
Metagenomic Investigation of Pathogenic RNA Viruses Causing Diarrhea in Sika Deer Fawns


Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Collection of Fecal Samples and Processing
2.3. RNA Isolation
2.4. Viral Metagenomic Library Construction and High-Throughput Sequencing
2.5. Bioinformatics Analysis
2.6. Real-Time PCR
2.7. Isolation and Identification of Rotavirus
2.8. Whole-Genome Sequencing and Assembly of the Rotavirus Genome
2.9. Phylogenetic Analysis
3. Results
3.1. Quality Control Analysis of Viral Metagenomic Data
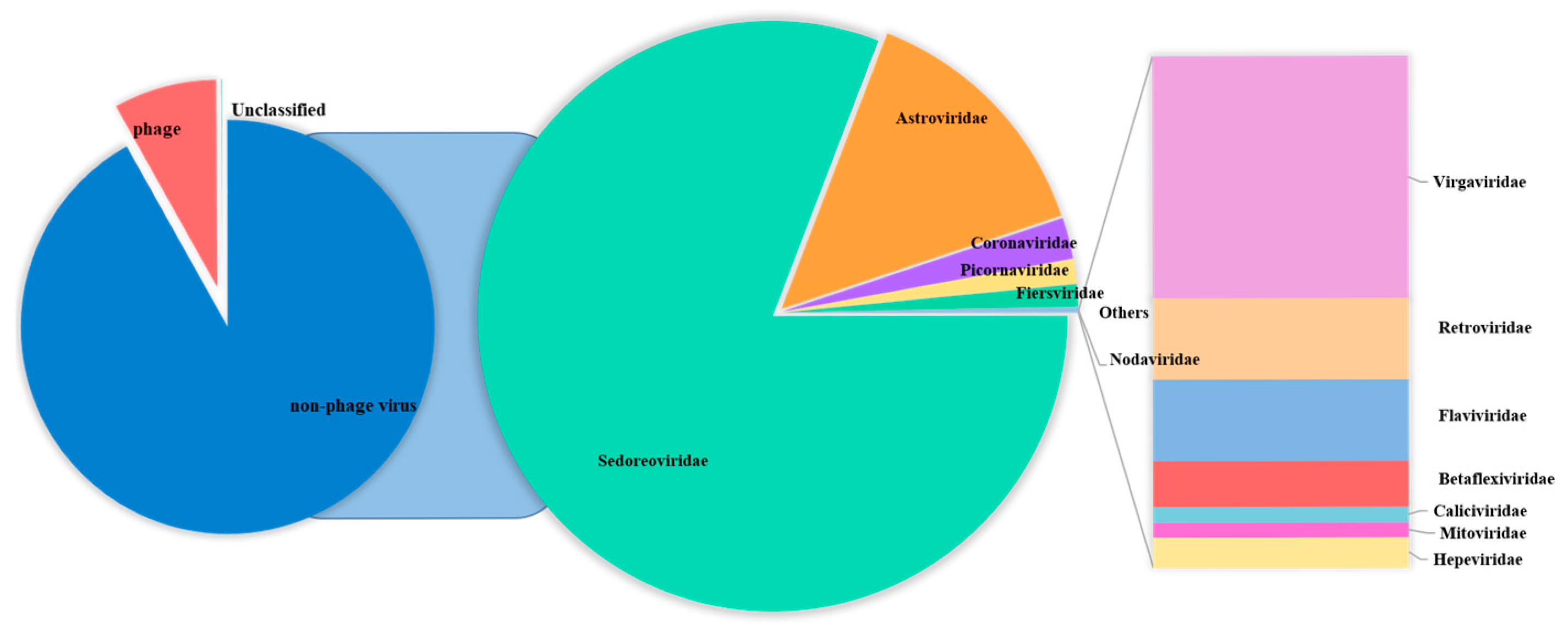
3.2. Diversity Analysis of Enteric RNA Viruses in Fawns
3.3. Epidemiological Investigation of Enteric Pathogens in Fawns
3.4. In Vitro Identification of Rotavirus
3.5. Genetype Analysis of Rotavirus
3.6. Genetic Evolutionary Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lv, Y.; Tong, Z.; Liu, J.; Zhang, Z.; Wang, C.; Zeng, Y.; Liu, P.; Zong, X.; Chen, G.; Chen, H.; et al. Molecular Characterization and Pathogenicity Analysis of Porcine Rotavirus A. Viruses 2024, 16, 1842. [Google Scholar] [CrossRef]
- Wang, L.P.; Zhou, S.X.; Wang, X.; Lu, Q.B.; Shi, L.S.; Ren, X.; Zhang, H.Y.; Wang, Y.F.; Lin, S.H.; Zhang, C.H.; et al. Etiological, epidemiological, and clinical features of acute diarrhea in China. Nat. Commun. 2021, 12, 2464. [Google Scholar] [CrossRef] [PubMed]
- Troeger, C.E.; Khalil, I.A.; Blacker, B.F.; Biehl, M.H.; Albertson, S.B.; Zimsen, S.R.M.; Rao, P.C.; Abate, D.; Ahmadi, A.; Ahmed, M.L.C.; et al. Quantifying risks and interventions that have affected the burden of diarrhoea among children younger than 5 years: An analysis of the Global Burden of Disease Study 2017. Lancet Infect. Dis. 2020, 20, 37–59. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, A.; Foroozanfard, F.; Tamadon, M.R. Evaluation of water and electrolytes disorders in severe acute diarrhea patients treated by WHO protocol in eight large hospitals in Tehran; a nephrology viewpoint. J. Ren. Inj. Prev. 2017, 6, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Pop, M.; Walker, A.W.; Paulson, J.; Lindsay, B.; Antonio, M.; Hossain, M.A.; Oundo, J.; Tamboura, B.; Mai, V.; Astrovskaya, I.; et al. Diarrhea in young children from low-income countries leads to large-scale alterations in intestinal microbiota composition. Genome Biol. 2014, 15, R76. [Google Scholar] [CrossRef]
- Guerrant, R.L.; DeBoer, M.D.; Moore, S.R.; Scharf, R.J.; Lima, A.A. The impoverished gut--a triple burden of diarrhoea, stunting and chronic disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 220–229. [Google Scholar] [CrossRef]
- Ma, W.; Gong, Z.; Abu-Sbeih, H.; Peng, Y.; Peng, F.; Zou, F.; Charabaty, A.; Okhuysen, P.C.; McQuade, J.L.; Altan, M.; et al. Outcomes of Immune Checkpoint Inhibitor-related Diarrhea or Colitis in Cancer Patients With Superimposed Gastrointestinal Infections. Am. J. Clin. Oncol. 2021, 44, 402–408. [Google Scholar] [CrossRef]
- Sparasci, O.A.; Rizzo, F.; Marchino, M.; Cerutti, F.; Guglielmetti, C.; Grosjacques, F.; Muratore, E.; Sona, B.; Biolatti, P.; Peletto, S.; et al. Influenza D Virus in Cattle and Swine in Piedmont Region, North-Western Italy. Int. J. Infect. Dis. 2022, 116, S92. [Google Scholar] [CrossRef]
- Bartels, C.J.; Holzhauer, M.; Jorritsma, R.; Swart, W.A.; Lam, T.J. Prevalence, prediction and risk factors of enteropathogens in normal and non-normal faeces of young Dutch dairy calves. Prev. Vet. Med. 2010, 93, 162–169. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, H.K.; Moon, H.J.; Song, D.S.; Rho, S.M.; Han, J.Y.; Nguyen, V.G.; Park, B.K. Molecular detection of porcine kobuviruses in pigs in Korea and their association with diarrhea. Arch. Virol. 2010, 155, 1803–1811. [Google Scholar] [CrossRef]
- Dall Agnol, A.M.; Lorenzetti, E.; Leme, R.A.; Ladeia, W.A.; Mainardi, R.M.; Bernardi, A.; Headley, S.A.; Freire, R.L.; Pereira, U.P.; Alfieri, A.F.; et al. Severe outbreak of bovine neonatal diarrhea in a dairy calf rearing unit with multifactorial etiology. Braz. J. Microbiol. 2021, 52, 2547–2553. [Google Scholar] [CrossRef]
- Zhao, W.; Xu, J.; Xiao, M.; Cao, J.; Jiang, Y.; Huang, H.; Zheng, B.; Shen, Y. Prevalence and Characterization of Cryptosporidium Species and Genotypes in Four Farmed Deer Species in the Northeast of China. Front. Vet. Sci. 2020, 7, 430. [Google Scholar] [CrossRef] [PubMed]
- Brunauer, M.; Roch, F.F.; Conrady, B. Prevalence of Worldwide Neonatal Calf Diarrhoea Caused by Bovine Rotavirus in Combination with Bovine Coronavirus, Escherichia coli K99 and Cryptosporidium spp.: A Meta-Analysis. Animals 2021, 11, 1014. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, L.; Zheng, Y.; Zhang, J.; Guo, B.; Yoon, K.J.; Gauger, P.C.; Harmon, K.M.; Main, R.G.; Li, G. Metagenomic analysis of the RNA fraction of the fecal virome indicates high diversity in pigs infected by porcine endemic diarrhea virus in the United States. Virol. J. 2018, 15, 95. [Google Scholar] [CrossRef]
- Trujillo-Ortega, M.E.; Beltrán-Figueroa, R.; García-Hernández, M.E.; Juárez-Ramírez, M.; Sotomayor-González, A.; Hernández-Villegas, E.N.; Becerra-Hernández, J.F.; Sarmiento-Silva, R.E. Isolation and characterization of porcine epidemic diarrhea virus associated with the 2014 disease outbreak in Mexico: Case report. BMC Vet. Res. 2016, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Trovão, N.S.; Shepherd, F.K.; Herzberg, K.; Jarvis, M.C.; Lam, H.C.; Rovira, A.; Culhane, M.R.; Nelson, M.I.; Marthaler, D.G. Evolution of rotavirus C in humans and several domestic animal species. Zoonoses Public. Health 2019, 66, 546–557. [Google Scholar] [CrossRef]
- Qiu, M.; Li, S.; Xiao, Y.; Li, J.; Zhang, Y.; Li, X.; Feng, B.; Li, C.; Lin, H.; Zhu, J.; et al. Pathogenic and metagenomic evaluations reveal the correlations of porcine epidemic diarrhea virus, porcine kobuvirus and porcine astroviruses with neonatal piglet diarrhea. Microb. Pathog. 2022, 170, 105703. [Google Scholar] [CrossRef]
- Dhakal, T.; Kim, T.S.; Kim, S.H.; Tiwari, S.; Kim, J.Y.; Jang, G.S.; Lee, D.H. Distribution of sika deer (Cervus nippon) and the bioclimatic impact on their habitats in South Korea. Sci. Rep. 2023, 13, 19040. [Google Scholar] [CrossRef]
- Rehbein, S.; Lindner, T.; Visser, M.; Lutz, W.; Reindl, H. Distribution, prevalence, and intensity of Sarcocystis infections in sika deer (Cervus nippon) of free-ranging populations in Germany and Austria. Parasitol. Res. 2022, 121, 2079–2086. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, S.; Du, R.; Wang, Q.; Sun, C.; Wang, N.; Zhang, P.; Zhang, L. Isolation and identification of a bovine viral diarrhea virus from sika deer in china. Virol. J. 2011, 8, 83. [Google Scholar] [CrossRef]
- Passler, T.; Ditchkoff, S.S.; Walz, P.H. Bovine Viral Diarrhea Virus (BVDV) in White-Tailed Deer (Odocoileus virginianus). Front. Microbiol. 2016, 7, 945. [Google Scholar] [CrossRef] [PubMed]
- das Neves, C.G.; Johansson Wensman, J.; Nymo, I.H.; Skjerve, E.; Alenius, S.; Tryland, M. Pestivirus Infections in Semi-Domesticated Eurasian Tundra Reindeer (Rangifer tarandus tarandus): A Retrospective Cross-Sectional Serological Study in Finnmark County, Norway. Viruses 2019, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, W.G.; Gois, B.V.A.; Pinheiro, K.D.C.; Aragão, A.O.; Queiroz, A.L.C.; da Silva, A.L.; Folador, A.C.; Ramos, R.T.J. Viral Metagenomics Reveals Widely Diverse Viral Community of Freshwater Amazonian Lake. Front. Public. Health 2022, 10, 869886. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.H.; Lin, S.C.; Hsu, Y.H.; Chen, S.Y. The Human Virome: Viral Metagenomics, Relations with Human Diseases, and Therapeutic Applications. Viruses 2022, 14, 278. [Google Scholar] [CrossRef]
- Coutinho, F.H.; Silveira, C.B.; Gregoracci, G.B.; Thompson, C.C.; Edwards, R.A.; Brussaard, C.P.D.; Dutilh, B.E.; Thompson, F.L. Marine viruses discovered via metagenomics shed light on viral strategies throughout the oceans. Nat. Commun. 2017, 8, 15955. [Google Scholar] [CrossRef]
- Li, Y.; Altan, E.; Reyes, G.; Halstead, B.; Deng, X.; Delwart, E. Virome of Bat Guano from Nine Northern California Roosts. J. Virol. 2021, 95, e01713-20. [Google Scholar] [CrossRef]
- George, D.R.; Edris, W.; Hanson, R.; Gilman, F. The art of medicine Medicinal plants-the next generation. Lancet 2016, 387, 220–221. [Google Scholar] [CrossRef]
- Day, J.M.; Ballard, L.L.; Duke, M.V.; Scheffler, B.E.; Zsak, L. Metagenomic analysis of the turkey gut RNA virus community. Virol. J. 2010, 7, 313. [Google Scholar] [CrossRef]
- Wang, C.; Li, A.; Shi, Q.; Yu, Z. Metagenomic next-generation sequencing clinches diagnosis of leishmaniasis. Lancet 2021, 397, 1213. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Park, S.-H.; Saif, L.J.; Jeong, C.; Lim, G.-K.; Park, S.-I.; Kim, H.-H.; Park, S.-J.; Kim, Y.-J.; Jeong, J.-H.; Kang, M.-I.; et al. Molecular Characterization of Novel G5 Bovine Rotavirus Strains. J. Clin. Microbiol. 2006, 44, 4101–4112. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Shen, H.; Zhao, S.; Chen, S.; Zhu, P.; Lin, W.; Chen, F. Isolation and Pathogenicity Analysis of a G5P[23] Porcine Rotavirus Strain. Viruses 2023, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, O.; Peregrin, A.T.; Persson, K.; Kordasti, S.; Uhnoo, I.; Svensson, L. Role of the enteric nervous system in the fluid and electrolyte secretion of rotavirus diarrhea. Science 2000, 287, 491–495. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kumar, S.; Tamura, K.; Nei, M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief. Bioinform. 2004, 5, 150–163. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Ackermann, H.-W.; Anany, H.; Blasdel, B.; Connerton, I.F.; Goulding, D.; Griffiths, M.W.; Hooton, S.P.; Kutter, E.M.; Kropinski, A.M.; et al. A suggested new bacteriophage genus: “Viunalikevirus”. Arch. Virol. 2012, 157, 2035–2046. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Siddell, S.G.; Walker, P.J.; Lefkowitz, E.J.; Mushegian, A.R.; Adams, M.J.; Dutilh, B.E.; Gorbalenya, A.E.; Harrach, B.; Harrison, R.L.; Junglen, S.; et al. Additional changes to taxonomy ratified in a special vote by the International Committee on Taxonomy of Viruses (October 2018). Arch Virol 2019, 164, 943–946. [Google Scholar] [CrossRef]
- Callanan, J.; Stockdale, S.R.; Adriaenssens, E.M.; Kuhn, J.H.; Rumnieks, J.; Pallen, M.J.; Shkoporov, A.N.; Draper, L.A.; Ross, R.P.; Hill, C. Leviviricetes: Expanding and restructuring the taxonomy of bacteria-infecting single-stranded RNA viruses. Microb. Genom. 2021, 7, 000686. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P.; Members Of The International Committee On The Taxonomy Of Viruses Study Group; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.-J.; Okamoto, H.; Vander Poel, W.H.M.; Purdy, M.A. Consensus proposals forclassification of thefamily Hepeviridae. J. Gen. Virol. 2014, 95, 2223–2232, Corrigendum in J. Gen. Virol. 2015, 96, 1191–1192. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Zirkel, F.; Kurth, A.; van Cleef, K.W.R.; Drosten, C.; van Rij, R.P.; Junglen, S. A unique nodavirus with novel features: Mosinovirus expresses two subgenomic RNAs, a capsid gene of unknown origin, and a suppressor of the antiviral RNA interference pathway. J. Virol. 2014, 88, 13447–13459. [Google Scholar] [CrossRef]
- Vinjé, J.; Estes, M.K.; Esteves, P.; Green, K.Y.; Katayama, K.; Knowles, N.J.; L’Homme, Y.; Martella, V.; Vennema, H.; White, P.A. ICTV Virus Taxonomy Profile: Caliciviridae. J. Gen. Virol. 2019, 100, 1469–1470. [Google Scholar] [CrossRef]
- Williams, S.H.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.; et al. Viral Diversity of House Mice in New York City. mBio 2018, 9, e01354-17. [Google Scholar] [CrossRef]
- Attoui, H.; Jaafar, F.M.; Belhouchet, M.; de Micco, P.; de Lamballerie, X.; Brussaard, C.P.D. Micromonas pusilla reovirus: A new member of the family Reoviridae assigned to a novel proposed genus (Mimoreovirus). J. Gen. Virol. 2006, 87, 1375–1383. [Google Scholar] [CrossRef]
- Attoui, H.; Fang, Q.; Jaafar, F.M.; Cantaloube, J.-F.; Biagini, P.; de Micco, P.; de Lamballerie, X. Common evolutionary origin of aquareoviruses and orthoreoviruses revealed by genome characterization of Golden shiner reovirus, Grass carp reovirus, Striped bass reovirus and golden ide reovirus (genus Aquareovirus, family Reoviridae). J. Gen. Virol. 2002, 83, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Zhuang, Z.; Lu, J.; Wang, H.; Wang, X.; Yang, S.; Ji, L.; Shen, Q.; Zhang, W.; Shan, T. Metagenomic survey of viral diversity obtained from feces of piglets with diarrhea. Heliyon 2024, 10, e25616. [Google Scholar] [CrossRef]
- Kwok, K.T.T.; Nieuwenhuijse, D.F.; Phan, M.V.T.; Koopmans, M.P.G. Virus Metagenomics in Farm Animals: A Systematic Review. Viruses 2020, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, H.Y.; Choi, E.W.; Kim, D. Causative agents and epidemiology of diarrhea in Korean native calves. J. Vet. Sci. 2019, 20, e64. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gómara, M.; Maes, P.; Patton, J.T.; et al. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, M.A.; Sattar, A.; Khalid, K.; Khalid, A.R.; Mahmood, M.S.; Aleem, M.T.; Ashraf, K.; Aslam, F.; Alouffi, A.; Mohammed, A.; et al. Molecular and Morphological Characterization of Eimeria crandallis Isolated from Deer (Cervidae) in Different Captive Animals. Life 2022, 12, 1621. [Google Scholar] [CrossRef]
- Majhdi, F.; Minocha, H.C.; Kapil, S. Isolation and characterization of a coronavirus from elk calves with diarrhea. J. Clin. Microbiol. 1997, 35, 2937–2942. [Google Scholar] [CrossRef]
- Tsunemitsu, H.; el-Kanawati, Z.R.; Smith, D.R.; Reed, H.H.; Saif, L.J. Isolation of coronaviruses antigenically indistinguishable from bovine coronavirus from wild ruminants with diarrhea. J. Clin. Microbiol. 1995, 33, 3264–3269. [Google Scholar] [CrossRef]
- Lu, J.T.; Campeau, P.M.; Lee, B.H. Genotype-phenotype correlation--promiscuity in the era of next-generation sequencing. N. Engl. J. Med. 2014, 371, 593–596. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, Q.; Yang, P.; Zhang, C.; Mortuza, S.M.; Xue, Z.; Ning, K.; Zhang, Y. Fueling ab initio folding with marine metagenomics enables structure and function predictions of new protein families. Genome Biol. 2019, 20, 229. [Google Scholar] [CrossRef]
- Niazian, M. Application of genetics and biotechnology for improving medicinal plants. Planta 2019, 249, 953–973. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Jia, R.; Xu, M.; Liu, P.; Su, L.; Cao, L.; Zhu, X.; Lu, L.; Xu, J. Emergence and high prevalence of unusual rotavirus G8P[8] strains in outpatients with acute gastroenteritis in Shanghai, China. J. Med. Virol. 2024, 96, e29368. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Y.; Yang, L.; Li, J.; Li, P.; Yang, C.; Jia, L.; Qiu, S.; Song, H.; Li, P. Genomic analysis of an acute gastroenteritis outbreak caused by rotavirus C in Hebei, China. Virol. J. 2024, 21, 242. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Otto, P.H.; Ciarlet, M.; Desselberger, U.; Van Ranst, M.; Johne, R. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch. Virol. 2012, 157, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Jesudason, T.; Sharomi, O.; Fleetwood, K.; Cheuk, A.L.; Bermudez, M.; Schirrmacher, H.; Hauck, C.; Matthijnssens, J.; Hungerford, D.; Tordrup, D.; et al. Systematic literature review and meta-analysis on the prevalence of rotavirus genotypes in Europe and the Middle East in the post-licensure period. Hum. Vaccin. Immunother. 2024, 20, 2389606. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Z.; Jiang, J.; He, B.; He, S.; Tu, C.; Guo, Y.; Gong, W. Outbreak of piglet diarrhea associated with a new reassortant porcine rotavirus B. Vet. Microbiol. 2024, 288, 109947. [Google Scholar] [CrossRef]
- Mebus, C.A.; Stair, E.L.; Rhodes, M.B.; Twiehaus, M.J. Pathology of neonatal calf diarrhea induced by a coronavirus-like agent. Vet. Pathol. 1973, 10, 45–64. [Google Scholar] [CrossRef]
- Beuttemmuller, E.A.; Alfieri, A.F.; Headley, S.A.; Alfieri, A.A. Brazilian strain of bovine respiratory coronavirus is derived from dual enteric and respiratory tropism. Genet. Mol. Res. 2017, 16, gmr16029580. [Google Scholar] [CrossRef]
- Amoroso, M.G.; Lucifora, G.; Degli Uberti, B.; Serra, F.; De Luca, G.; Borriello, G.; De Domenico, A.; Brandi, S.; Cuomo, M.C.; Bove, F.; et al. Fatal Interstitial Pneumonia Associated with Bovine Coronavirus in Cows from Southern Italy. Viruses 2020, 12, 1331. [Google Scholar] [CrossRef]
- Amer, H.M. Bovine-like coronaviruses in domestic and wild ruminants. Anim. Health Res. Rev. 2018, 19, 113–124. [Google Scholar] [CrossRef]
- Lorusso, A.; Desario, C.; Mari, V.; Campolo, M.; Lorusso, E.; Elia, G.; Martella, V.; Buonavoglia, C.; Decaro, N. Molecular characterization of a canine respiratory coronavirus strain detected in Italy. Virus Res. 2009, 141, 96–100. [Google Scholar] [CrossRef]
- Zhang, X.M.; Herbst, W.; Kousoulas, K.G.; Storz, J. Biological and genetic characterization of a hemagglutinating coronavirus isolated from a diarrhoeic child. J. Med. Virol. 1994, 44, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Tamamoto, C.; Seino, N.; Suzuki, M.; Kaji, K.; Takahashi, H.; Inokuma, H. Detection of Ehrlichia muris DNA from sika deer (Cervus nippon yesoensis) in Hokkaido, Japan. Vet. Parasitol. 2007, 150, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Che, L.-F.; Wu, Q.X.; Zhao, Z.M.; Zhu, Q.F.; Jin, X.L. A survey of BVDV, BCoV and BRV infections in sika deer in some areas of Shaanxi. Spec. Econ. Anim. Plants 2022, 25, 1–5. [Google Scholar]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.C. The clinical manifestations of bovine viral diarrhea infection. Vet. Clin. North. Am. Food Anim. Pract. 1995, 11, 425–445. [Google Scholar] [CrossRef]
- Moennig, V.; Liess, B. Pathogenesis of intrauterine infections with bovine viral diarrhea virus. Vet. Clin. North. Am. Food Anim. Pract. 1995, 11, 477–487. [Google Scholar] [CrossRef]
- Tesfaye, A.; Omer, A.; Hussein, A.; Garoma, A.; Guyassa, C.; Paeshuyse, J.; Tolera, T.S. Seroprevalence of Bovine Viral Diarrhea Virus in Local Borana Cattle Breed and Camels (Camelus dromedarius) in Ethiopia. Vet. Med. 2021, 12, 141–148. [Google Scholar] [CrossRef]
- Choe, S.; Lim, S.I.; Park, G.N.; Song, S.; Shin, J.; Kim, K.S.; Hyun, B.H.; Kim, J.H.; An, D.J. Prevalence of Bovine Viral Diarrhea Virus Infections in Pigs on Jeju Island, South Korea, from 2009-2019 and Experimental Infection of Pigs with BVDV Strains Isolated from Cattle. Vet. Sci. 2022, 9, 146. [Google Scholar] [CrossRef]
- Krametter-Froetscher, R.; Duenser, M.; Preyler, B.; Theiner, A.; Benetka, V.; Moestl, K.; Baumgartner, W. Pestivirus infection in sheep and goats in West Austria. Vet. J. 2010, 186, 342–346. [Google Scholar] [CrossRef]
- Cantu, A.; Ortega, S.J.; Mosqueda, J.; Garcia-Vazquez, Z.; Henke, S.E.; George, J.E. Prevalence of infectious agents in free-ranging white-tailed deer in northeastern Mexico. J. Wildl. Dis. 2008, 44, 1002–1007. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Clean Reads | All Tran Num | Clean Bases | Error Rate | Q20 | Q30 | GC Content |
|---|---|---|---|---|---|---|---|
| A1 | 37,291,842 | 56,013 | 5.59 G | 0.02 | 98.56 | 95.57 | 40 |
| A2 | 45,535,848 | 84,558 | 6.83 G | 0.02 | 98.63 | 95.75 | 44.53 |
| A4 | 36,317,604 | 53,317 | 5.45 G | 0.02 | 98.47 | 95.34 | 47.47 |
| A3 | 34,308,390 | 54,727 | 5.15 G | 0.02 | 98.43 | 95.22 | 39.22 |
| A5 | 40,418,916 | 42,393 | 6.06 G | 0.02 | 98.42 | 95.15 | 37.46 |
| Family/Groups | A1 | A2 | A3 | A4 | A5 | Total |
|---|---|---|---|---|---|---|
| Sedoreoviridae | 95.21% | 78.00% | 93.85% | 17.55% | 96.82% | 82.10% |
| Astroviridae | 3.932% | 20.27% | 5.582% | 61.17% | 2.779% | 14.25% |
| Coronaviridae | 0.007009% | 0.01029% | 0.004904% | 15.90% | 0.1652% | 2.279% |
| Caliciviridae | 0 | 0 | 0.0007864% | 0.009401% | 0.00004804% | 0.001481% |
| Retroviridae | 0.003899% | 0.01039% | 0.006507% | 0.02781% | 0.001106% | 0.007843% |
| Nodaviridae | 0.001326% | 0 | 0 | 0 | 0 | 0.0003038% |
| Hepeviridae | 0.001023% | 0.004980% | 0.0006945% | 0.0004869% | 0.0003797% | 0.001258% |
| Flaviviridae | 0.0003907% | 0.01831% | 0.006307% | 0.01925% | 0.003013% | 0.007497% |
| Picornaviridae | 0.8440% | 1.690% | 0.5484% | 5.318% | 0.2328% | 1.353% |
| Pathogens | Positive Samples | Negative Samples | Positive Rate/% |
|---|---|---|---|
| BRV | 212 | 148 | 58.89 |
| BCoV | 214 | 146 | 59.44 |
| BVDV | 78 | 282 | 21.67 |
| BRV and BCoV | 97 | 263 | 26.94 |
| BRV and BVDV | 5 | 355 | 1.39 |
| BCoV and BVDV | 13 | 347 | 3.61 |
| BRV, BCoV and BVDV | 58 | 302 | 16.11 |
| Gene | Length (bp) | Coding Region (bp) | Genotype | Virus with the Highest Identity | Identity (%) | GenBank ID |
|---|---|---|---|---|---|---|
| VP1 | 3302 | 3267 | R2 | Human-wt/HUN/BP1062 | 99.76 | PV158729 |
| VP2 | 2690 | 2260 | C2 | Human-wt/MWI/MW2-181_B | 100 | PV158730 |
| VP3 | 2508 | 2508 | M2 | Cow-tc/China/SCMY2 | 99.48 | PV158731 |
| VP4 | 2362 | 2088 | P[1] | Cow-tc/CHN/SDA2/G6P[1] | 99.83 | PV158732 |
| VP6 | 1352 | 1215 | I2 | RV/Bovine/HM26 | 96.60 | PV158733 |
| VP7 | 1062 | 981 | G6 | Yak-tc/China/F8 | 96.81 | PV158734 |
| NSP1 | 1579 | 1476 | A3 | Yak-tc/CHN/HY-1 | 100.00 | PV158735 |
| NSP2 | 1055 | 996 | N2 | Bovine/C73 | 97.43 | PV158736 |
| NSP3 | 1074 | 966 | T6 | Lamb/CHN/LLR | 98.24 | PV158737 |
| NSP4 | 749 | 567 | E2 | Bovine/HM26 | 98.40 | PV158738 |
| NSP5 | 597 | 597 | H3 | Cow-tc/JPN/GB12-22 | 98.32 | PV158739 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Wang, Q.; Cao, R.; Li, Y.; Liu, Z.; Xue, Z.; Wang, X.; Liu, Z. Metagenomic Investigation of Pathogenic RNA Viruses Causing Diarrhea in Sika Deer Fawns. Viruses 2025, 17, 803. https://doi.org/10.3390/v17060803
Wang W, Wang Q, Cao R, Li Y, Liu Z, Xue Z, Wang X, Liu Z. Metagenomic Investigation of Pathogenic RNA Viruses Causing Diarrhea in Sika Deer Fawns. Viruses. 2025; 17(6):803. https://doi.org/10.3390/v17060803
Chicago/Turabian StyleWang, Weiyang, Qilin Wang, Runlai Cao, Yacong Li, Ziyu Liu, Zhuqing Xue, Xiaoxu Wang, and Zhijie Liu. 2025. "Metagenomic Investigation of Pathogenic RNA Viruses Causing Diarrhea in Sika Deer Fawns" Viruses 17, no. 6: 803. https://doi.org/10.3390/v17060803
APA StyleWang, W., Wang, Q., Cao, R., Li, Y., Liu, Z., Xue, Z., Wang, X., & Liu, Z. (2025). Metagenomic Investigation of Pathogenic RNA Viruses Causing Diarrhea in Sika Deer Fawns. Viruses, 17(6), 803. https://doi.org/10.3390/v17060803