

The Evolving Role of Zika Virus Envelope Protein in Viral Entry and Pathogenesis

 , , and
, , and 
Abstract
1. Introduction
1.1. Zika Virus (ZIKV): A Brief History
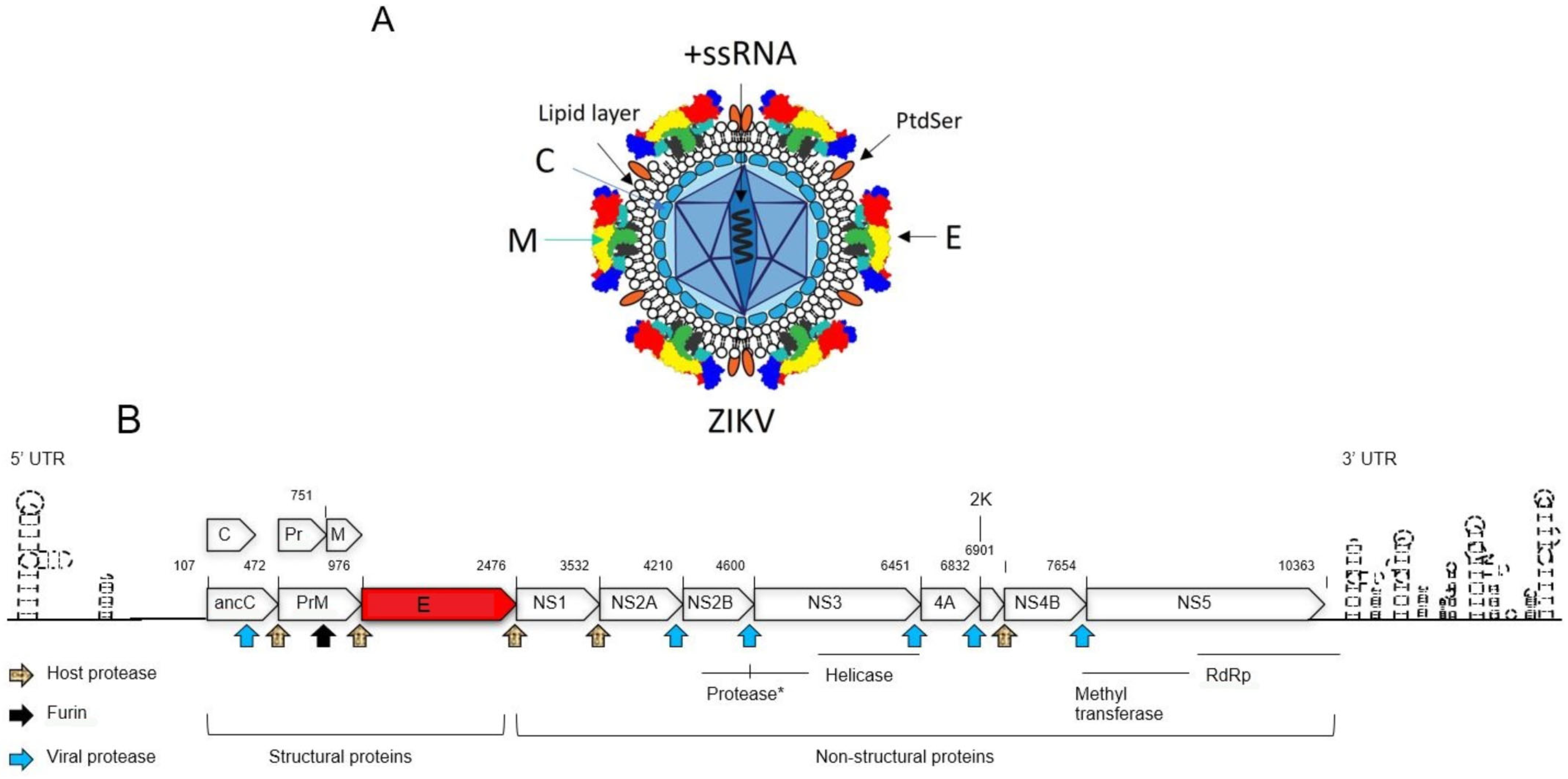
1.2. The Structure and Genomic Organization of ZIKV
1.3. The Viral Infection Cycle
2. ZIKV Envelope Protein
2.1. The Structure of ZIKV E Protein
2.2. Post-Translational Modification of ZIKV E Protein
2.2.1. Glycosylation of ZIKV E Protein
2.2.2. Protonation of ZIKV E Protein
2.2.3. Phosphatidylserine on ZIKV E Protein
2.2.4. Consideration of Epistatic Interaction
2.3. Distinctive Features of E Proteins Between Ancestral African and Epidemic ZIKV
3. Viral Entry Mediated by ZIKV E Protein
3.1. The Cellular Receptors
3.2. ZIKV E Protein-Mediated Virus Attachment to Host Cells
3.2.1. The Importance of E Protein Structure
3.2.2. The Role of N154 Glycosylation
3.2.3. The Role of PtdSer
3.3. The Role of ZIKV E Protein in Endocytosis and Viral Fusion
4. ZIKV E Protein-Mediated Immune Response and Evasion
5. Neutralizing Antibodies, Antibody-Dependent Enhancement, and Vaccine Development
5.1. Neutralizing Antibodies (nAbs)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neutralizing Antibody | Target Site | Specificity | Reference |
|---|---|---|---|
| ZV-2, ZV-48, ZV-54, ZV-64, ZV-67 | DIII or DIII LR | ZIKV | [132,149] |
| ZIKV-116 | DIII | ZIKV | [131,149] |
| Z004, Z006, Z021 | DIII LR | ZIKV and DENV1 | [134] |
| HA-12, 1C-11, 2F-8, 1D-9, 1D-11 | DIII | ZIKV Asian lineage | [135] |
| B11F | DI and DIII | ZIKV | [136] |
| ZKA190 | DI-DIII linker, DIII LR | ZIKV | [137] |
| MZ1/MZ4/MZ24 | DI/DIII linker region | ZIKV, DENV1-4 | [138] |
| ZKA185 | DII | ZIKV | [140] |
| 2A10G6 | DII (FL) | ZIKV, DENV1-4 and WNV | [44,141] |
| AZ1p, AZ6m | DII(FL) | ZIKV, YFV, and DENV | [142] |
| 3E8, 5F8, 5G3, 8A2, 9C3 | Linear Epitope on GL | ZIKV Asian lineage | [143] |
| A11, A42 | GL | ZIKV | [144] |
| Z3L1, Z23, Z20 | DI, DII, or DIII | ZIKV | [145] |
| rhMZ—Group A, B, C, D | EDE | ZIKV | [146] |
| EDE1-B10, EDE1-C8, EDE1-C10 | EDE | ZIKV and DENV 1-4 | [69,139,147] |
| ZIKV-117 | EDE | ZIKV | [149,150] |
5.2. Antibody-Dependent Enhancement (ADE) and Vaccine Development
6. Anti-ZIKV E Protein Inhibitors
6.1. Direct E Protein Inhibitors
6.2. Viral Attachment Inhibitors
6.3. Viral Fusion Inhibitors
6.4. Host Restriction Factors
| Compound Name | Effect | IC50/EC50 (µM) | CC50 (µM) | Cell /Model | Reference |
|---|---|---|---|---|---|
| Direct E Protein Inhibitors | |||||
| F1065-0358 | Bind to the DI and DIII regions and interfere with the E protein trimerization during | 14 | 200 | Vero | [175] |
| Gossypol | Bind to DIII | 3.75 ± 0.01 | 14.17 ± 0.74 | Vero E6 | [177] |
| EGCG | Bind to DI and DII | na | na | Vero E6 | [178] |
| PGG | Interacts with charged residues of glycosylated E protein | 4.1 | 114 | Vero B4 | [179] |
| Polysaccharides (PGG, parishin and stevioside) | Bind to E protein | na | na | Docking analysis | [180] |
| Apigenin | Bind to DIII | >100 | na | Vero | [181] |
| Baicalin | Bind to E protein | 14 | na | Vero | [181,182] |
| Atranorin | Bind to DI and DIII | 11.9 | >50 | SNB-19 | [123] |
| Palmatine | Interact with E protein | na | na | Vero | [183] |
| Harringtonine | Bind to E Protein | 0.287 | >10 | Vero | [184] |
| Viral Attachment Inhibitors | |||||
| Cabozantinib (R428, TP-0903, and BMS-777607) | Inhibit AXL receptor | na | na | hCMEC/D3/HUVECs | [185] |
| ZINC33683341 | Bind to primary receptors | na | na | Vero | [187] |
| Curcumin | Viral attachment and entry by abrogating the function of viral envelope proteins | 1.9 | 11.6 | Vero | [188,189] |
| Viral Fusion Inhibitors | |||||
| Seven compounds | Prevent E-mediated membrane fusion | 0.9–19.3 | 5.2–>100 | Vero | [190] |
| Ev37 | Prevent viral membrane–endosomal membrane at low pH | na | 116.3 | Huh-7 | [191] |
| Atovaquone | Block E-mediated membrane fusion | 2.1 | na | Vero/MDCK/C6/36 | [194] |
| Peptide Z2 | Disrupt E conformational changes | 1.75 | na | C57BL/6BHK21 | [195,196] |
| P5 | Change E protein conformation at low pH | 3.27 | na | Vero/AG6 | [197] |
| Host Restriction Factors | |||||
| LAMR1 | Attenuate E protein ubiquitination | na | na | HeLa/HEK293T | [198] |
| Hpa | Attenuates ZIKV infection by destabilizing the E protein | na | na | MEF | [199] |
| Viperin | Restrict a wide range of viruses including ZIKV | na | na | Huh7 | [200] |
| USP38 | Attenuates K48- and K63-linked polyubiquitination of E protein | na | na | HeLa/HEK293 | [201] |
7. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| a.a. | Amino acids |
| ADE | Antibody-dependent enhancement |
| BBB | Blood-brain barriers |
| CDE | Clathrin-dependent endocytosis |
| CHIKV | Chikungunya virus |
| CIE | Clathrin-independent endocytosis |
| CPE | Cytopathic effect |
| CNS | Central nervous system |
| DC | Dendritic cell |
| DENV | Dengue virus |
| dsRNA | Double stranded RNA |
| EDE | E dimer epitope |
| EGCG | Epigallocatechin gallate |
| EM | Electron microscopy |
| ER | Endoplasmic reticulum |
| FL | Fusion loop |
| GBS | Guillain–Barré syndrome |
| GL | Glycan loop |
| HAVcr-1 | Hepatitis A virus cellular receptor 1 |
| hBMEC | Human brain microvascular endothelial cell |
| hEC | Human epithelial cell |
| hNPC | Human neural progenitor cell |
| hUVEC | Human umbilical vein endothelial cells |
| IFN | Interferon |
| IRF3 | Interferon regulatory factor 3 |
| ISGS | Interferon stimulated genes |
| JEV | Japanese Encephalitis virus |
| LAMR1 | Laminin receptor 1 |
| LR | Lateral ridge |
| mAb | Monoclonal antibody |
| MEF | Mouse Embryonic Fibroblasts |
| NAb | Neutralizing antibody |
| NPCs | Neural progenitor stem cells |
| NSCs | Neural Stem Cells |
| PAMP | Pathogen-associated molecular pattern |
| PGG | Pentagalloylglucose |
| PRRs | Pattern recognition receptors |
| PtdSer | Phosphatidylserine |
| RdRP | RNA-dependent RNA polymerase |
| RIG-1 | RIG-1-like receptor |
| RLRs | RIG-I like receptors |
| ROS | Reactive oxygen species |
| sfRNA | Subgenomic flavivirus RNA |
| TAM | Tyro3, Axl and Mer |
| TGN | Trans-Golgi network |
| TM | Trans-membrane |
| TLR | Toll-like receptors |
| TOR | Target of rapamycin |
| WHO | World health organization |
| WNV | West Nile virus |
| YFV | Yellow Fever Virus |
| ZIKV | Zika virus |
References
- Lee, I.; Bos, S.; Li, G.; Wang, S.; Gadea, G.; Despres, P.; Zhao, R.Y. Probing Molecular Insights into Zika Virus-Host Interactions. Viruses 2018, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika Virus: A New Virus Isolated from Aedes mosquitoes in Uganda. Nature 1952, 229, 735–736. [Google Scholar]
- Bell, T.M.; Field, E.J.; Narang, H.K. Zika virus infection of the central nervous system of mice. Arch. Gesamte Virusforsch. 1971, 35, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W. Zika virus. II. Pathogenicity and physical properties. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 521–534. [Google Scholar] [CrossRef]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Hayes, E.B. Zika virus outside Africa. Emerg. Infect. Dis. 2009, 15, 1347–1350. [Google Scholar] [CrossRef]
- Foy, B.D.; Kobylinski, K.C.; Chilson Foy, J.L.; Blitvich, B.J.; Travassos da Rosa, A.; Haddow, A.D.; Lanciotti, R.S.; Tesh, R.B. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg. Infect. Dis. 2011, 17, 880–882. [Google Scholar] [CrossRef]
- Mead, P.S.; Hills, S.L.; Brooks, J.T. Zika virus as a sexually transmitted pathogen. Curr. Opin. Infect. Dis. 2018, 31, 39–44. [Google Scholar] [CrossRef]
- Musso, D.; Nhan, T.; Robin, E.; Roche, C.; Bierlaire, D.; Zisou, K.; Shan Yan, A.; Cao-Lormeau, V.M.; Broult, J. Potential for Zika virus transmission through blood transfusion demonstrated during an outbreak in French Polynesia, November 2013 to February 2014. Euro. Surveill. 2014, 19, 20761. [Google Scholar] [CrossRef]
- Macnamara, F.N. Zika virus: A report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 1954, 48, 139–145. [Google Scholar] [CrossRef]
- Marchette, N.J.; Garcia, R.; Rudnick, A. Isolation of Zika virus from Aedes aegypti mosquitoes in Malaysia. Am. J. Trop. Med. Hyg. 1969, 18, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.G.; Ksiazek, T.G.; Suhandiman; Triwibowo. Zika virus, a cause of fever in Central Java, Indonesia. Trans. R. Soc. Trop. Med. Hyg. 1981, 75, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Darwish, M.A.; Hoogstraal, H.; Roberts, T.J.; Ahmed, I.P.; Omar, F. A sero-epidemiological survey for certain arboviruses (Togaviridae) in Pakistan. Trans. R. Soc. Trop. Med. Hyg. 1983, 77, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef]
- Gulland, A. Zika virus is a global public health emergency, declares WHO. BMJ 2016, 352, i657. [Google Scholar] [CrossRef]
- Heukelbach, J.; Alencar, C.H.; Kelvin, A.A.; de Oliveira, W.K.; Pamplona de Goes Cavalcanti, L. Zika virus outbreak in Brazil. J. Infect. Dev. Ctries. 2016, 10, 116–120. [Google Scholar] [CrossRef]
- Yadav, P.D.; Niyas, V.K.M.; Arjun, R.; Sahay, R.R.; Shete, A.M.; Sapkal, G.N.; Pawar, S.D.; Patil, D.Y.; Gupta, N.; Abraham, P. Detection of Zika virus disease in Thiruvananthapuram, Kerala, India 2021 during the second wave of COVID-19 pandemic. J. Med. Virol. 2022, 94, 2346. [Google Scholar] [CrossRef]
- Yu, X.; Cheng, G. Adaptive Evolution as a Driving Force of the Emergence and Re-Emergence of Mosquito-Borne Viral Diseases. Viruses 2022, 14, 435. [Google Scholar] [CrossRef]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 A resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef]
- Harris, E.; Holden, K.L.; Edgil, D.; Polacek, C.; Clyde, K. Molecular biology of flaviviruses. Novartis Found. Symp. 2006, 277, 23–39, discussion 40, 71–23, 251–253. [Google Scholar] [PubMed]
- Li, G.; Poulsen, M.; Fenyvuesvolgyi, C.; Yashiroda, Y.; Yoshida, M.; Simard, J.M.; Gallo, R.C.; Zhao, R.Y. Characterization of cytopathic factors through genome-wide analysis of the Zika viral proteins in fission yeast. Proc. Natl. Acad. Sci. USA 2017, 114, E376–E385. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Huang, X.-Y.; Liu, Z.-Y.; Zhang, F.; Zhu, X.-L.; Yu, J.-Y.; Ji, X.; Xu, Y.-P.; Li, G.; Li, C.J.S. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Nambala, P.; Su, W.-C.J.F.i.m. Role of Zika virus prM protein in viral pathogenicity and use in vaccine development. Front. Microbiol. 2018, 9, 1797. [Google Scholar] [CrossRef]
- Amberg, S.M.; Nestorowicz, A.; McCourt, D.W.; Rice, C.M. NS2B-3 proteinase-mediated processing in the yellow fever virus structural region: In vitro and in vivo studies. J. Virol. 1994, 68, 3794–3802. [Google Scholar] [CrossRef]
- Lobigs, M.; Lee, E.; Ng, M.L.; Pavy, M.; Lobigs, P. A flavivirus signal peptide balances the catalytic activity of two proteases and thereby facilitates virus morphogenesis. Virology 2010, 401, 80–89. [Google Scholar] [CrossRef]
- Li, G.; Bos, S.; Tsetsarkin, K.A.; Pletnev, A.G.; Despres, P.; Gadea, G.; Zhao, R.Y. The Roles of prM-E Proteins in Historical and Epidemic Zika Virus-mediated Infection and Neurocytotoxicity. Viruses 2019, 11, 157. [Google Scholar] [CrossRef]
- Stadler, K.; Allison, S.L.; Schalich, J.; Heinz, F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997, 71, 8475–8481. [Google Scholar] [CrossRef]
- Elshuber, S.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J. Gen. Virol. 2003, 84, 183–191. [Google Scholar] [CrossRef]
- Majowicz, S.A.; Narayanan, A.; Moustafa, I.M.; Bator, C.M.; Hafenstein, S.L.; Jose, J.J.n.V. Zika virus M protein latches and locks the E protein from transitioning to an immature state after prM cleavage. NPJ Viruses 2023, 1, 4. [Google Scholar] [CrossRef]
- Faye, O.; Freire, C.C.; Iamarino, A.; Faye, O.; de Oliveira, J.V.; Diallo, M.; Zanotto, P.M.; Sall, A.A. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl. Trop. Dis. 2014, 8, e2636. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Piramanayagam, S.; Natarajan, J.J.V.G. A review on structural genomics approach applied for drug discovery against three vector-borne viral diseases: Dengue, Chikungunya and Zika. Virus Genes 2022, 58, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R.J.S. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Hui, L.; Nie, Y.; Tefsen, B.; Wu, Y.J.S.C.L.S. ZIKV viral proteins and their roles in virus-host interactions. Sci. China Life Sci. 2021, 64, 709–719. [Google Scholar] [CrossRef]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.J.C.r. Ultrastructural characterization of Zika virus replication factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef]
- Tabata, T.; Petitt, M.; Puerta-Guardo, H.; Michlmayr, D.; Wang, C.; Fang-Hoover, J.; Harris, E.; Pereira, L. Zika Virus Targets Different Primary Human Placental Cells, Suggesting Two Routes for Vertical Transmission. Cell Host Microbe 2016, 20, 155–166. [Google Scholar] [CrossRef]
- de Paula Freitas, B.; de Oliveira Dias, J.R.; Prazeres, J.; Sacramento, G.A.; Ko, A.I.; Maia, M.; Belfort, R., Jr. Ocular Findings in Infants With Microcephaly Associated With Presumed Zika Virus Congenital Infection in Salvador, Brazil. JAMA Ophthalmol. 2016, 134, 529–535. [Google Scholar] [CrossRef]
- Sager, G.; Gabaglio, S.; Sztul, E.; Belov, G.A.J.V. Role of host cell secretory machinery in Zika virus life cycle. Viruses 2018, 10, 559. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, R.; Shen, H.; Wang, M.; Yin, Z.; Cheng, A. Structures and functions of the envelope glycoprotein in flavivirus infections. Viruses 2017, 9, 338. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, G.F. Structural biology of the Zika virus. Trends Biochem. Sci. 2017, 42, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Barrows, N.J.; Campos, R.K.; Liao, K.-C.; Prasanth, K.R.; Soto-Acosta, R.; Yeh, S.-C.; Schott-Lerner, G.; Pompon, J.; Sessions, O.M.; Bradrick, S.S. Biochemistry and molecular biology of flaviviruses. Chem. Rev. 2018, 118, 4448–4482. [Google Scholar] [CrossRef] [PubMed]
- Frumence, E.; Haddad, J.G.; Vanwalscappel, B.; Andries, J.; Decotter, J.; Viranaicken, W.; Gadea, G.; Desprès, P. Immune Reactivity of a 20-mer Peptide Representing the Zika E Glycan Loop Involves the Antigenic Determinants E-152/156/158. Viruses 2020, 12, 1258. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Song, J.; Lu, X.; Deng, Y.Q.; Musyoki, A.M.; Cheng, H.; Zhang, Y.; Yuan, Y.; Song, H.; Haywood, J.; et al. Structures of the Zika Virus Envelope Protein and Its Complex with a Flavivirus Broadly Protective Antibody. Cell Host Microbe 2016, 19, 696–704. [Google Scholar] [CrossRef]
- Routhu, N.K.; Lehoux, S.D.; Rouse, E.A.; Bidokhti, M.R.M.; Giron, L.B.; Anzurez, A.; Reid, S.P.; Abdel-Mohsen, M.; Cummings, R.D.; Byrareddy, S.N. Glycosylation of Zika Virus is Important in Host-Virus Interaction and Pathogenic Potential. Int. J. Mol. Sci. 2019, 20, 5206. [Google Scholar] [CrossRef]
- Hu, T.; Wu, Z.; Wu, S.; Chen, S.; Cheng, A. The key amino acids of E protein involved in early flavivirus infection: Viral entry. Virol. J. 2021, 18, 136. [Google Scholar] [CrossRef]
- Chakraborty, S. Computational analysis of perturbations in the post-fusion Dengue virus envelope protein highlights known epitopes and conserved residues in the Zika virus. F1000Research 2016, 5, 1150. [Google Scholar] [CrossRef]
- Bressanelli, S.; Stiasny, K.; Allison, S.L.; Stura, E.A.; Duquerroy, S.; Lescar, J.; Heinz, F.X.; Rey, F.A. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004, 23, 728–738. [Google Scholar] [CrossRef]
- Lee, J.W.-M.; Chu, J.J.-H.; Ng, M.-L. Quantifying the specific binding between West Nile virus envelope domain III protein and the cellular receptor αVβ3 integrin. J. Biol. Chem. 2006, 281, 1352–1360. [Google Scholar] [CrossRef]
- Agrelli, A.; de Moura, R.R.; Crovella, S.; Brandão, L.A.C. ZIKA virus entry mechanisms in human cells. Infect. Genet. Evol. 2019, 69, 22–29. [Google Scholar] [CrossRef]
- Qu, P.; Zhang, W.; Li, D.; Zhang, C.; Liu, Q.; Zhang, X.; Wang, X.; Dai, W.; Xu, Y.; Leng, Q. Insect cell-produced recombinant protein subunit vaccines protect against Zika virus infection. Antivir. Res. 2018, 154, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.; Kim, K.; Lee, H.J.; Jung, Y.J.; Park, J.; Hahn, T.W. Vaccination with a Zika virus envelope domain III protein induces neutralizing antibodies and partial protection against Asian genotype in immunocompetent mice. Trop. Med. Health 2022, 50, 91. [Google Scholar] [CrossRef]
- Gallichotte, E.N.; Dinnon, K.H., 3rd; Lim, X.N.; Ng, T.S.; Lim, E.X.Y.; Menachery, V.D.; Lok, S.M.; Baric, R.S. CD-loop Extension in Zika Virus Envelope Protein Key for Stability and Pathogenesis. J. Infect. Dis. 2017, 216, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Carbaugh, D.L.; Lazear, H.M. Flavivirus envelope protein glycosylation: Impacts on viral infection and pathogenesis. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Zhang, J.; Chen, Z.; Pan, W.; Chen, Z.; Yan, Y.; Dai, J. Glycosylation of viral proteins: Implication in virus–host interaction and virulence. Virulence 2022, 13, 670–683. [Google Scholar] [CrossRef]
- Carbaugh, D.L.; Baric, R.S.; Lazear, H.M. Envelope Protein Glycosylation Mediates Zika Virus Pathogenesis. J. Virol. 2019, 93, e00113-19. [Google Scholar] [CrossRef]
- Bos, S.; Viranaicken, W.; Frumence, E.; Li, G.; Despres, P.; Zhao, R.Y.; Gadea, G. The Envelope Residues E152/156/158 of Zika Virus Influence the Early Stages of Virus Infection in Human Cells. Cells 2019, 8, 1444. [Google Scholar] [CrossRef]
- Lei, Y.; Yu, H.; Dong, Y.; Yang, J.; Ye, W.; Wang, Y.; Chen, W.; Jia, Z.; Xu, Z.; Li, Z.J.P.O. Characterization of N-glycan structures on the surface of mature dengue 2 virus derived from insect cells. PLoS ONE 2015, 10, e0132122. [Google Scholar] [CrossRef]
- Pralow, A.; Nikolay, A.; Leon, A.; Genzel, Y.; Rapp, E.; Reichl, U.J.S.R. Site-specific N-glycosylation analysis of animal cell culture-derived Zika virus proteins. Sci. Rep. 2021, 11, 5147. [Google Scholar] [CrossRef]
- Gong, D.; Zhang, T.H.; Zhao, D.; Du, Y.; Chapa, T.J.; Shi, Y.; Wang, L.; Contreras, D.; Zeng, G.; Shi, P.Y.; et al. High-Throughput Fitness Profiling of Zika Virus E Protein Reveals Different Roles for Glycosylation during Infection of Mammalian and Mosquito Cells. iScience 2018, 1, 97–111. [Google Scholar] [CrossRef]
- Sun, J.; Li, Y.; Liu, P.; Lin, J. Study of the mechanism of protonated histidine-induced conformational changes in the Zika virus dimeric envelope protein using accelerated molecular dynamic simulations. J. Mol. Graph. Model. 2017, 74, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Moller-Tank, S.; Maury, W. Phosphatidylserine receptors: Enhancers of enveloped virus entry and infection. Virology 2014, 468–470, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Song, D.H.; Garcia, G., Jr.; Situ, K.; Chua, B.A.; Hong, M.L.O.; Do, E.A.; Ramirez, C.M.; Harui, A.; Arumugaswami, V.; Morizono, K. Development of a blocker of the universal phosphatidylserine- and phosphatidylethanolamine-dependent viral entry pathways. Virology 2021, 560, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.; Sansom, M.S. The Role of the Membrane in the Structure and Biophysical Robustness of the Dengue Virion Envelope. Structure 2016, 24, 375–382. [Google Scholar] [CrossRef]
- Ghosh Roy, S. TAM receptors: A phosphatidylserine receptor family and its implications in viral infections. Int. Rev. Cell Mol. Biol. 2020, 357, 81–122. [Google Scholar] [CrossRef]
- Tang, D.; Wang, Y.; Dong, X.; Yuan, Y.; Kang, F.; Tian, W.; Wang, K.; Li, H.; Qi, S. Scramblases and virus infection. BioEssays 2022, 44, 2100261. [Google Scholar] [CrossRef]
- Chua, B.A.; Ngo, J.A.; Situ, K.; Morizono, K. Roles of phosphatidylserine exposed on the viral envelope and cell membrane in HIV-1 replication. Cell Commun. Signal. 2019, 17, 132. [Google Scholar] [CrossRef]
- Berneck, B.S.; Rockstroh, A.; Fertey, J.; Grunwald, T.; Ulbert, S.J.V. A recombinant Zika virus envelope protein with mutations in the conserved fusion loop leads to reduced antibody cross-reactivity upon vaccination. Vaccines 2020, 8, 603. [Google Scholar] [CrossRef]
- Kikawa, C.; Cartwright-Acar, C.H.; Stuart, J.B.; Contreras, M.; Levoir, L.M.; Evans, M.J.; Bloom, J.D.; Goo, L. The effect of single mutations in Zika virus envelope on escape from broadly neutralizing antibodies. J. Virol. 2023, 97, e0141423. [Google Scholar] [CrossRef]
- Ma, X.; Yuan, Z.; Yi, Z. Identification and characterization of key residues in Zika virus envelope protein for virus assembly and entry. Emerg. Microbes Infect. 2022, 11, 1604–1620. [Google Scholar] [CrossRef]
- Sourisseau, M.; Lawrence, D.J.P.; Schwarz, M.C.; Storrs, C.H.; Veit, E.C.; Bloom, J.D.; Evans, M.J. Deep mutational scanning comprehensively maps how Zika envelope protein mutations affect viral growth and antibody escape. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Esser-Nobis, K.; Aarreberg, L.D.; Roby, J.A.; Fairgrieve, M.R.; Green, R.; Gale, M., Jr. Comparative Analysis of African and Asian Lineage-Derived Zika Virus Strains Reveals Differences in Activation of and Sensitivity to Antiviral Innate Immunity. J. Virol. 2019, 93, e00640-19. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Herrlinger, S.; Zhu, Y.N.; Yang, M.; Goodfellow, F.; Stice, S.L.; Qi, X.P.; Brindley, M.A.; Chen, J.F. The African Zika virus MR-766 is more virulent and causes more severe brain damage than current Asian lineage and dengue virus. Development 2017, 144, 4114–4124. [Google Scholar] [CrossRef] [PubMed]
- Anfasa, F.; Siegers, J.Y.; van der Kroeg, M.; Mumtaz, N.; Stalin Raj, V.; de Vrij, F.M.S.; Widagdo, W.; Gabriel, G.; Salinas, S.; Simonin, Y.; et al. Phenotypic Differences between Asian and African Lineage Zika Viruses in Human Neural Progenitor Cells. mSphere 2017, 2, e00292-17. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Du, S.; Shan, C.; Nie, K.; Zhang, R.; Li, X.F.; Zhang, R.; Wang, T.; Qin, C.F.; et al. Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 2017, 545, 482–486. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Bianchini, G.; Sanchez-Baracaldo, P. TreeViewer: Flexible, modular software to visualise and manipulate phylogenetic trees. Ecol. Evol. 2024, 14, e10873. [Google Scholar] [CrossRef]
- Faria, N.R.; Azevedo Rdo, S.; Kraemer, M.U.; Souza, R.; Cunha, M.S.; Hill, S.C.; Theze, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef]
- Haddow, A.D.; Schuh, A.J.; Yasuda, C.Y.; Kasper, M.R.; Heang, V.; Huy, R.; Guzman, H.; Tesh, R.B.; Weaver, S.C. Genetic characterization of Zika virus strains: Geographic expansion of the Asian lineage. PLoS Negl. Trop. Dis. 2012, 6, e1477. [Google Scholar] [CrossRef]
- Beaver, J.T.; Lelutiu, N.; Habib, R.; Skountzou, I. Evolution of Two Major Zika Virus Lineages: Implications for Pathology, Immune Response, and Vaccine Development. Front. Immunol. 2018, 9, 1640. [Google Scholar] [CrossRef]
- Bos, S.; Viranaicken, W.; Turpin, J.; El-Kalamouni, C.; Roche, M.; Krejbich-Trotot, P.; Despres, P.; Gadea, G. The structural proteins of epidemic and historical strains of Zika virus differ in their ability to initiate viral infection in human host cells. Virology 2018, 516, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Frumence, E.; Viranaicken, W.; Bos, S.; Alvarez-Martinez, M.T.; Roche, M.; Arnaud, J.D.; Gadea, G.; Despres, P. A Chimeric Zika Virus between Viral Strains MR766 and BeH819015 Highlights a Role for E-glycan Loop in Antibody-mediated Virus Neutralization. Vaccines 2019, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, A.; Dai, L.; Contreras, D.; Sinha, S.; Sun, R.; Arumugaswami, V. Comparative analysis of protein evolution in the genome of pre-epidemic and epidemic Zika virus. Infect. Genet. Evol. 2017, 51, 74–85. [Google Scholar] [CrossRef]
- Goo, L.; DeMaso, C.R.; Pelc, R.S.; Ledgerwood, J.E.; Graham, B.S.; Kuhn, R.J.; Pierson, T.C. The Zika virus envelope protein glycan loop regulates virion antigenicity. Virology 2018, 515, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Annamalai, A.S.; Pattnaik, A.; Sahoo, B.R.; Muthukrishnan, E.; Natarajan, S.K.; Steffen, D.; Vu, H.L.X.; Delhon, G.; Osorio, F.A.; Petro, T.M.; et al. Zika Virus Encoding Non-Glycosylated Envelope Protein is Attenuated and Defective in Neuroinvasion. J. Virol. 2017, 91, e01348-17. [Google Scholar] [CrossRef]
- Bowen, J.R.; Zimmerman, M.G.; Suthar, M.S. Taking the defensive: Immune control of Zika virus infection. Virus Res. 2018, 254, 21–26. [Google Scholar] [CrossRef]
- Mesci, P.; Macia, A.; LaRock, C.N.; Tejwani, L.; Fernandes, I.R.; Suarez, N.A.; Zanotto, P.M.d.A.; Beltrão-Braga, P.C.B.; Nizet, V.; Muotri, A.R. Modeling neuro-immune interactions during Zika virus infection. Hum. Mol. Genet. 2018, 27, 41–52. [Google Scholar] [CrossRef]
- Sheridan, M.A.; Yunusov, D.; Balaraman, V.; Alexenko, A.P.; Yabe, S.; Verjovski-Almeida, S.; Schust, D.J.; Franz, A.W.; Sadovsky, Y.; Ezashi, T. Vulnerability of primitive human placental trophoblast to Zika virus. Proc. Natl. Acad. Sci. USA 2017, 114, E1587–E1596. [Google Scholar] [CrossRef]
- Li, C.; Xu, D.; Ye, Q.; Hong, S.; Jiang, Y.; Liu, X.; Zhang, N.; Shi, L.; Qin, C.F.; Xu, Z. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell 2016, 19, 672. [Google Scholar] [CrossRef]
- Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.-M. Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Zwernik, S.D.; Adams, B.H.; Raymond, D.A.; Warner, C.M.; Kassam, A.B.; Rovin, R.A.; Akhtar, P. AXL receptor is required for Zika virus strain MR-766 infection in human glioblastoma cell lines. Mol. Ther. Oncolytics 2021, 23, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Al Kafri, N.; Ahnström, J.; Teraz-Orosz, A.; Chaput, L.; Singh, N.; Villoutreix, B.O.; Hafizi, S. The first laminin G-like domain of protein S is essential for binding and activation of Tyro3 receptor and intracellular signalling. Biochem. Biophys. Rep. 2022, 30, 101263. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Burstyn-Cohen, T. TAM receptors and the clearance of apoptotic cells. Ann. N. Y. Acad. Sci. 2010, 1209, 23–29. [Google Scholar] [CrossRef]
- Espino, A.; Gouilly, J.; Chen, Q.; Colin, P.; Guerby, P.; Izopet, J.; Amara, A.; Tabiasco, J.; Al-Daccak, R.; El Costa, H. The mechanisms underlying the immune control of zika virus infection at the maternal-fetal interface. Front. Immunol. 2022, 13, 1000861. [Google Scholar] [CrossRef]
- Komarasamy, T.V.; Adnan, N.A.A.; James, W.; Balasubramaniam, V. Zika Virus Neuropathogenesis: The Different Brain Cells, Host Factors and Mechanisms Involved. Front. Immunol. 2022, 13, 773191. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F. Biology of Zika virus infection in human skin cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef]
- Richard, A.S.; Shim, B.-S.; Kwon, Y.-C.; Zhang, R.; Otsuka, Y.; Schmitt, K.; Berri, F.; Diamond, M.S.; Choe, H. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc. Natl. Acad. Sci. USA 2017, 114, 2024–2029. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Kondratowicz, A.S.; Davey, R.A.; Rennert, P.D.; Maury, W. Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. J. Virol. 2013, 87, 8327–8341. [Google Scholar] [CrossRef]
- Estevez-Herrera, J.; Perez-Yanes, S.; Cabrera-Rodriguez, R.; Marquez-Arce, D.; Trujillo-Gonzalez, R.; Machado, J.D.; Madrid, R.; Valenzuela-Fernandez, A. Zika Virus Pathogenesis: A Battle for Immune Evasion. Vaccines 2021, 9, 294. [Google Scholar] [CrossRef]
- Eder, J.; Zijlstra-Willems, E.; Koen, G.; Kootstra, N.A.; Wolthers, K.C.; Geijtenbeek, T.B.J.F.i.I. Transmission of Zika virus by dendritic cell subsets in skin and vaginal mucosa. Cell 2023, 14, 1125565. [Google Scholar] [CrossRef] [PubMed]
- Jaimipuk, T.; Sachdev, S.; Yoksan, S.; Thepparit, C. A Small-Plaque Isolate of the Zika Virus with Envelope Domain III Mutations Affect Viral Entry and Replication in Mammalian but Not Mosquito Cells. Viruses 2022, 14, 480. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S. MEPPitope: Spatial, electrostatic and secondary structure perturbations in the post-fusion Dengue virus envelope protein highlights known epitopes and conserved residues in the Zika virus. F1000Research 2016, 5, 1150. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, D.; Kuhn, R.J. Zika virus structure, maturation, and receptors. J. Infect. Dis. 2017, 216, S935–S944. [Google Scholar] [CrossRef]
- Fontes-Garfias, C.R.; Shan, C.; Luo, H.; Muruato, A.E.; Medeiros, D.B.A.; Mays, E.; Xie, X.; Zou, J.; Roundy, C.M.; Wakamiya, M. Functional analysis of glycosylation of Zika virus envelope protein. Cell Rep. 2017, 21, 1180–1190. [Google Scholar] [CrossRef]
- Mossenta, M.; Marchese, S.; Poggianella, M.; Slon Campos, J.L.; Burrone, O.R. Role of N-glycosylation on Zika virus E protein secretion, viral assembly and infectivity. Biochem. Biophys. Res. Commun. 2017, 492, 579–586. [Google Scholar] [CrossRef]
- Morizono, K.; Chen, I.S. Role of phosphatidylserine receptors in enveloped virus infection. J. Virol. 2014, 88, 4275–4290. [Google Scholar] [CrossRef]
- Perera-Lecoin, M.; Meertens, L.; Carnec, X.; Amara, A. Flavivirus entry receptors: An update. Viruses 2013, 6, 69–88. [Google Scholar] [CrossRef]
- Li, M.; Zhang, D.; Li, C.; Zheng, Z.; Fu, M.; Ni, F.; Liu, Y.; Du, T.; Wang, H.; Griffin, G.E.J.F.I.M. Characterization of Zika virus endocytic pathways in human glioblastoma cells. Front. Microbiol. 2020, 11, 242. [Google Scholar] [CrossRef]
- Owczarek, K.; Chykunova, Y.; Jassoy, C.; Maksym, B.; Rajfur, Z.; Pyrc, K. Zika virus: Mapping and reprogramming the entry. Cell Commun. Signal. 2019, 17, 41. [Google Scholar] [CrossRef]
- Rinkenberger, N.; Schoggins, J.W. Comparative analysis of viral entry for Asian and African lineages of Zika virus. Virology 2019, 533, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Bi, S.; Zhao, K.; Yu, C. Inhibition of Mer and Axl receptor tyrosine kinases leads to increased apoptosis and improved chemosensitivity in human neuroblastoma. Biochem. Biophys. Res. Commun. 2015, 457, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Tappe, D.; Perez-Giron, J.V.; Zammarchi, L.; Rissland, J.; Ferreira, D.F.; Jaenisch, T.; Gomez-Medina, S.; Gunther, S.; Bartoloni, A.; Munoz-Fontela, C.; et al. Cytokine kinetics of Zika virus-infected patients from acute to reconvalescent phase. Med. Microbiol. Immunol. 2016, 205, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Galliez, R.M.; Spitz, M.; Rafful, P.P.; Cagy, M.; Escosteguy, C.; Germano, C.S.; Sasse, E.; Goncalves, A.L.; Silveira, P.P.; Pezzuto, P.; et al. Zika Virus Causing Encephalomyelitis Associated With Immunoactivation. Open Forum Infect. Dis. 2016, 3, ofw203. [Google Scholar] [CrossRef]
- Ornelas Pereira, I.; Santelli, A.; Leite, P.L.; Attell, J.; Bertolli, J.; Kotzky, K.; Araujo, W.N.; Peacock, G. Parental Stress in Primary Caregivers of Children with Evidence of Congenital Zika Virus Infection in Northeastern Brazil. Matern. Child Health J. 2021, 25, 360–367. [Google Scholar] [CrossRef]
- Figueiredo, C.P.; Barros-Aragao, F.G.Q.; Neris, R.L.S.; Frost, P.S.; Soares, C.; Souza, I.N.O.; Zeidler, J.D.; Zamberlan, D.C.; de Sousa, V.L.; Souza, A.S.; et al. Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat. Commun. 2019, 10, 3890. [Google Scholar] [CrossRef]
- Yang, M.; Sun, H.; Lai, H.; Hurtado, J.; Chen, Q. Plant-produced Zika virus envelope protein elicits neutralizing immune responses that correlate with protective immunity against Zika virus in mice. Plant. Biotechnol. J. 2018, 16, 572–580. [Google Scholar] [CrossRef]
- Shin, M.; Kim, K.; Lee, H.J.; Lee, R.; Jung, Y.J.; Park, J.; Hahn, T.W. Zika virus baculovirus-expressed envelope protein elicited humoral and cellular immunity in immunocompetent mice. Sci. Rep. 2022, 12, 660. [Google Scholar] [CrossRef]
- Lunardelli, V.A.S.; de Souza Apostolico, J.; Souza, H.F.S.; Coirada, F.C.; Martinho, J.A.; Astray, R.M.; Boscardin, S.B.; Rosa, D.S. ZIKV-envelope proteins induce specific humoral and cellular immunity in distinct mice strains. Sci. Rep. 2022, 12, 15733. [Google Scholar] [CrossRef]
- Hu, H.; Feng, Y.; He, M.L. Targeting Type I Interferon Induction and Signaling: How Zika Virus Escapes from Host Innate Immunity. Int. J. Biol. Sci. 2023, 19, 3015–3028. [Google Scholar] [CrossRef]
- Anglero-Rodriguez, Y.I.; MacLeod, H.J.; Kang, S.; Carlson, J.S.; Jupatanakul, N.; Dimopoulos, G. Aedes aegypti Molecular Responses to Zika Virus: Modulation of Infection by the Toll and Jak/Stat Immune Pathways and Virus Host Factors. Front. Microbiol. 2017, 8, 2050. [Google Scholar] [CrossRef]
- Strange, D.P.; Jiyarom, B.; Pourhabibi Zarandi, N.; Xie, X.; Baker, C.; Sadri-Ardekani, H.; Shi, P.-Y.; Verma, S. Axl promotes zika virus entry and modulates the antiviral state of human sertoli cells. mBio 2019, 10, e01372-19. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.G.; Wang, H.Y.; Wang, X.H.; Yang, T.; Zhang, X.M.; Feng, C.L.; Zhao, W.M.; Tang, W. Atranorin inhibits Zika virus infection in human glioblastoma cell line SNB-19 via targeting Zika virus envelope protein. Phytomedicine 2024, 125, 155343. [Google Scholar] [CrossRef] [PubMed]
- Mellors, J.; Tipton, T.; Longet, S.; Carroll, M. Viral Evasion of the Complement System and Its Importance for Vaccines and Therapeutics. Front. Immunol. 2020, 11, 1450. [Google Scholar] [CrossRef] [PubMed]
- Malekshahi, Z.; Schiela, B.; Bernklau, S.; Banki, Z.; Wurzner, R.; Stoiber, H. Interference of the Zika Virus E-Protein With the Membrane Attack Complex of the Complement System. Front. Immunol. 2020, 11, 569549. [Google Scholar] [CrossRef]
- Wen, D.; Li, S.; Dong, F.; Zhang, Y.; Lin, Y.; Wang, J.; Zou, Z.; Zheng, A. N-glycosylation of viral E protein is the determinant for vector midgut invasion by flaviviruses. mBio 2018, 9, e00046-18. [Google Scholar] [CrossRef]
- Dussupt, V.; Modjarrad, K.; Krebs, S.J. Landscape of monoclonal antibodies targeting zika and dengue: Therapeutic solutions and critical insights for vaccine development. Front. Immunol. 2021, 11, 621043. [Google Scholar] [CrossRef]
- Rey, F.A.; Stiasny, K.; Vaney, M.C.; Dellarole, M.; Heinz, F.X. The bright and the dark side of human antibody responses to flaviviruses: Lessons for vaccine design. EMBO Rep. 2018, 19, 206–224. [Google Scholar] [CrossRef]
- Gallichotte, E.N.; Young, E.F.; Baric, T.J.; Yount, B.L.; Metz, S.W.; Begley, M.C.; de Silva, A.M.; Baric, R.S. Role of Zika Virus Envelope Protein Domain III as a Target of Human Neutralizing Antibodies. mBio 2019, 10, e01485-19. [Google Scholar] [CrossRef]
- Yang, C.; Gong, R.; de Val, N. Development of Neutralizing Antibodies against Zika Virus Based on Its Envelope Protein Structure. Virol. Sin. 2019, 34, 168–174. [Google Scholar] [CrossRef]
- Zhao, H.; Xu, L.; Bombardi, R.; Nargi, R.; Deng, Z.; Errico, J.M.; Nelson, C.A.; Dowd, K.A.; Pierson, T.C.; Crowe, J.E.; et al. Mechanism of differential Zika and dengue virus neutralization by a public antibody lineage targeting the DIII lateral ridge. J. Exp. Med. 2020, 217, e20191792. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Fernandez, E.; Dowd, K.A.; Speer, S.D.; Platt, D.J.; Gorman, M.J.; Govero, J.; Nelson, C.A.; Pierson, T.C.; Diamond, M.S. Structural basis of Zika virus-specific antibody protection. Cell 2016, 166, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Van Rompay, K.K.A.; Coffey, L.L.; Kapoor, T.; Gazumyan, A.; Keesler, R.I.; Jurado, A.; Peace, A.; Agudelo, M.; Watanabe, J.; Usachenko, J. A combination of two human monoclonal antibodies limits fetal damage by Zika virus in macaques. Proc. Natl. Acad. Sci. USA 2020, 117, 7981–7989. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Bozzacco, L.; Keeffe, J.R.; Khouri, R.; Olsen, P.C.; Gazumyan, A.; Schaefer-Babajew, D.; Avila-Rios, S.; Nogueira, L.; Patel, R. Recurrent potent human neutralizing antibodies to Zika virus in Brazil and Mexico. Cell 2017, 169, 597–609. [Google Scholar] [CrossRef]
- Kim, S.I.; Kim, S.; Shim, J.M.; Lee, H.J.; Chang, S.Y.; Park, S.; Min, J.Y.; Park, W.B.; Oh, M.D.; Kim, S.; et al. Neutralization of Zika virus by E protein domain III-Specific human monoclonal antibody. Biochem. Biophys. Res. Commun. 2021, 545, 33–39. [Google Scholar] [CrossRef]
- Graham, S.D.; Tu, H.A.; McElvany, B.D.; Graham, N.R.; Grinyo, A.; Davidson, E.; Doranz, B.J.; Diehl, S.A.; de Silva, A.M.; Markmann, A.J. A Novel Antigenic Site Spanning Domains I and III of the Zika Virus Envelope Glycoprotein Is the Target of Strongly Neutralizing Human Monoclonal Antibodies. J. Virol. 2021, 95, e02423-20. [Google Scholar] [CrossRef]
- Stettler, K.; Beltramello, M.; Espinosa, D.A.; Graham, V.; Cassotta, A.; Bianchi, S.; Vanzetta, F.; Minola, A.; Jaconi, S.; Mele, F. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 2016, 353, 823–826. [Google Scholar] [CrossRef]
- Dussupt, V.; Sankhala, R.S.; Gromowski, G.D.; Donofrio, G.; De La Barrera, R.A.; Larocca, R.A.; Zaky, W.; Mendez-Rivera, L.; Choe, M.; Davidson, E. Potent Zika and dengue cross-neutralizing antibodies induced by Zika vaccination in a dengue-experienced donor. Nat. Med. 2020, 26, 228–235. [Google Scholar] [CrossRef]
- Barba-Spaeth, G.; Dejnirattisai, W.; Rouvinski, A.; Vaney, M.C.; Medits, I.; Sharma, A.; Simon-Loriere, E.; Sakuntabhai, A.; Cao-Lormeau, V.M.; Haouz, A.; et al. Structural basis of potent Zika-dengue virus antibody cross-neutralization. Nature 2016, 536, 48–53. [Google Scholar] [CrossRef]
- Wang, J.; Bardelli, M.; Espinosa, D.A.; Pedotti, M.; Ng, T.S.; Bianchi, S.; Simonelli, L.; Lim, E.X.Y.; Foglierini, M.; Zatta, F.; et al. A Human Bi-specific Antibody against Zika Virus with High Therapeutic Potential. Cell 2017, 171, 229–241.e25. [Google Scholar] [CrossRef]
- Deng, Y.-Q.; Dai, J.-X.; Ji, G.-H.; Jiang, T.; Wang, H.-J.; Yang, H.-o.; Tan, W.-L.; Liu, R.; Yu, M.; Ge, B.-X. A broadly flavivirus cross-neutralizing monoclonal antibody that recognizes a novel epitope within the fusion loop of E protein. PLoS ONE 2011, 6, e16059. [Google Scholar] [CrossRef] [PubMed]
- França, R.; Silva, J.M.; Rodrigues, L.S.; Sokolowskei, D.; Brigido, M.M.; Maranhão, A.Q. New Anti-Flavivirus Fusion Loop Human Antibodies with Zika Virus-Neutralizing Potential. Int. J. Mol. Sci. 2022, 23, 7805. [Google Scholar] [CrossRef] [PubMed]
- Qu, P.; Zhang, C.; Li, M.; Ma, W.; Xiong, P.; Liu, Q.; Zou, G.; Lavillette, D.; Yin, F.; Jin, X. A new class of broadly neutralizing antibodies that target the glycan loop of Zika virus envelope protein. Cell Discov. 2020, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- McLaury, A.R.; Haun, B.K.; To, A.; Mayerlen, L.; Medina, L.O.; Lai, C.Y.; Wong, T.A.S.; Nakano, E.; Strange, D.; Aquino, D.; et al. Characterization of Two Highly Specific Monoclonal Antibodies Targeting the Glycan Loop of the Zika Virus Envelope Protein. Viral Immunol. 2024, 37, 167–175. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, H.; Liu, X.; Dai, L.; Ma, T.; Qi, J.; Wong, G.; Peng, R.; Liu, S.; Li, J. Molecular determinants of human neutralizing antibodies isolated from a patient infected with Zika virus. Sci. Transl. Med. 2016, 8, 369ra179. [Google Scholar] [CrossRef]
- Sankhala, R.S.; Dussupt, V.; Donofrio, G.; Gromowski, G.D.; Rafael, A.; Larocca, R.A.; Mendez-Rivera, L.; Lee, A.; Choe, M.; Zaky, W. Zika-specific neutralizing antibodies targeting inter-dimer envelope epitopes. Cell Rep. 2023, 42, 112942. [Google Scholar] [CrossRef]
- Zhang, S.; Kostyuchenko, V.A.; Ng, T.S.; Lim, X.N.; Ooi, J.S.G.; Lambert, S.; Tan, T.Y.; Widman, D.G.; Shi, J.; Baric, R.S.; et al. Neutralization mechanism of a highly potent antibody against Zika virus. Nat. Commun. 2016, 7, 13679. [Google Scholar] [CrossRef]
- Fernandez, E.; Dejnirattisai, W.; Cao, B.; Scheaffer, S.M.; Supasa, P.; Wongwiwat, W.; Esakky, P.; Drury, A.; Mongkolsapaya, J.; Moley, K.H.; et al. Human antibodies to the dengue virus E-dimer epitope have therapeutic activity against Zika virus infection. Nat. Immunol. 2017, 18, 1261–1269. [Google Scholar] [CrossRef]
- Sapparapu, G.; Fernandez, E.; Kose, N.; Bin, C.; Fox, J.M.; Bombardi, R.G.; Zhao, H.; Nelson, C.A.; Bryan, A.L.; Barnes, T.; et al. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 2016, 540, 443–447. [Google Scholar] [CrossRef]
- Hasan, S.S.; Miller, A.; Sapparapu, G.; Fernandez, E.; Klose, T.; Long, F.; Fokine, A.; Porta, J.C.; Jiang, W.; Diamond, M.S. A human antibody against Zika virus crosslinks the E protein to prevent infection. Nat. Commun. 2017, 8, 14722. [Google Scholar] [CrossRef]
- Sariol, C.A.; Nogueira, M.L.; Vasilakis, N. A Tale of Two Viruses: Does Heterologous Flavivirus Immunity Enhance Zika Disease? Trends Microbiol. 2018, 26, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Langerak, T.; Mumtaz, N.; Tolk, V.I.; van Gorp, E.C.M.; Martina, B.E.; Rockx, B.; Koopmans, M.P.G. The possible role of cross-reactive dengue virus antibodies in Zika virus pathogenesis. PLoS Pathog. 2019, 15, e1007640. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.A.S.; de Oliveira-Filho, E.F.; Fernandes Iv, A.; Brito, C.A.A.; Marques, E.T.A.; Tenório, M.C.; Gil, L.H.G.V. Previous dengue or Zika virus exposure can drive to infection enhancement or neutralisation of other flaviviruses. Mem. Inst. Oswaldo Cruz 2019, 114, e190098. [Google Scholar] [CrossRef] [PubMed]
- Lessler, J.; Chaisson, L.H.; Kucirka, L.M.; Bi, Q.; Grantz, K.; Salje, H.; Carcelen, A.C.; Ott, C.T.; Sheffield, J.S.; Ferguson, N.M. Assessing the global threat from Zika virus. Science 2016, 353, aaf8160. [Google Scholar] [CrossRef]
- Castanha, P.M.S.; Marques, E.T.A. A glimmer of hope: Recent updates and future challenges in Zika vaccine development. Viruses 2020, 12, 1371. [Google Scholar] [CrossRef]
- Malafa, S.; Medits, I.; Aberle, J.H.; Aberle, S.W.; Haslwanter, D.; Tsouchnikas, G.; Wolfel, S.; Huber, K.L.; Percivalle, E.; Cherpillod, P.; et al. Impact of flavivirus vaccine-induced immunity on primary Zika virus antibody response in humans. PLoS Negl. Trop. Dis. 2020, 14, e0008034. [Google Scholar] [CrossRef]
- Montecillo-Aguado, M.R.; Montes-Gomez, A.E.; Garcia-Cordero, J.; Corzo-Gomez, J.; Vivanco-Cid, H.; Mellado-Sanchez, G.; Munoz-Medina, J.E.; Gutierrez-Castaneda, B.; Santos-Argumedo, L.; Gonzalez-Bonilla, C.; et al. Cross-Reaction, Enhancement, and Neutralization Activity of Dengue Virus Antibodies against Zika Virus: A Study in the Mexican Population. J. Immunol. Res. 2019, 2019, 7239347. [Google Scholar] [CrossRef]
- Wang, W.H.; Urbina, A.N.; Wu, C.C.; Lin, C.Y.; Thitithanyanont, A.; Assavalapsakul, W.; Lu, P.L.; Chen, Y.H.; Wang, S.F. An epidemiological survey of the current status of Zika and the immune interaction between dengue and Zika infection in Southern Taiwan. Int. J. Infect. Dis. 2020, 93, 151–159. [Google Scholar] [CrossRef]
- Bonheur, A.N.; Thomas, S.; Soshnick, S.H.; McGibbon, E.; Dupuis, A.P., 2nd; Hull, R.; Slavinski, S.; Del Rosso, P.E.; Weiss, D.; Hunt, D.T.; et al. A fatal case report of antibody-dependent enhancement of dengue virus type 1 following remote Zika virus infection. BMC Infect. Dis. 2021, 21, 749. [Google Scholar] [CrossRef]
- Bardina, S.V.; Bunduc, P.; Tripathi, S.; Duehr, J.; Frere, J.J.; Brown, J.A.; Nachbagauer, R.; Foster, G.A.; Krysztof, D.; Tortorella, D.; et al. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 2017, 356, 175–180. [Google Scholar] [CrossRef]
- Crooks, C.M.; Weiler, A.M.; Rybarczyk, S.L.; Bliss, M.I.; Jaeger, A.S.; Murphy, M.E.; Simmons, H.A.; Mejia, A.; Fritsch, M.K.; Hayes, J.M.; et al. Previous exposure to dengue virus is associated with increased Zika virus burden at the maternal-fetal interface in rhesus macaques. PLoS Negl. Trop. Dis. 2021, 15, e0009641. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dai, L.; Song, H.; Gao, G.F. Structures of Zika Virus E & NS1: Relations with Virus Infection and Host Immune Responses. Adv. Exp. Med. Biol. 2018, 1062, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Wolford, R.W.; Schaefer, T.J. Zika Virus. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Figueiredo, L.T. The Brazilian flaviviruses. Microbes Infect. 2000, 2, 1643–1649. [Google Scholar] [CrossRef] [PubMed]
- Anez, G.; Rios, M. Dengue in the United States of America: A worsening scenario? BioMed Res. Int. 2013, 2013, 678645. [Google Scholar] [CrossRef]
- Chang, H.H.; Huber, R.G.; Bond, P.J.; Grad, Y.H.; Camerini, D.; Maurer-Stroh, S.; Lipsitch, M. Systematic analysis of protein identity between Zika virus and other arthropod-borne viruses. Bull. World Health Organ. 2017, 95, 517–525I. [Google Scholar] [CrossRef]
- Sekaran, S.D.; Ismail, A.A.; Thergarajan, G.; Chandramathi, S.; Rahman, S.K.H.; Mani, R.R.; Jusof, F.F.; Lim, Y.A.L.; Manikam, R. Host immune response against DENV and ZIKV infections. Front. Cell. Infect. Microbiol. 2022, 12, 975222. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Supasa, P.; Wongwiwat, W.; Rouvinski, A.; Barba-Spaeth, G.; Duangchinda, T.; Sakuntabhai, A.; Cao-Lormeau, V.-M.; Malasit, P.; Rey, F.A.; et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat. Immunol. 2016, 17, 1102–1108. [Google Scholar] [CrossRef]
- Kribs, C.; Greenhalgh, D.J.J.o.M.B. Impact of tetravalent dengue vaccination with screening, ADE, and altered infectivity on single-serotype dengue and Zika transmission. J. Math. Biol. 2023, 86, 85. [Google Scholar] [CrossRef]
- Xu, M.; Zuest, R.; Velumani, S.; Tukijan, F.; Toh, Y.X.; Appanna, R.; Tan, E.Y.; Cerny, D.; MacAry, P.; Wang, C.I.; et al. A potent neutralizing antibody with therapeutic potential against all four serotypes of dengue virus. NPJ Vaccines 2017, 2, 2. [Google Scholar] [CrossRef]
- Kam, Y.-W.; Lee, C.Y.-P.; Teo, T.-H.; Howland, S.W.; Amrun, S.N.; Lum, F.-M.; See, P.; Kng, N.Q.-R.; Huber, R.G.; Xu, M.-H. Cross-reactive dengue human monoclonal antibody prevents severe pathologies and death from Zika virus infections. JCI Insight 2017, 2, e92428. [Google Scholar] [CrossRef]
- Sun, H.; Yang, M.; Lai, H.; Neupane, B.; Teh, A.Y.; Jugler, C.; Ma, J.K.; Steinkellner, H.; Bai, F.; Chen, Q. A Dual-Approach Strategy to Optimize the Safety and Efficacy of Anti-Zika Virus Monoclonal Antibody Therapeutics. Viruses 2023, 15, 1156. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Malik, Y.S.; Singh, R.K.; Chaicumpa, W. Modulation of Dengue/Zika Virus Pathogenicity by Antibody-Dependent Enhancement and Strategies to Protect Against Enhancement in Zika Virus Infection. Front. Immunol. 2018, 9, 597. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.A.; Pierson, T.C. Antibody-mediated neutralization of flaviviruses: A reductionist view. Virology 2011, 411, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Prosser, O.; Kumar, P.; Tuplin, A.; Giri, R. Small molecule inhibitors possibly targeting the rearrangement of Zika virus envelope protein. Antivir. Res. 2020, 182, 104876. [Google Scholar] [CrossRef]
- Ci, Y.; Yao, B.; Yue, K.; Yang, Y.; Xu, C.; Li, D.F.; Qin, C.F.; Shi, L. Bortezomib inhibits ZIKV/DENV by interfering with viral polyprotein cleavage via the ERAD pathway. Cell Chem. Biol. 2023, 30, 527–539.e5. [Google Scholar] [CrossRef]
- Gao, Y.; Tai, W.; Wang, N.; Li, X.; Jiang, S.; Debnath, A.K.; Du, L.; Chen, S. Identification of Novel Natural Products as Effective and Broad-Spectrum Anti-Zika Virus Inhibitors. Viruses 2019, 11, 1019. [Google Scholar] [CrossRef]
- Carneiro, B.M.; Batista, M.N.; Braga, A.C.S.; Nogueira, M.L.; Rahal, P. The green tea molecule EGCG inhibits Zika virus entry. Virology 2016, 496, 215–218. [Google Scholar] [CrossRef]
- Behrendt, P.; Perin, P.; Menzel, N.; Banda, D.; Pfaender, S.; Alves, M.P.; Thiel, V.; Meuleman, P.; Colpitts, C.C.; Schang, L.M. Pentagalloylglucose, a highly bioavailable polyphenolic compound present in Cortex moutan, efficiently blocks hepatitis C virus entry. Antivir. Res. 2017, 147, 19–28. [Google Scholar] [CrossRef]
- Sharma, N.; Kumar, P.; Giri, R. Polysaccharides like pentagalloylglucose, parishin a and stevioside inhibits the viral entry by binding the Zika virus envelope protein. J. Biomol. Struct. Dyn. 2021, 39, 6008–6020. [Google Scholar] [CrossRef]
- Sangeetha, K.; Martin-Acebes, M.A.; Saiz, J.C.; Meena, K.S. Molecular docking and antiviral activities of plant derived compounds against zika virus. Microb. Pathog. 2020, 149, 104540. [Google Scholar] [CrossRef]
- Oo, A.; Teoh, B.T.; Sam, S.S.; Bakar, S.A.; Zandi, K. Baicalein and baicalin as Zika virus inhibitors. Arch. Virol. 2019, 164, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-J.; Lu, J.-W.; Huang, Y.-L.; Lai, Z.-Z. Palmatine inhibits Zika virus infection by disrupting virus binding, entry, and stability. Biochem. Biophys. Res. Commun. 2019, 518, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.-Z.; Ho, Y.-J.; Lu, J.-W. Harringtonine inhibits Zika virus infection through multiple mechanisms. Molecules 2020, 25, 4082. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; DeLalio, L.J.; Isakson, B.E.; Wang, T.T. AXL-Mediated Productive Infection of Human Endothelial Cells by Zika Virus. Circ. Res. 2016, 119, 1183–1189. [Google Scholar] [CrossRef]
- Zhang, B.; Yu, J.; Zhu, G.; Huang, Y.; Zhang, K.; Xiao, X.; He, W.; Yuan, J.; Gao, X. Dapoxetine, a Selective Serotonin Reuptake Inhibitor, Suppresses Zika Virus Infection In Vitro. Molecules 2023, 28, 8142. [Google Scholar] [CrossRef]
- Fernando, S.; Fernando, T.; Stefanik, M.; Eyer, L.; Ruzek, D. An Approach for Zika Virus Inhibition Using Homology Structure of the Envelope Protein. Mol. Biotechnol. 2016, 58, 801–806. [Google Scholar] [CrossRef]
- Mounce, B.C.; Cesaro, T.; Carrau, L.; Vallet, T.; Vignuzzi, M. Curcumin inhibits Zika and chikungunya virus infection by inhibiting cell binding. Antivir. Res. 2017, 142, 148–157. [Google Scholar] [CrossRef]
- Kim, M.; Choi, H.; Kim, Y.B. Therapeutic targets and biological mechanisms of action of curcumin against Zika virus: In silico and in vitro analyses. Eur. J. Pharmacol. 2021, 904, 174144. [Google Scholar] [CrossRef]
- Pitts, J.; Hsia, C.Y.; Lian, W.; Wang, J.; Pfeil, M.P.; Kwiatkowski, N.; Li, Z.; Jang, J.; Gray, N.S.; Yang, P.L. Identification of small molecule inhibitors targeting the Zika virus envelope protein. Antivir. Res. 2019, 164, 147–153. [Google Scholar] [CrossRef]
- Li, F.; Lang, Y.; Ji, Z.; Xia, Z.; Han, Y.; Cheng, Y.; Liu, G.; Sun, F.; Zhao, Y.; Gao, M. A scorpion venom peptide Ev37 restricts viral late entry by alkalizing acidic organelles. J. Biol. Chem. 2019, 294, 182–194. [Google Scholar] [CrossRef]
- Clark, H.L.; Minns, M.S.; Sun, Y.; de Jesus, T.; Ghannoum, M.G.; Pearlman, E. Atovaquone impairs growth of Aspergillus and Fusarium keratitis isolates by modulating mitochondrial function and zinc homeostasis. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.L.; Moss, D.M.; Shone, A.E.; Lalloo, D.G.; Fisher, N.; O’Neill, P.M.; Ward, S.A.; Biagini, G.A. Antimalarial pharmacology and therapeutics of atovaquone. J. Antimicrob. Chemother. 2013, 68, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Ichinohe, T.; Watanabe, A.; Kobayashi, A.; Zhang, R.; Song, J.; Kawaguchi, Y.; Matsuda, Z.; Inoue, J.-i. The antimalarial compound atovaquone inhibits Zika and dengue virus infection by blocking E protein-mediated membrane fusion. Viruses 2020, 12, 1475. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liang, R.; Gao, Y.; Li, Y.; Deng, X.; Xiang, R.; Zhang, Y.; Ying, T.; Jiang, S.; Yu, F. Development of Small-Molecule Inhibitors Against Zika Virus Infection. Front. Microbiol. 2019, 10, 2725. [Google Scholar] [CrossRef]
- Yu, Y.; Deng, Y.Q.; Zou, P.; Wang, Q.; Dai, Y.; Yu, F.; Du, L.; Zhang, N.N.; Tian, M.; Hao, J.N.; et al. A peptide-based viral inactivator inhibits Zika virus infection in pregnant mice and fetuses. Nat. Commun. 2017, 8, 15672. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Y.; Wang, S.; Sun, J.; Wang, P.; Xin, Q.; Zhang, L.; Xiao, G.; Wang, W. Antiviral activity of peptide inhibitors derived from the protein E stem against Japanese encephalitis and Zika viruses. Antivir. Res. 2017, 141, 140–149. [Google Scholar] [CrossRef]
- Hu, D.; Wang, Y.; Li, A.; Li, Q.; Wu, C.; Shereen, M.A.; Huang, S.; Wu, K.; Zhu, Y.; Wang, W. LAMR1 restricts Zika virus infection by attenuating the envelope protein ubiquitination. Virulence 2021, 12, 1795–1807. [Google Scholar] [CrossRef]
- Ling, J.; Morente, S.F.; Lundkvist, Å.; Li, J. Heparanase attenuates Zika virus infection by destabilizing the viral envelope protein. bioRxiv 2025. [Google Scholar] [CrossRef]
- Van der Hoek, K.H.; Eyre, N.S.; Shue, B.; Khantisitthiporn, O.; Glab-Ampi, K.; Carr, J.M.; Gartner, M.J.; Jolly, L.A.; Thomas, P.Q.; Adikusuma, F.; et al. Viperin is an important host restriction factor in control of Zika virus infection. Sci. Rep. 2017, 7, 4475. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Q.; Hu, D.; Gao, D.; Wang, W.; Wu, K.; Wu, J. USP38 Inhibits Zika Virus Infection by Removing Envelope Protein Ubiquitination. Viruses 2021, 13, 2099. [Google Scholar] [CrossRef]
- Shan, C.; Xie, X.; Muruato, A.E.; Rossi, S.L.; Roundy, C.M.; Azar, S.R.; Yang, Y.; Tesh, R.B.; Bourne, N.; Barrett, A.D. An infectious cDNA clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe 2016, 19, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Annamalai, A.S.; Pattnaik, A.; Sahoo, B.R.; Guinn, Z.P.; Bullard, B.L.; Weaver, E.A.; Steffen, D.; Natarajan, S.K.; Petro, T.M.; Pattnaik, A.K. An Attenuated Zika Virus Encoding Non-Glycosylated Envelope (E) and Non-Structural Protein 1 (NS1) Confers Complete Protection against Lethal Challenge in a Mouse Model. Vaccines 2019, 7, 112. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Portela Catani, J.P.; Mc Cafferty, S.; Couck, L.; Van Den Broeck, W.; Gorlé, N.; Vandenbroucke, R.E.; Devriendt, B.; Ulbert, S.; Cnops, L. Immunogenicity and protection efficacy of a naked self-replicating mRNA-based Zika virus vaccine. Vaccines 2019, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Contreras, M.; Stuart, J.B.; Levoir, L.M.; Belmont, L.; Goo, L. Defining the impact of flavivirus envelope protein glycosylation site mutations on sensitivity to broadly neutralizing antibodies. mBio 2024, 15, e03048-23. [Google Scholar] [CrossRef]
- Feng, Y.J.F.i.P. Recent advances in the study of zika virus structure, drug targets, and inhibitors. Front. Pharmacol. 2024, 15, 1418516. [Google Scholar] [CrossRef]
- Khan, E.; Jindal, H.; Mishra, P.; Suvvari, T.K.; Jonna, S. The 2021 Zika outbreak in Uttar Pradesh state of India: Tackling the emerging public health threat. Trop. Dr. 2022, 52, 474–478. [Google Scholar] [CrossRef]
- Regla-Nava, J.A.; Wang, Y.T.; Fontes-Garfias, C.R.; Liu, Y.; Syed, T.; Susantono, M.; Gonzalez, A.; Viramontes, K.M.; Verma, S.K.; Kim, K.; et al. A Zika virus mutation enhances transmission potential and confers escape from protective dengue virus immunity. Cell Rep. 2022, 39, 110655. [Google Scholar] [CrossRef]
- Chan, K.W.K.; Bifani, A.M.; Watanabe, S.; Choy, M.M.; Ooi, E.E.; Vasudevan, S.G. Tissue-specific expansion of Zika virus isogenic variants drive disease pathogenesis. eBioMedicine 2023, 91, 104570. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Gonzalez-Orozco, M.; Rajsbaum, R. Pathogenesis of Zika Virus Infection. Annu. Rev. Pathol. 2023, 18, 181–203. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roozitalab, A.; Zhang, J.; Zhang, C.; Tang, Q.; Zhao, R.Y. The Evolving Role of Zika Virus Envelope Protein in Viral Entry and Pathogenesis. Viruses 2025, 17, 817. https://doi.org/10.3390/v17060817
Roozitalab A, Zhang J, Zhang C, Tang Q, Zhao RY. The Evolving Role of Zika Virus Envelope Protein in Viral Entry and Pathogenesis. Viruses. 2025; 17(6):817. https://doi.org/10.3390/v17060817
Chicago/Turabian StyleRoozitalab, Ashkan, Jiantao Zhang, Chenyu Zhang, Qiyi Tang, and Richard Y. Zhao. 2025. "The Evolving Role of Zika Virus Envelope Protein in Viral Entry and Pathogenesis" Viruses 17, no. 6: 817. https://doi.org/10.3390/v17060817
APA StyleRoozitalab, A., Zhang, J., Zhang, C., Tang, Q., & Zhao, R. Y. (2025). The Evolving Role of Zika Virus Envelope Protein in Viral Entry and Pathogenesis. Viruses, 17(6), 817. https://doi.org/10.3390/v17060817