Dynamics of Pathological and Virological Findings During Experimental Calpox Virus Infection of Common Marmosets (Callithrix jacchus)


 ,
,
Abstract
1. Introduction
2. Material and Methods
2.1. Experimental Animals
2.2. Virus, Infection and Necropsy
2.3. Microscopy
2.4. Histologic and Immunohistochemistry Scoring
2.5. Microbiological Examinations
2.6. Isolation of Calpox Virus DNA from Tissues
2.7. Isolation of Calpox Virus DNA from Blood
2.8. Analysis of Viral Load by Real-Time PCR
2.9. Quantification of Infectious Virus by Endpoint Dilution Assay
3. Results
3.1. Clinical Findings
3.2. Gross Necropsy Findings
3.3. Histopathology and Immunohistochemistry
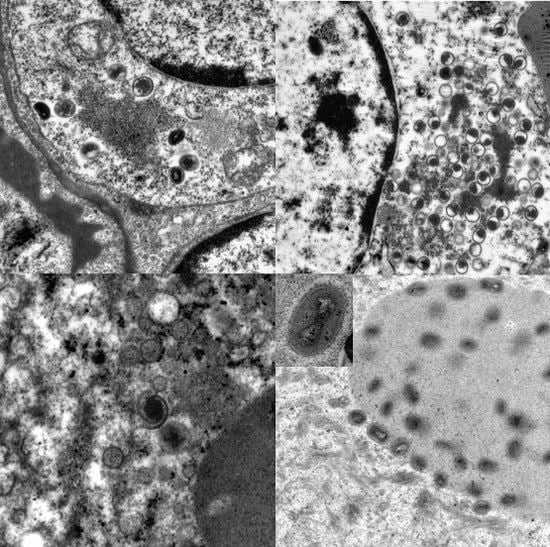
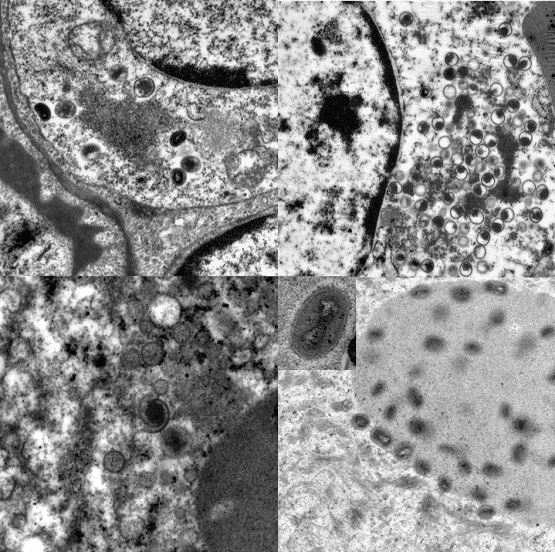
3.4. Transmission Electron Microscopy
3.5. Microbiological Examination
3.6. Calpox Virus Load in Different Tissues
3.7. Calpox Viral Load in Blood
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fenner, F. A successful eradication campaign. Global eradication of smallpox. Rev. Infect. Dis. 1982, 4, 916–930. [Google Scholar] [CrossRef] [PubMed]
- Breman, J.G.; Henderson, D.A. Poxvirus dilemmas-monkeypox, smallpox, and biologic terrorism. N. Engl. J. Med. 1998, 339, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.D.; Bokor, G. Bioterrorism: Pathogens as weapons. J. Pharm. Pract. 2012, 25, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Campe, H.; Zimmermann, P.; Glos, K.; Bayer, M.; Bergemann, H.; Dreweck, C.; Graf, P.; Weber, B.K.; Meyer, H.; Buttner, M.; et al. Cowpox virus transmission from pet rats to humans, Germany. Emerg. Infect. Dis. 2009, 15, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov, S.N. An increasing danger of zoonotic orthopoxvirus infections. PLoS Pathog. 2013, 9, e1003756. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.D.; Melski, J.W.; Graham, M.B.; Regnery, R.L.; Sotir, M.J.; Wegner, M.V.; Kazmierczak, J.J.; Stratman, E.J.; Li, Y.; Fairley, J.A.; et al. The detection of monkeypox in humans in the Western Hemisphere. N. Engl. J. Med. 2004, 350, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, P.W.; Radonic, A.; Kurth, A.; Nitsche, A. Genome-wide comparison of cowpox viruses reveals a new clade related to variola virus. PLoS ONE 2013, 8, e79953. [Google Scholar] [CrossRef] [PubMed]
- Rimoin, A.W.; Mulembakani, P.M.; Johnston, S.C.; Lloyd Smith, J.O.; Kisalu, N.K.; Kinkela, T.L.; Blumberg, S.; Thomassen, H.A.; Pike, B.L.; Fair, J.N.; et al. Major increase in human monkeypox incidence 30 years after smallpox vaccination campaigns cease in the Democratic Republic of Congo. Proc. Natl. Acad. Sci. USA 2010, 107, 16262–16267. [Google Scholar] [CrossRef] [PubMed]
- Karem, K.L.; Reynolds, M.; Hughes, C.; Braden, Z.; Nigam, P.; Crotty, S.; Glidewell, J.; Ahmed, R.; Amara, R.; Damon, I.K. Monkeypox-induced immunity and failure of childhood smallpox vaccination to provide complete protection. Clin. Vaccine Immunol. 2007, 14, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Update: Multistate outbreak of monkeypox--Illinois, Indiana, Kansas, Missouri, Ohio, and Wisconsin, 2003. Morb. Mortal. Wkly. Rep. 2003, 52, 642–646. [Google Scholar]
- Wolfs, T.F.; Wagenaar, J.A.; Niesters, H.G.; Osterhaus, A.D. Rat-to-human transmission of Cowpox infection. Emerg. Infect. Dis. 2002, 8, 1495–1496. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Kurth, A.; Hessler, F.; Kramp, H.; Gokel, M.; Hoffmann, R.; Kuczka, A.; Nitsche, A. Cowpox virus infection in pet rat owners: Not always immediately recognized. Dtsch. Arzteblatt Int. 2009, 106, 329–334. [Google Scholar]
- Baxby, D.; Bennett, M.; Getty, B. Human cowpox 1969–1993: A review based on 54 cases. Br. J. Dermatol. 1994, 131, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Kurth, A.; Wibbelt, G.; Gerber, H.P.; Petschaelis, A.; Pauli, G.; Nitsche, A. Rat-to-elephant-to-human transmission of cowpox virus. Emerg. Infect. Dis. 2008, 14, 670–671. [Google Scholar] [CrossRef] [PubMed]
- Eis-Hübinger, A.M.; Gerritzen, A.; Schneweis, K.E.; Pfeiff, B.; Pullmann, H.; Mayr, A.; Czerny, C.P. Fatal cowpox-like virus infection transmitted by cat. Lancet 1990, 336, 880. [Google Scholar] [CrossRef]
- Czerny, C.P.; Eis-Hübinger, A.M.; Mayr, A.; Schneweis, K.E.; Pfeiff, B. Animal poxviruses transmitted from cat to man: Current event with lethal end. Zent. Vet. B 1991, 38, 421–431. [Google Scholar] [CrossRef]
- Vorou, R.M.; Papavassiliou, V.G.; Pierroutsakos, I.N. Cowpox virus infection: An emerging health threat. Curr. Opin. Infect. Dis. 2008, 21, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Blackford, S.; Roberts, D.L.; Thomas, P.D. Cowpox infection causing a generalized eruption in a patient with atopic dermatitis. Br. J. Dermatol. 1993, 129, 628–629. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D.S.; Emerson, G.L.; Li, Y.; Sammons, S.; Olson, V.; Frace, M.; Nakazawa, Y.; Czerny, C.P.; Tryland, M.; Kolodziejek, J.; et al. Chasing Jenner’s vaccine: Revisiting cowpox virus classification. PLoS ONE 2011, 6, e23086. [Google Scholar] [CrossRef] [PubMed]
- Ninove, L.; Domart, Y.; Vervel, C.; Voinot, C.; Salez, N.; Raoult, D.; Meyer, H.; Capek, I.; Zandotti, C.; Charrel, R.N. Cowpox virus transmission from pet rats to humans, France. Emerg. Infect. Dis. 2009, 15, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.; El-Gharib, I.; Rasehd, A.; Farag, F.; Emara, M. Human cowpox infection in Sharkia Governorate. Egypt. Int. J. Dermatol. 2001, 40, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Yeruham, I.; Nyska, A.; Abraham, A.; Algazi, R. Occurrence of cowpox-like lesions in cattle in Israel. Rev. Elev. Med. Vet. Pays. Trop. 1996, 49, 299–302. [Google Scholar] [PubMed]
- Essbauer, S.; Pfeffer, M.; Meyer, H. Zoonotic poxviruses. Vet. Microbiol. 2010, 140, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Baxby, D. Cowpox. J. Med. Microbiol. 1996, 45, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Wisser, J.; Pilaski, J.; Strauss, G.; Meyer, H.; Burck, G.; Truyen, U.; Rudolph, M.; Frölich, K. Cowpox virus infection causing stillbirth in an Asian elephant (Elephas maximus). Vet. Rec. 2001, 149, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Kalthoff, D.; Bock, W.I.; Huhn, F.; Beer, M.; Hoffmann, B. Fatal cowpox virus infection in cotton-top tamarins (Saguinus oedipus) in Germany. Vector Borne Zoonotic Dis. 2014, 14, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, P.M.; Henttonen, H.; Hoffmann, B.; Kallio, E.R.; Korthase, C.; Laakkonen, J.; Niemimaa, J.; Palva, A.; Schlegel, M.; Ali, H.S.; et al. Orthopox virus infections in Eurasian wild rodents. Vector Borne Zoonotic Dis. 2011, 11, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Begon, M.; Hazel, S.M.; Baxby, D.; Bown, K.; Cavanagh, R.; Chantrey, J.; Jones, T.; Bennett, M. Transmission dynamics of a zoonotic pathogen within and between wildlife host species. Proc. Biol. Sci. R. Soc. 1999, 266, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Crouch, A.C.; Baxby, D.; McCracken, C.M.; Gaskell, R.M.; Bennett, M. Serological evidence for the reservoir hosts of cowpox virus in British wildlife. Epidemiol. Infect. 1995, 115, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Hutson, C.L.; Damon, I.K. Monkeypox virus infections in small animal models for evaluation of anti-poxvirus agents. Viruses 2010, 2, 2763–2776. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.L.; Nichols, D.K.; Martinez, M.J.; Raymond, J.W. Animal models of orthopoxvirus infection. Vet. Pathol. 2010, 47, 852–870. [Google Scholar] [CrossRef] [PubMed]
- Smee, D.F. Orthopoxvirus inhibitors that are active in animal models: An update from 2008 to 2012. Future Virol. 2013, 8, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Cann, J.A.; Jahrling, P.B.; Hensley, L.E.; Wahl-Jensen, V. Comparative pathology of smallpox and monkeypox in man and macaques. J. Comp. Pathol. 2013, 148, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Mätz-Rensing, K.; Kaup, F.-J. Non-human primate models of orthopoxvirus infections. Vet. Sci. 2014, 1, 40–62. [Google Scholar] [CrossRef]
- Nalca, A.; Livingston, V.A.; Garza, N.L.; Zumbrun, E.E.; Frick, O.M.; Chapman, J.L.; Hartings, J.M. Experimental infection of cynomolgus macaques (Macaca fascicularis) with aerosolized monkeypox virus. PLoS ONE 2010, 5, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Safronetz, D.; Geisbert, T.W.; Feldmann, H. Animal models for highly pathogenic emerging viruses. Curr. Opin. Virol. 2013, 3, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, R.S. Should remaining stockpiles of smallpox virus (variola) be destroyed? Emerg. Infect. Dis. 2011, 17, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.M.; Poland, G.A. Why not destroy the remaining smallpox virus stocks? Vaccine 2011, 29, 2823–2824. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.B. Breaking the deadlock over destruction of the smallpox virus stocks. Biosecur. Bioterror. 2011, 9, 55–67. [Google Scholar] [CrossRef] [PubMed]
- McFadden, G. Killing a killer: What next for smallpox? PLoS Pathog. 2010, 6, e1000727. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S. "Forgotten" NIH smallpox virus languishes on death row. Nature 2014, 514, 544. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.F.; Dyall, J.; Ragland, D.R.; Huzella, L.; Byrum, R.; Jett, C.; St Claire, M.; Smith, A.L.; Paragas, J.; Blaney, J.E.; et al. Comparative analysis of monkeypox virus infection of cynomolgus macaques by the intravenous or intrabronchial inoculation route. J. Virol. 2011, 85, 2112–2125. [Google Scholar] [CrossRef] [PubMed]
- Seet, B.T.; Johnston, J.B.; Brunetti, C.R.; Barrett, J.W.; Everett, H.; Cameron, C.; Sypula, J.; Nazarian, S.H.; Lucas, A.; McFadden, G. Poxviruses and immune evasion. Ann. Rev. Immunol. 2003, 21, 377–423. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; St Claire, M.; Yellayi, S.; Bollinger, L.; Jahrling, P.B.; Paragas, J.; Blaney, J.E.; Johnson, R.F. Intrabronchial inoculation of cynomolgus macaques with cowpox virus. J. Gen. Virol. 2012, 93, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Kramski, M.; Mätz-Rensing, K.; Stahl-Hennig, C.; Kaup, F.J.; Nitsche, A.; Pauli, G.; Ellerbrok, H. A novel highly reproducible and lethal nonhuman primate model for orthopox virus infection. PLoS ONE 2010, 5, e10412. [Google Scholar] [CrossRef] [PubMed]
- Mätz-Rensing, K.; Ellerbrok, H.; Ehlers, B.; Pauli, G.; Floto, A.; Alex, M.; Czerny, C.P.; Kaup, F.J. Fatal poxvirus outbreak in a colony of New World monkeys. Vet. Pathol. 2006, 43, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Pilaski, J.; Rösen, A.; Darai, G. Comparative analysis of the genomes of orthopoxviruses isolated from elephant, rhinoceros, and okapi by restriction enzymes. Arch Virol. 1986, 88, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Mätz-Rensing, K.; Stahl-Hennig, C.; Kramski, M.; Pauli, G.; Ellerbrok, H.; Kaup, F.J. The pathology of experimental poxvirus infection in common marmosets (Callithrix jacchus): Further characterization of a new primate model for orthopoxvirus infections. J. Comp. Pathol. 2012, 146, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.F.; Yellayi, S.; Cann, J.A.; Johnson, A.; Smith, A.L.; Paragas, J.; Jahrling, P.B.; Blaney, J.E. Cowpox virus infection of cynomolgus macaques as a model of hemorrhagic smallpox. Virology 2011, 418, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Fenner, F.; Henderson, D.A.; Arita, I.; Jezek, Z.; Ladnyi, I.D. Pathogenesis, pathology and immunology of smallpox. In Smallpox and Its Eradication; WHO: Geneva, Switzerland, 1988; pp. 121–168. [Google Scholar]
- Barnewall, R.E.; Fisher, D.A.; Robertson, A.B.; Vales, P.A.; Knostman, K.A.; Bigger, J.E. Inhalational monkeypox virus infection in cynomolgus macaques. Front. Cell. Infect. Microbiol. 2012, 2, 117. [Google Scholar] [CrossRef] [PubMed]
- Bienenstock, J.; McDermott, M.R. Bronchus- and nasal-associated lymphoid tissues. Immunol. Rev. 2005, 206, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yuen, P.W.; Lam, J.K. Intranasal DNA vaccine for protection against respiratory infectious diseases: The delivery perspectives. Pharmaceutics 2014, 6, 378–415. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.; Martinez, M.; Smee, D.F.; Kefauver, D.; Thompson, E.; Huggins, J.W. Cidofovir protects mice against lethal aerosol or intranasal cowpox virus challenge. J. Infect. Dis. 2000, 181, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A. Histopathogenesis of mousepox. I. Respiratory infection. Br. J. Exp. Pathol. 1962, 43, 451–461. [Google Scholar] [PubMed]
- Breithaupt, A.; Kalthoff, D.; Deutskens, F.; König, P.; Hoffmann, B.; Beer, M.; Meyer, H.; Teifke, J.P. Clinical course and pathology in rats (Rattus norvegicus) after experimental cowpox virus infection by percutaneous and intranasal application. Vet. Pathzol. 2012, 49, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, U.; van de Poel, H.; van den Ingh, T.S. Necrotizing pneumonia in a cat caused by an orthopox virus. J. Comp. Pathol. 1999, 121, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.C.; Boulter, E.A.; Westwood, J.C.; Randles, J. Experimental respiratory infection with poxviruses. II. Pathological studies. Br. J. Exp. Pathol. 1966, 47, 466–471. [Google Scholar] [PubMed]
- Schöniger, S.; Chan, D.L.; Hollinshead, M.; Humm, K.; Smith, G.L.; Beard, P.M. Cowpox virus pneumonia in a domestic cat in Great Britain. Vet. Rec. 2007, 160, 522–523. [Google Scholar] [CrossRef] [PubMed]
- Westwood, J.C.; Boulter, E.A.; Bowen, E.T.; Maber, H.B. Experimental respiratory infection with poxviruses. I. Clinical virological and epidemiological studies. Br. J. Exp. Pathol. 1966, 47, 453–465. [Google Scholar] [PubMed]
- Councilman, W.; Magrath, G.; WR, B. The pathological anatomy and histology of variola. J. Med. Res. 1904, 11, 12–132. [Google Scholar] [PubMed]
- Martinez, M.J.; Bray, M.P.; Huggins, J.W. A mouse model of aerosol-transmitted orthopoxviral disease: Morphology of experimental aerosol-transmitted orthopoxviral disease in a cowpox virus-BALB/c mouse system. Arch. Pathol. Lab. Med. 2000, 124, 362–377. [Google Scholar] [PubMed]
- Bowden, T.R.; Babiuk, S.L.; Parkyn, G.R.; Copps, J.S.; Boyle, D.B. Capripoxvirus tissue tropism and shedding: A quantitative study in experimentally infected sheep and goats. Virology 2008, 371, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Embury-Hyatt, C.; Babiuk, S.; Manning, L.; Ganske, S.; Bowden, T.R.; Boyle, D.B.; Copps, J. Pathology and viral antigen distribution following experimental infection of sheep and goats with capripoxvirus. J. Comp. Pathol. 2012, 146, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Pauli, G.; Blumel, J.; Burger, R.; Drosten, C.; Groner, A.; Gurtler, L.; Heiden, M.; Hildebrandt, M.; Jansen, B.; Montag-Lessing, T.; et al. Orthopox viruses: Infections in humans. Transfus. Med. Hemother. 2010, 37, 351–364. [Google Scholar] [PubMed]
- Zaucha, G.M.; Jahrling, P.B.; Geisbert, T.W.; Swearengen, J.R.; Hensley, L. The pathology of experimental aerosolized monkeypox virus infection in cynomolgus monkeys (Macaca fascicularis). Lab. Investig. 2001, 81, 1581–1600. [Google Scholar] [CrossRef] [PubMed]
- Wahl-Jensen, V.; Cann, J.A.; Rubins, K.H.; Huggins, J.W.; Fisher, R.W.; Johnson, A.J.; de Kok-Mercado, F.; Larsen, T.; Raymond, J.L.; Hensley, L.E.; et al. Progression of pathogenic events in cynomolgus macaques infected with variola virus. PLoS ONE 2011, 6, e24832. [Google Scholar] [CrossRef] [PubMed]
- Nagata, N.; Saijo, M.; Kataoka, M.; Ami, Y.; Suzaki, Y.; Sato, Y.; Iwata-Yoshikawa, N.; Ogata, M.; Kurane, I.; Morikawa, S.; et al. Pathogenesis of fulminant monkeypox with bacterial sepsis after experimental infection with West African monkeypox virus in a cynomolgus monkey. Int. J. Clin. Exp. Pathol. 2014, 7, 4359–4370. [Google Scholar] [PubMed]
- Nash, P.; Barrett, J.; Cao, J.X.; Hota-Mitchell, S.; Lalani, A.S.; Everett, H.; Xu, X.M.; Robichaud, J.; Hnatiuk, S.; Ainslie, C.; et al. Immunomodulation by viruses: The myxoma virus story. Immunol. Rev. 1999, 168, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.J.; Rushton, J.; Dekonenko, A.; Chand, H.S.; Olson, G.K.; Hutt, J.A.; Pickup, D.; Lyons, C.R.; Lipscomb, M.F. Cowpox virus inhibits human dendritic cell immune function by nonlethal, nonproductive infection. Virology 2011, 412, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; McFadden, G. Technical knockout: Understanding poxvirus pathogenesis by selectively deleting viral immunomodulatory genes. Cell. Microbiol. 2004, 6, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.P.; Turner, P.C.; Ali, A.N.; Crenshaw, B.C.; Moyer, R.W. The effects of serpin gene mutations on the distinctive pathobiology of cowpox and rabbitpox virus following intranasal inoculation of Balb/c mice. Virology 1993, 197, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Pickup, D.J.; Ink, B.S.; Hu, W.; Ray, C.A.; Joklik, W.K. Hemorrhage in lesions caused by cowpox virus is induced by a viral protein that is related to plasma protein inhibitors of serine proteases. Proc. Natl. Acad. Sci. USA 1986, 83, 7698–7702. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Veldhuis Kroeze, E.J.; Reperant, L.A.; Richard, M.; Kuiken, T. Influenza virus and endothelial cells: A species specific relationship. Front. Microbiol. 2014, 5, 653. [Google Scholar] [CrossRef] [PubMed]
- Jahrling, P.B.; Hensley, L.E.; Martinez, M.J.; Leduc, J.W.; Rubins, K.H.; Relman, D.A.; Huggins, J.W. Exploring the potential of variola virus infection of cynomolgus macaques as a model for human smallpox. Proc. Natl. Acad. Sci. USA 2004, 101, 15196–15200. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Puig, J.M.; Sanchez, L.; Roy, G.; Blasco, R. Susceptibility of different leukocyte cell types to Vaccinia virus infection. Virol. J. 2004, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Breman, J.G.; Henderson, D.A. Diagnosis and management of smallpox. N. Engl. J. Med. 2002, 346, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Schriewer, J.; Buller, R.M.; Owens, G. Mouse models for studying orthopoxvirus respiratory infections. Methods Mol. Biol. 2004, 269, 289–308. [Google Scholar] [PubMed]
- Fenner, F.; Buller, R.M. Mousepox. In Viral Pathogenesis; Nathansan, N., Ed.; Lippincott-Raven: Philadelphia, PA, USA, 1997; pp. 535–553. [Google Scholar]
- Smee, D.F.; Bailey, K.W.; Wong, M.H.; Sidwell, R.W. Effects of cidofovir on the pathogenesis of a lethal vaccinia virus respiratory infection in mice. Antivir. Res. 2001, 52, 55–62. [Google Scholar] [CrossRef]
- Williamson, J.D.; Reith, R.W.; Jeffrey, L.J.; Arrand, J.R.; Mackett, M. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J. Gen. Virol. 1990, 71, 2761–2767. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal No. a | Age (Years) | Gender | Time of Death (dpi) b |
|---|---|---|---|
| 1 | 5 | m | 3 |
| 2 | 6 | m | 3 |
| 3 | 6 | m | 3 |
| 4 | 3 | f | 5 |
| 5 | 4 | f | 5 |
| 6 | 4 | m | 5 |
| 7 | 3 | m | 7 |
| 8 | 4 | f | 7 |
| 9 | 3 | f | 7 |
| 10 | 4 | f | 10 |
| 11 | 2 | f | 10 |
| 12 | 3 | f | 10 |
| 13 | 5 | m | 12 |
| 14 | 5 | m | 12 |
| 15 | 5 | m | 12 |
| 16 | 5 | f | 12 |
| 17 | 5 | f | 12 |
| 18 | 5 | m | 13 |
| 19 | 5 | m | 14 |
| 20 | 5 | m | 14 |
| 21 | 4 | f | 16 |
| dpi | 3 a | 5 | 7 | 10 | 12 | 12–16 |
|---|---|---|---|---|---|---|
| N | 3 | 3 | 3 | 3 | 3 | 6 |
| Gross lesion | ||||||
| Splenic hyperplasia | 2 | 2 | 1 | 3 | 3 | 2 |
| Hyperplasia of submandibular lymph node | 1 | 1 | 0 | 2 | 3 | 6 |
| Cutaneous pox lesions | 0 | 0 | 0 | 0 | 2 | 6 |
| Hemorrhagic pneumonia | 0 | 0 | 0 | 0 | 1 | 2 |
| Mucosal petechiae on urinary bladder | 0 | 0 | 0 | 0 | 1 | 3 |
| Histological lesion | ||||||
| Hyperplasia of nasal ciliated epithelium | 2 | 1/2 b | 1/1 b | 1/2 b | 2/2 b | 3/3 b |
| Lung Hyperplasia of bronchial epithelium | 1 | 0 | 0 | 0 | 2 | - |
| interstitial pneumonia | 0 | 0 | 0 | 0 | 1 | 4 |
| Spleen Follicular hyperplasia | 1 | 2 | 0 | 3 | 1 | - |
| Necrotizing splenitis with depletion | 0 | 0 | 0 | 0 | 2 | 6 |
| Submandibular lymph node Follicular hyperplasia | 3 | 3 | 1/1 b | 3 | - | 1/4 b |
| lymphadenitis with depletion | - | - | - | - | 3 | 3/4 b |
| Vesicular ulcerative dermatitis | 0 | 0 | 0 | 0 | 2 | 6 |
| Hemorrhagic/edematous cystitis | 0 | 0 | 0 | 1 | 1 | 3/5 b |
| Liver Hepatocellular degeneration | 2 | 3 | 3 | 3 | - | - |
| Necrotizing hepatitis | - | - | - | - | 2 | 5 |
| dpi | Animal | Nasal Epithelium | Buccal Mucosa | Subm. LN | Trachea | Lung | Spleen | Liver | Urinary Bladder | Skin |
|---|---|---|---|---|---|---|---|---|---|---|
| HE/IHC | HE/IHC | HE/IHC | HE/IHC | HE/IHC | HE/IHC | HE/IHC | HE/IHC | HE/IHC | ||
| 3 | 1 | 0/− | 0/− | 1/− | 0/− | 1/+ | 0/− | 1/− | 0/− | 2/− |
| 3 | 2 | 1/(+) | 0/− | 1/− | 1/+ | 1/+ | 0/− | 0/− | 0/− | 0/− |
| 3 | 3 | 2/++ | 0/− | 1/− | 0/− | 1/+ | 1/− | 1/− | 0/− | 0/− |
| 5 | 4 | 0/− | 0/− | 1/− | 0/− | 0/− | 1/− | 1/− | 0/− | 0/− |
| 5 | 5 | na | 1/− | 1/− | 0/− | 1/− | 1/− | 1/− | 1/− | 0/− |
| 5 | 6 | 1/(+) | 0/− | 1/− | 0/− | 1/− | 0/− | 1/− | 0/− | 0/− |
| 7 | 7 | na | 0/− | na | 0/− | 0/− | 0/− | 1/− | 0/− | 0/− |
| 7 | 8 | 1/+ | 0/− | 1/+ | 0/− | 0/− | 0/+ | 1/− | 1/− | 0/− |
| 7 | 9 | na | 0/− | na | 0/− | 1/+ | 0/− | 1/− | 0/− | 0/− |
| 10 | 10 | 1/− | 0/− | 1/− | 1/− | 1/+ | 1/− | 2/− | 2/− | 0/− |
| 10 | 11 | na | 1/− | 1/+ | 2/− | 1/− | 1/+ | 1/+ | 0/− | 0/− |
| 10 | 12 | 0/− | 0/− | 1/++ | 0/− | 1/− | 1/− | 1/− | 0/− | 0/− |
| 12 | 13 | 1/− | 0/− | 2/(+) | 0/− | 1/+ | 1/+ | 2/+ | 1/− | 3/+++ |
| 12 | 14 | na | 0/− | 3/++ | 0/− | 1/+ | 2/+ | 1/− | 0/− | 0/− |
| 12 | 15 | 3/+++ | 2/++ | 2/++ | 0/− | 2/++ | 2/+++ | 3/++ | 2/++ | 3/++ |
| 12 | 16 | 2/+ | 3/++ | 3/++ | 0/− | 2/(+) | 2/+ | 2/+++ | 3/+ | 3/++ |
| 12 | 17 | na | 1/+ | 2/++ | 0/− | 1/+ | 3/+ | 2/++ | 0/− | 3/++ |
| 13 | 18 | 1/+ | 1/+ | na | 2/++ | 1/++ | 3/+++ | 2/++ | 0/(+) | 3/++ |
| 14 | 19 | na | 3/++ | 3/+++ | 0/− | 0/(+) | 3/+++ | 1/+ | 0/− | 3/+++ |
| 14 | 20 | 2/++ | 0/− | na | 0/− | 2/++ | 1/(+) | 2/− | na | 3/+++ |
| 16 | 21 | na | 3/+++ | 2/+++ | 0/− | 1/+ | 3/+++ | 2/+++ | 1/− | 3/+++ |
| Animal # | Necropsy dpi * | Calpox DNA Copies/µL Blood on Day of Necropsy |
|---|---|---|
| 1 | 3 | neg |
| 2 | 3 | neg |
| 3 | 3 | 115 |
| 4 | 5 | neg |
| 5 | 5 | neg |
| 6 | 5 | neg |
| 7 | 7 | neg |
| 8 | 7 | 14 |
| 9 | 7 | 55 |
| 10 | 10 | neg |
| 11 | 10 | 1.0 × 103 |
| 12 | 10 | 83 |
| 13 | 12 | 3.6 × 105 |
| 14 | 12 | 4.5 × 104 |
| 15 | 12 | 8.4 × 105 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitt, A.; Gan, L.L.; Abd El Wahed, A.; Shi, T.; Ellerbrok, H.; Kaup, F.-J.; Stahl-Hennig, C.; Mätz-Rensing, K. Dynamics of Pathological and Virological Findings During Experimental Calpox Virus Infection of Common Marmosets (Callithrix jacchus). Viruses 2017, 9, 363. https://doi.org/10.3390/v9120363
Schmitt A, Gan LL, Abd El Wahed A, Shi T, Ellerbrok H, Kaup F-J, Stahl-Hennig C, Mätz-Rensing K. Dynamics of Pathological and Virological Findings During Experimental Calpox Virus Infection of Common Marmosets (Callithrix jacchus). Viruses. 2017; 9(12):363. https://doi.org/10.3390/v9120363
Chicago/Turabian StyleSchmitt, Anne, Li Lin Gan, Ahmed Abd El Wahed, Tingchuan Shi, Heinz Ellerbrok, Franz-Josef Kaup, Christiane Stahl-Hennig, and Kerstin Mätz-Rensing. 2017. "Dynamics of Pathological and Virological Findings During Experimental Calpox Virus Infection of Common Marmosets (Callithrix jacchus)" Viruses 9, no. 12: 363. https://doi.org/10.3390/v9120363
APA StyleSchmitt, A., Gan, L. L., Abd El Wahed, A., Shi, T., Ellerbrok, H., Kaup, F.-J., Stahl-Hennig, C., & Mätz-Rensing, K. (2017). Dynamics of Pathological and Virological Findings During Experimental Calpox Virus Infection of Common Marmosets (Callithrix jacchus). Viruses, 9(12), 363. https://doi.org/10.3390/v9120363