
Immunostimulatory Gene Therapy Using Oncolytic Viruses as Vehicles

{kind=link}
{kind=link}
Abstract
:1. Cancer Immunotherapy
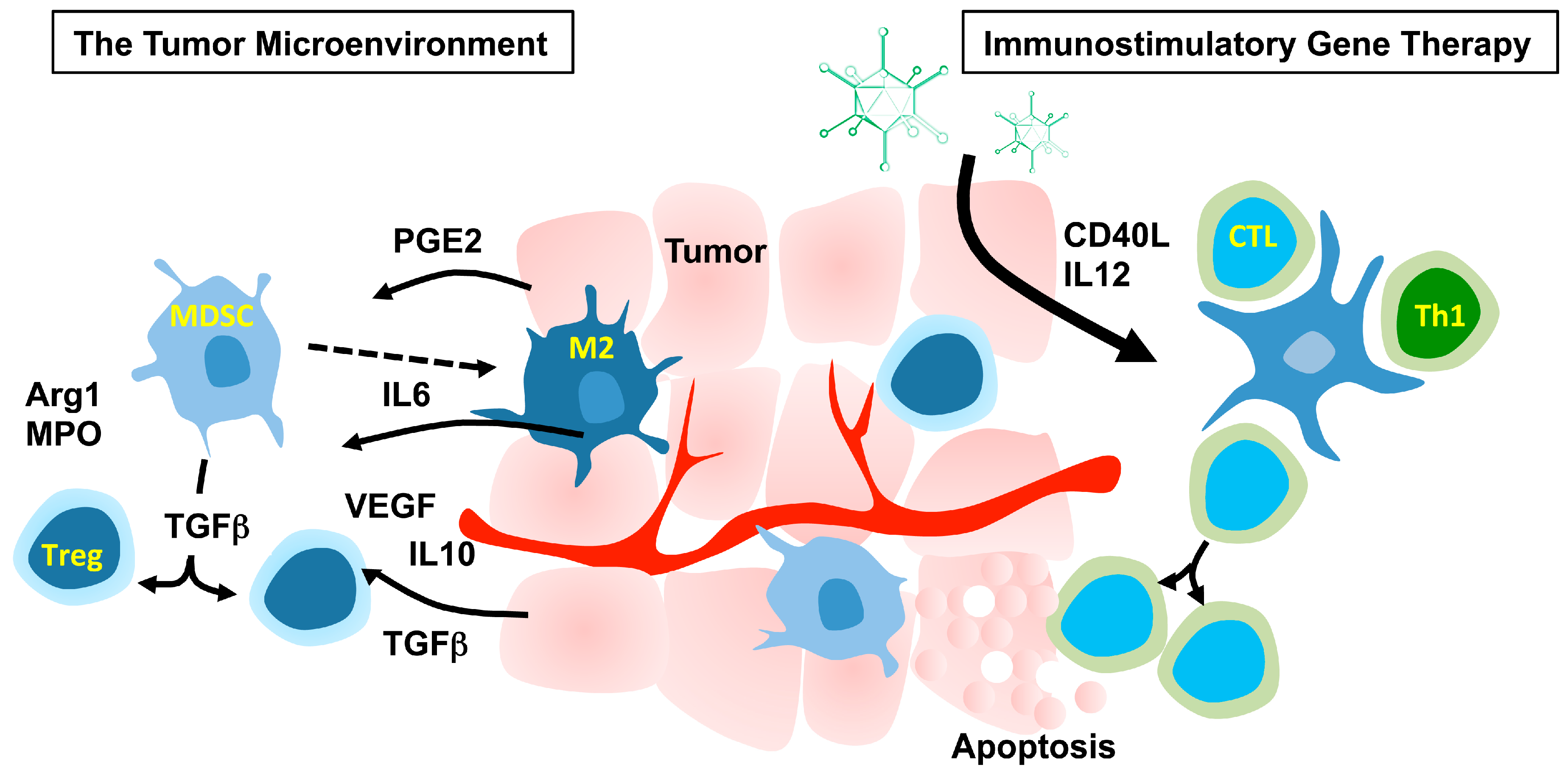
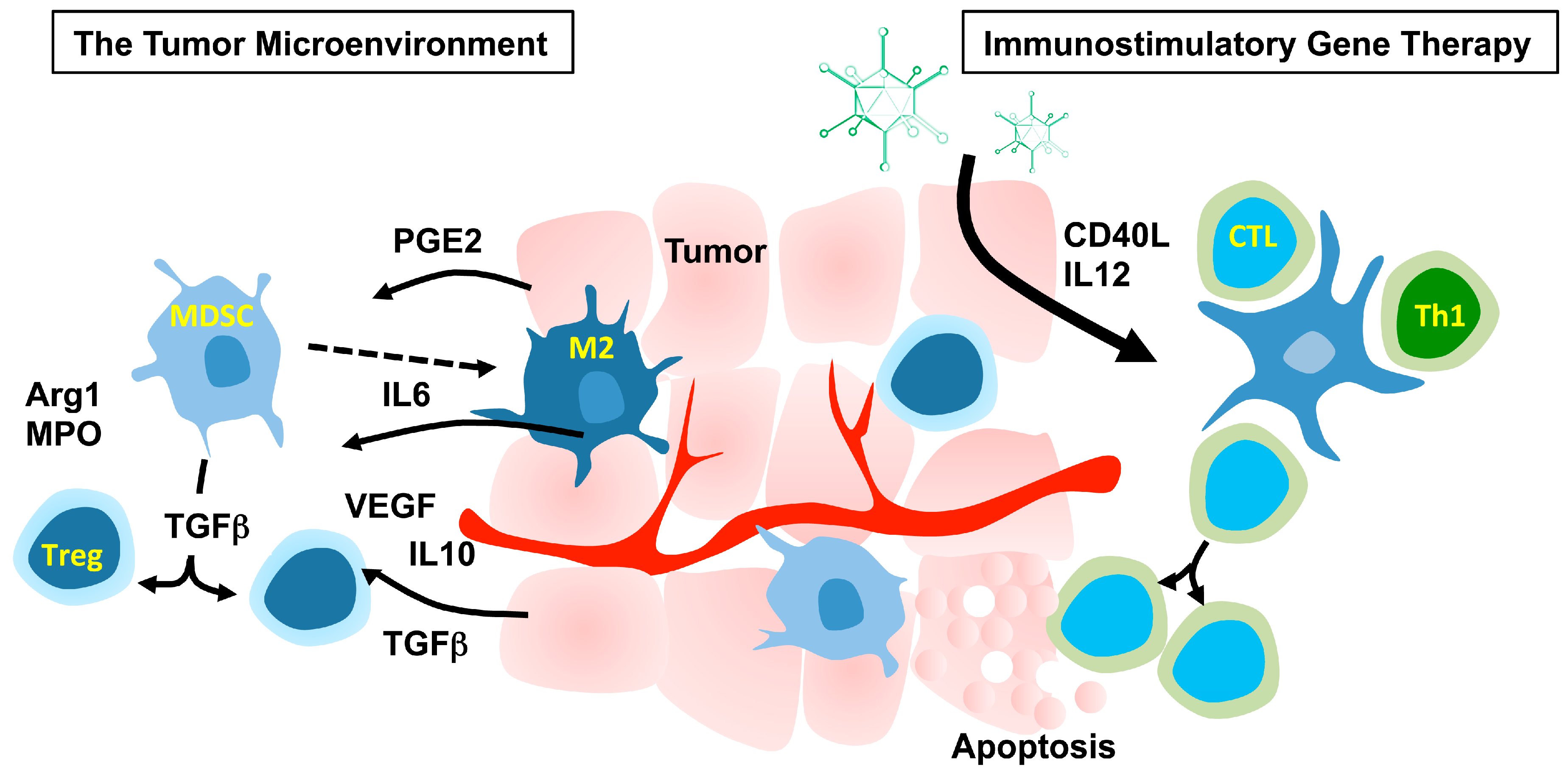
2. Immunostimulatory Gene Therapy
2.1. The Tumor Microenvironment
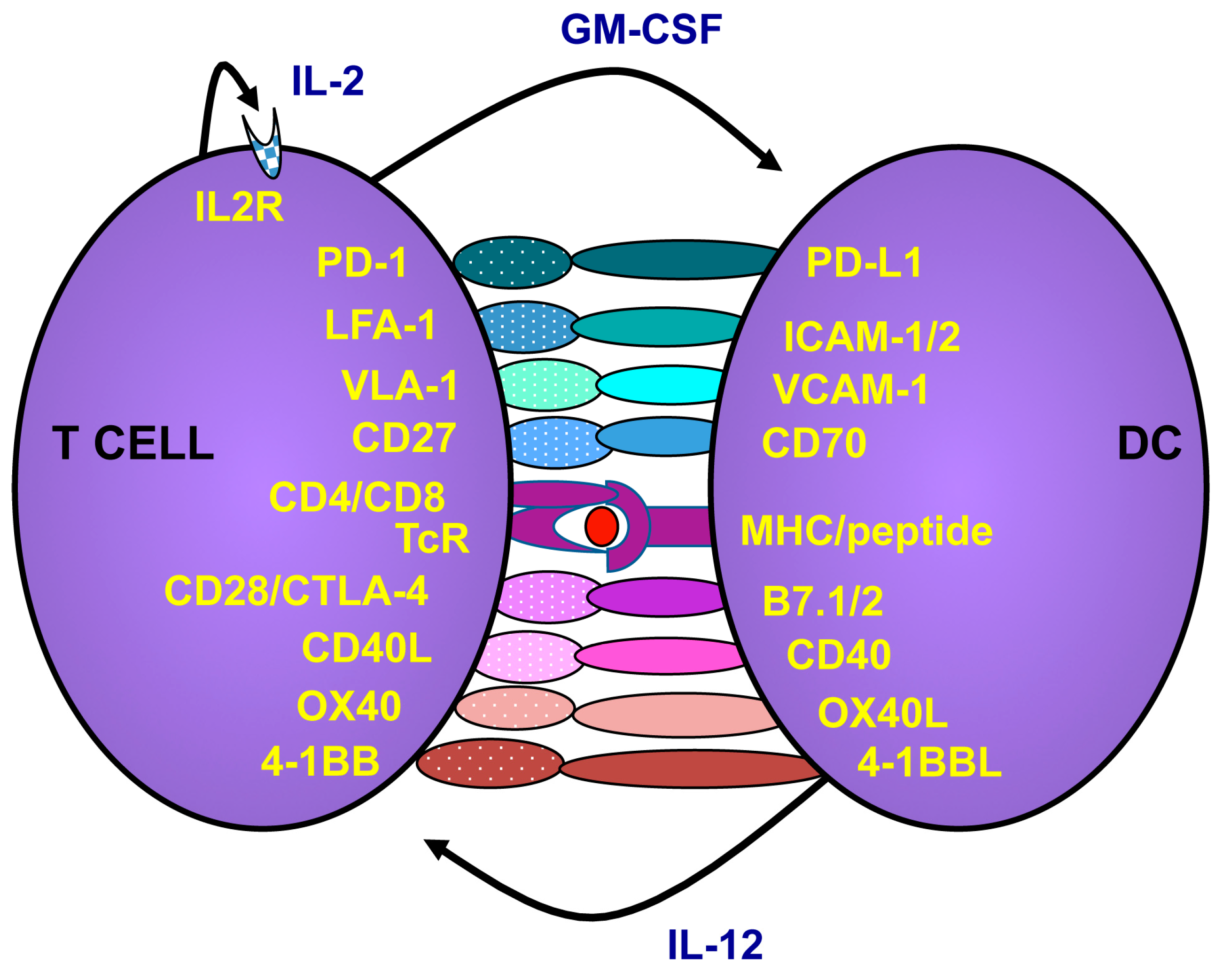
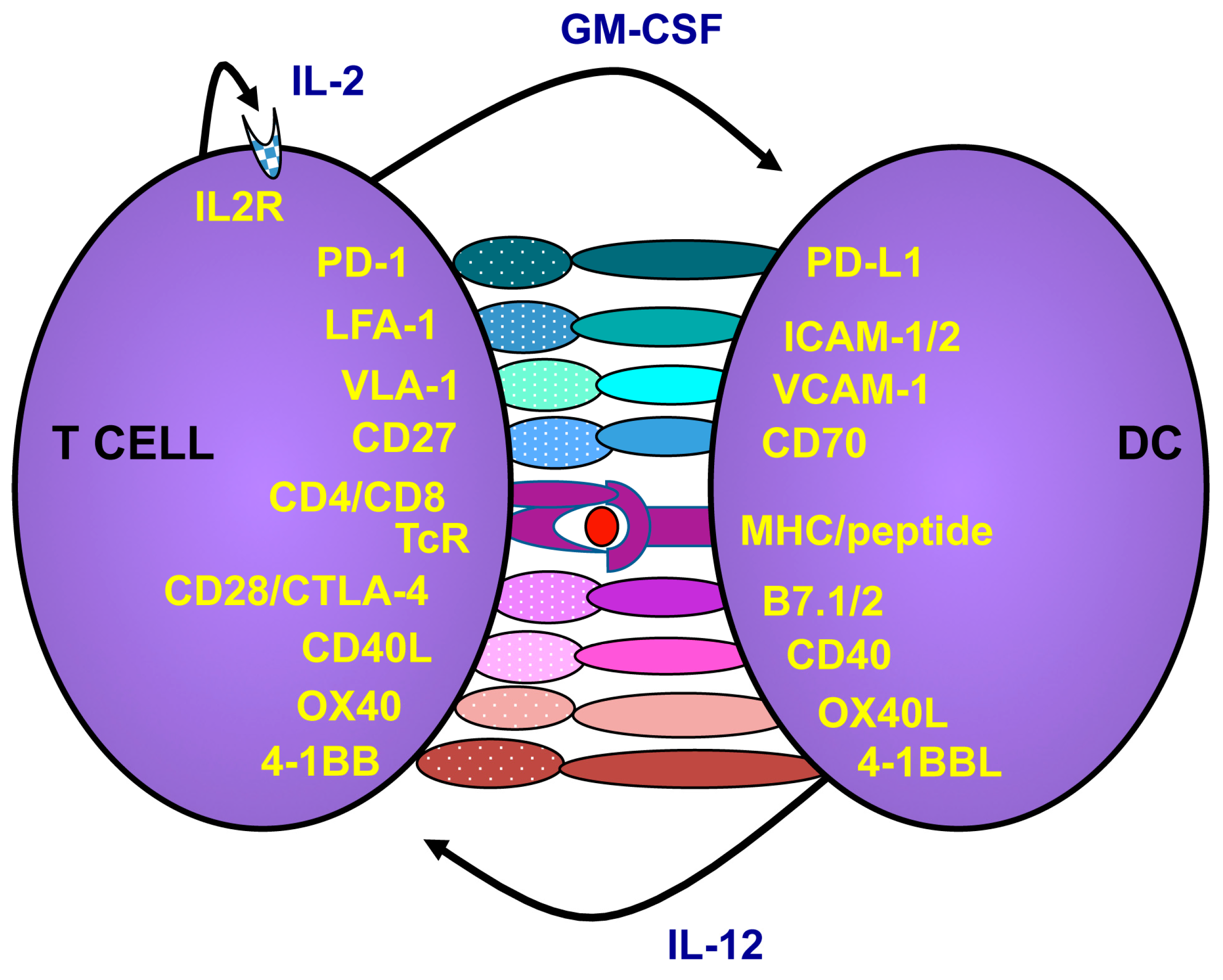
2.2. How to Activate and Maintain Th1 Responses
2.3. Targeting the Microenvironment to Promote Cancer Immunotherapy
2.4. Oncolytic Adenoviruses as Immunostimulatory Gene Vehicles
3. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Redelman-Sidi, G.; Glickman, M.S.; Bochner, B.H. The mechanism of action of BCG therapy for bladder cancer—A current perspective. Nat. Rev. Urol. 2014, 11, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Medina-Echeverz, J.; Aranda, F.; Berraondo, P. Myeloid-derived cells are key targets of tumor immunotherapy. Oncoimmunology 2014, 3, e28398. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Regulatory T cell subsets in human cancer: Are they regulating for or against tumor progression? Cancer Immunol. Immunother. 2014, 63, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive cell therapy for patients with metastatic melanoma: Evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectvely treated with autologous T cells expressing and anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Ribas, A. The evolution of checkpoint blockade as a cancer therapy: What’s here, what’s next? Curr. Opin. Immunol. 2015, 33, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Eggermont, A.; Sautes-Fridman, C.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Oncolytic viruses for cancer therapy. Oncoimmunology 2013, 2, e24612. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, M.E.; Brown, M.P.; Brenner, M.K. Antitumor responses induced by transgenic expression of CD40 ligand. Hum. Gene Ther. 1997, 8, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Addison, C.L.; Bramson, J.L.; Hitt, M.M.; Muller, W.J.; Gauldie, J.; Graham, F.L. Intratumoral coinjection of adenoviral vectors expressing IL-2 and IL-12 results in enhanced frequency of regression of injected and untreated distal tumors. Gene Ther. 1998, 5, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, F.; Signorelli, P.; Giovarelli, M.; Musiani, P.; Modesti, A.; Brunda, M.J.; Colombo, M.P.; Forni, G. Antitumor efficacy of adenocarcinoma cells engineered to produce interleukin 12 (IL-12) or other cytokines compared with exogenous IL-12. J. Natl. Cancer Inst. 1997, 89, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Pützer, B.M.; Hitt, M.; Muller, W.J.; Emtage, P.; Gauldie, J.; Graham, F.L. Interleukin 12 and B7–1 costimulatory molecule expressed by an adenovirus vector act synergistically to facilitate tumor regression. Proc. Natl. Acad. Sci. USA 1997, 94, 10889–10894. [Google Scholar] [CrossRef] [PubMed]
- Couderc, B.; Zitvogel, L.; Douin-Echinard, V.; Djennane, L.; Tahara, H.; Favre, G.; Lotze, M.T.; Robbins, P.D. Enhancement of antitumor immunity by expression of CD70 (CD27 ligand) or CD154 (CD40 ligand) costimulatory molecules in tumor cells. Cancer Gene Ther. 1998, 5, 163–175. [Google Scholar] [PubMed]
- Takahashi, S.; Rousseau, R.F.; Yotnda, P.; Mei, Z.; Dotti, G.; Rill, D.; Hurwitz, R.; Marini, F.; Andreeff, M.; Brenner, M.K. Autologous antileukemic immune response induced by chronic lymphocytic leukemia B cells expressing the CD40 ligand and interleukin 2 transgenes. Hum. Gene Ther. 2001, 12, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Douin-Echinard, V.; Robbins, P.D.; Lotze, M.T.; Favre, G.; Couderc, B. Enhancement of anti-tumor immunity by injection of fibroblasts genetically engineered to produce IL-12 and to express CD70. Adv. Exp. Med. Biol. 1998, 451, 353–357. [Google Scholar] [PubMed]
- Ajith, T.A. Strategies used in the clinical trials of gene therapy for cancer. J. Exp. Ther. Oncol. 2015, 11, 33–39. [Google Scholar] [PubMed]
- Draghiciu, O.; Lubbers, J.; Nijman, H.W.; Daemen, T. Myeloid derived suppressor cells-An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology 2015, 4, e954829. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Assiri, A.M.; Broering, D.C. Complement and macrophage crosstalk during process of angiogenesis in tumor progression. J. Biomed. Sci. 2015, 22, 58. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Capone, M.; Urba, W.J.; Bifulco, C.B.; Botti, G.; Lugli, A.; Marincola, F.M.; Ciliberto, G.; Galon, J.; Fox, B.A. The additional facet of immunoscore: Immunoprofiling as a possible predictive tool for cancer treatment. J. Transl. Med. 2013, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Bakdash, G.; Sittig, S.P.; van Dijk, T.; Figdor, C.G.; de Vries, I.J. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front. Immunol. 2013, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, X.; Yang, Y. Innate Immune Response to Adenoviral Vectors Is Mediated by both Toll-Like Receptor-Dependent and –Independent pathways. J. Virol. 2007, 81, 3170–3180. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.G.; Blattman, J.N.; Kasturi, S.P.; Kelley, R.P.; Kaufman, D.R.; Lynch, D.M.; la Porte, A.; Simmons, N.L.; Clark, S.L.; Pulendran, B.; et al. Multiple innate immune pathways contribute to the immunogenicity of recombinant adenovirus vaccine vectors. J. Virol. 2011, 85, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Ahonen, C.L.; Doxsee, C.L.; McGurran, S.M.; Riter, T.R.; Wade, W.F.; Barth, R.J.; Vasilakos, J.P.; Noelle, R.J.; Kedl, R.M. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J. Exp. Med. 2004, 199, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Simons, J.W.; Jaffee, E.M.; Weber, C.E.; Levitsky, H.I.; Nelson, W.G.; Carducci, M.A.; Lazenby, A.J.; Cohen, L.K.; Finn, C.C.; Clift, S.M.; et al. Bioactivity of autologous irradiated renal cell carcinoma vaccines generated by ex vivo granulocyte-macrophage colony-stimulating factor gene transfer. Cancer Res. 1997, 57, 1537–1546. [Google Scholar] [PubMed]
- Loskog, A.; Dzojic, H.; Vikman, S.; Ninalga, C.; Essand, M.; Korsgren, O.; Totterman, T.H. Adenovirus CD40 ligand gene therapy counteracts immune escape mechanisms in the tumor Microenvironment. J. Immunol. 2004, 172, 7200–7205. [Google Scholar] [CrossRef] [PubMed]
- Poutou, J.; Bunuales, M.; Gonzalez-Aparicio, M.; Garcia-Aragoncillo, E.; Quetglas, J.I.; Casado, R.; Bravo-Perez, C.; Alzuguren, P.; Hernandez-Alcoceba, R. Safety and antitumor effect of oncolytic and helper-dependent adenoviruses expressing interleukin-12 variants in a hamster pancreatic cancer model. Gene Ther. 2015, 22, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Dzojic, H.; Loskog, A.; Tötterman, T.H.; Essand, M. Adenovirus-mediated CD40 ligand therapy induces tumor cell apoptosis and systemic immunity in the TRAMP-C2 mouse prostate cancer model. Prostate 2006, 66, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Loskog, A.; Björkland, A.; Brown, M.P.; Korsgren, O.; Malmström, P.U.; Tötterman, T.H. Potent antitumor effects of CD154 transduced tumor cells in experimental bladder cancer. J. Urol. 2001, 166, 1093–1097. [Google Scholar] [CrossRef]
- Diaconu, I.; Cerullo, V.; Hirvinen, M.L.; Escutenaire, S.; Ugolini, M.; Pesonen, S.K.; Bramante, S.; Parviainen, S.; Kanerva, A.; Loskog, A.S.; et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012, 72, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Quetglas, J.I.; Reboredo, M.; Dubrot, J.; Rodriguez-Madoz, J.R.; Mancheño, U.; Casales, E.; Riezu-Boj, J.I.; Ruiz-Guillen, M.; Ochoa, M.C.; et al. Strict requirement for vector-induced type I interferon in efficacious antitumor responses to virally encoded IL12. Cancer Res. 2015, 75, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.X.; Li, Z.J.; Zhang, Y.; Zhang, X.N.; Zhao, K.C.; Li, Y.G.; Zhang, M.M.; Yu, X.W.; Liu, M.Y.; Li, Y. Enhanced antitumor immunity is elicited by adenovirus-mediated gene transfer of CCL21 and IL-15 in murine colon carcinomas. Cell Immunol. 2014, 289, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Ellem, K.A.; O’Rourke, M.G.; Johnson, G.R.; Parry, G.; Misko, I.S.; Schmidt, C.W.; Parsons, P.G.; Burrows, S.R.; Cross, S.; Fell, A.; et al. A case report: Immune responses and clinical course of the first human use of granulocyte/macrophage-colony-stimulating-factor-transduced autologous melanoma cells for immunotherapy. Cancer Immunol. Immunother. 1997, 44, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Soiffer, R.; Hodi, F.S.; Haluska, F.; Jung, K.; Gillessen, S.; Singer, S.; Tanabe, K.; Duda, R.; Mentzer, S.; Jaklitsch, M.; et al. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J. Clin. Oncol. 2003, 21, 3343–3350. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Yotnda, P.; Rousseau, R.F.; Mei, Z.; Smith, S.; Rill, D.; Younes, A.; Brenner, M.K. Transgenic expression of CD40L and interleukin-2 induces an autologous antitumor immune response in patients with non-Hodgkin’s lymphoma. Cancer Gene Ther. 2001, 8, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, R.F.; Biagi, E.; Dutour, A.; Yvon, E.S.; Brown, M.P.; Lin, T.; Mei, Z.; Grilley, B.; Popek, E.; Heslop, H.E.; et al. Immunotherapy of high-risk acute leukemia with a recipient (autologous) vaccine expressing transgenic human CD40L and IL-2 after chemotherapy and allogeneic stem cell transplantation. Blood 2006, 107, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Wierda, W.G.; Cantwell, M.J.; Woods, S.J.; Rassenti, L.Z.; Prussak, C.E.; Kipps, T.J. CD40-ligand (CD154) gene therapy for chronic lymphocytic leukemia. Blood 2000, 96, 2917–2924. [Google Scholar] [PubMed]
- Vassilev, L.; Ranki, T.; Joensuu, T.; Jäger, E.; Karbach, J.; Wahle, C.; Partanen, K.; Kairemo, K.; Alanko, T.; Turkki, R.; et al. Repeated intratumoral administration of ONCOS-102 leads to systemic antitumor CD8+ T-cell response and robust cellular and transcriptional immune activation at tumor site in a patient with ovarian cancer. Oncoimmunology 2015, 4, e1017702. [Google Scholar] [CrossRef] [PubMed]
- Bramante, S.; Kaufmann, J.K.; Veckman, V.; Liikanen, I.; Nettelbeck, D.M.; Hemminki, O.; Vassilev, L.; Cerullo, V.; Oksanen, M.; Heiskanen, R.; et al. Treatment of melanoma with a serotype 5/3 chimeric oncolytic adenovirus coding for GM-CSF: Results in vitro, in rodents and in humans. Int. J. Cancer 2015, 137, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, O.; Parviainen, S.; Juhila, J.; Turkki, R.; Linder, N.; Lundin, J.; Kankainen, M.; Ristimäki, A.; Koski, A.; Liikanen, I.; et al. Immunological data from cancer patients treated with Ad5/3-E2F-Δ24-GMCSF suggests utility for tumor immunotherapy. Oncotarget 2015, 6, 4467–4481. [Google Scholar] [CrossRef] [PubMed]
- Malmström, P.U.; Loskog, A.S.; Lindqvist, C.A.; Mangsbo, S.M.; Fransson, M.; Wanders, A.; Gårdmark, T.; Tötterman, T.H. AdCD40L immunogene therapy for bladder carcinoma—The first phase I/IIa trial. Clin. Cancer Res. 2010, 16, 3279–3287. [Google Scholar] [CrossRef] [PubMed]
- Pesonen, S.; Diaconu, I.; Kangasniemi, L.; Ranki, T.; Kanerva, A.; Pesonen, S.K.; Gerdemann, U.; Leen, A.M.; Kairemo, K.; Oksanen, M.; et al. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: Assessment of safety and immunologic responses in patients. Cancer Res. 2012, 72, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.E.; Melo-Cardenas, J.; Urquiza, M.; Barajas-Gamboa, J.S.; Pakbaz, R.S.; Kipps, T.J. Gene immunotherapy of chronic lymphocytic leukemia: A phase I study of intranodally injected adenovirus expressing a chimeric CD154 molecule. Cancer Res. 2012, 72, 2937–2948. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Pagès, F.; Marincola, F.M.; Thurin, M.; Trinchieri, G.; Fox, B.A.; Gajewski, T.F.; Ascierto, P.A. The immune score as a new possible approach for the classification of cancer. J. Transl. Med. 2012, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Viaud, S.; Chaput, N.; Bracci, L.; Proietti, E.; Zitvogel, L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin. Immunopathol. 2011, 33, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Homma, Y.; Taniguchi, K.; Nakazawa, M.; Matsuyama, R.; Mori, R.; Takeda, K.; Ichikawa, Y.; Tanaka, K.; Endo, I. Changes in the immune cell population and cell proliferation in peripheral blood after gemcitabine-based chemotherapy for pancreatic cancer. Clin. Transl. Oncol. 2014, 16, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Plate, J.M.; Plate, A.E.; Shott, S.; Bograd, S.; Harris, J.E. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol. Immunother. 2005, 54, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.A.; Clements, D.; Dielschneider, R.; Helson, E.; Marcato, P.; Lee, P.W. Gemcitabine enhances the efficacy of reovirus-based oncotherapy through anti-tumour immunological mechanisms. Br. J. Cancer 2014, 110, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Draghiciu, O.; Nijman, H.W.; Hoogeboom, B.N.; Meijerhof, T.; Daemen, T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology 2015, 4, e989764. [Google Scholar] [CrossRef] [PubMed]
- Guislain, A.; Gadiot, J.; Kaiser, A.; Jordanova, E.S.; Broeks, A.; Sanders, J.; van Boven, H.; de Gruijl, T.D.; Haanen, J.B.; Bex, A.; et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol. Immunother. 2015, 64, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Christiansson, L.; Söderlund, S.; Mangsbo, S.; Hjorth-Hansen, H.; Höglund, M.; Markevärn, B.; Richter, J.; Stenke, L.; Mustjoki, S.; Loskog, A.; et al. The tyrosine kinase inhibitors imatinib and dasatinib reduce myeloid suppressor cells and release effector lymphocyte responses. Mol. Cancer Ther. 2015, 14, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Christiansson, L.; Söderlund, S.; Svensson, E.; Mustjoki, S.; Bengtsson, M.; Simonsson, B.; Olsson-Strömberg, U.; Loskog, A.S. Increased level of myeloid-derived suppressor cells, programmed death receptor ligand 1/programmed death receptor 1, and soluble CD25 in Sokal high risk chronic myeloid leukemia. PLoS ONE 2013, 8, e55818. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Galustian, C.; Meyer, B.; Labarthe, M.C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D.; et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2009, 58, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Nepstad, I.; Hauge, M.; Hatfield, K.J.; Reikvam, H. STAT3 as a possible therapeutic target in human malignancies: Lessons from acute myeloid leukemia. Expert Rev. Hematol. 2015, 8, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Liljenfeldt, L.; Dieterich, L.C.; Dimberg, A.; Mangsbo, S.M.; Loskog, A.S. CD40L gene therapy tilts the myeloid cell profile and promotes infiltration of activated lymphocytes. Cancer Gene Ther. 2014, 21, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Ellmark, P.; Mangsbo, S.M.; Furebring, C.; Tötterman, T.H.; Norlén, P. Kick-starting the cancer-immunity cycle by targeting CD40. Oncoimmunology 2015, 4, e1011484. [Google Scholar] [CrossRef] [PubMed]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Roche, F.; Ohlin, E.; Essand, M.; Claesson-Welsh, L. Histidine-Rich Glycoprotein (HRG): A Novel Gene-Therapy Effector for the Treatment of Cancer. Mol. Ther. 2014, 22, S243–S244. [Google Scholar] [CrossRef]
- Yadav, L.; Puri, N.; Rastogi, V.; Satpute, P.; Sharma, V. Tumour Angiogenesis and Angiogenic Inhibitors: A Review. J. Clin. Diagn. Res. 2015, 9, XE01–XE05. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Dimberg, A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J. 2015, 29, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Mangsbo, S.M.; Sandin, L.C.; Anger, K.; Korman, A.J.; Loskog, A.; Tötterman, T.H. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J. Immunother. 2010, 33, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Löskog, A.; et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Seregin, S.S.; Amalfitano, A. Overcoming pre-existing adenovirus immunity by genetic engineering of adenovirus-based vectors. Expert Opin. Biol. Ther. 2009, 9, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic immunotherapy: Dying the right way is a key to eliciting potent antitumor immunity. Front. Oncol. 2014, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Alemany, R. Chapter four—Design of improved oncolytic adenoviruses. Adv. Cancer Res. 2012, 115, 93–114. [Google Scholar] [PubMed]
- Rojas, J.J.; Guedan, S.; Searle, P.F.; Martinez-Quintanilla, J.; Gil-Hoyos, R.; Alcayaga-Miranda, F.; Cascallo, M.; Alemany, R. Minimal RB-responsive E1A promoter modification to attain potency, selectivity, and transgene-arming capacity in oncolytic adenoviruses. Mol. Ther. 2010, 18, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2792. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.I.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzales, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009, 34, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Marincola, F.M.; Atkins, M.B. What’s new in melanoma? Combiantion! J. Transl. Med. 2015, 13, 213–217. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loskog, A. Immunostimulatory Gene Therapy Using Oncolytic Viruses as Vehicles. Viruses 2015, 7, 5780-5791. https://doi.org/10.3390/v7112899
Loskog A. Immunostimulatory Gene Therapy Using Oncolytic Viruses as Vehicles. Viruses. 2015; 7(11):5780-5791. https://doi.org/10.3390/v7112899
Chicago/Turabian StyleLoskog, Angelica. 2015. "Immunostimulatory Gene Therapy Using Oncolytic Viruses as Vehicles" Viruses 7, no. 11: 5780-5791. https://doi.org/10.3390/v7112899
APA StyleLoskog, A. (2015). Immunostimulatory Gene Therapy Using Oncolytic Viruses as Vehicles. Viruses, 7(11), 5780-5791. https://doi.org/10.3390/v7112899