% of the maximum bioavailability | 11.9 | 12.4 (9.55–16.2) | 10.6 | 10.8 (8.89–14.8) |
| ωCL (%) | Interindividual variability of CL | 42.8 | 42.2 (35.9–47.4) | 25.9 | 25.2 (19.8–31.4) |
| ωQ (%) | Interindividual variability of Q | 23.9 | 23.5 (19.5–26.6) | 60.8 | 0.579 (0.357–0.727) |
| ωα (%) | Interindividual variability of α | 27.1 | 26.5 (21.3–31.8) | 27.0 | 23.6 (8.40–27.6) |
| ωβ (%) | Interindividual variability of β | 65.7 | 62.4 (47.3–76.3) | 24.0 | 26.2 (3.00–34.1) |
| Allometry | CL (L/h) | Vc (L) | Q (L/h) | Vp (L) | α | β | Fmax | F50 |
|---|---|---|---|---|---|---|---|---|
| Simple | 0.264 | 3.29 | 1.17 | 49.7 | 4.51 | 3.11 | 0.192 | 10.6 |
| Brain weight-corrected | 0.777 | 4.41 | 2.39 | 48.8 |
| Dose (mg) | Absolute Bioavailability |
|---|---|
| 25 | 0.0362 |
| 50 | 0.0321 |
| 100 | 0.0191 |
| 200 | 0.0140 |
| 400 | 0.00814 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.; Han, S.; Jeon, S.; Yim, D.-S. Quantitative Prediction of Human Pharmacokinetics and Pharmacodynamics of CKD519, a Potent Inhibitor of Cholesteryl Ester Transfer Protein (CETP). Pharmaceutics 2019, 11, 336. https://doi.org/10.3390/pharmaceutics11070336
Choi S, Han S, Jeon S, Yim D-S. Quantitative Prediction of Human Pharmacokinetics and Pharmacodynamics of CKD519, a Potent Inhibitor of Cholesteryl Ester Transfer Protein (CETP). Pharmaceutics. 2019; 11(7):336. https://doi.org/10.3390/pharmaceutics11070336
Chicago/Turabian StyleChoi, Suein, Seunghoon Han, Sangil Jeon, and Dong-Seok Yim. 2019. "Quantitative Prediction of Human Pharmacokinetics and Pharmacodynamics of CKD519, a Potent Inhibitor of Cholesteryl Ester Transfer Protein (CETP)" Pharmaceutics 11, no. 7: 336. https://doi.org/10.3390/pharmaceutics11070336
APA StyleChoi, S., Han, S., Jeon, S., & Yim, D.-S. (2019). Quantitative Prediction of Human Pharmacokinetics and Pharmacodynamics of CKD519, a Potent Inhibitor of Cholesteryl Ester Transfer Protein (CETP). Pharmaceutics, 11(7), 336. https://doi.org/10.3390/pharmaceutics11070336