
Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity



Abstract
1. Quasispecies and Lethal Mutagenesis
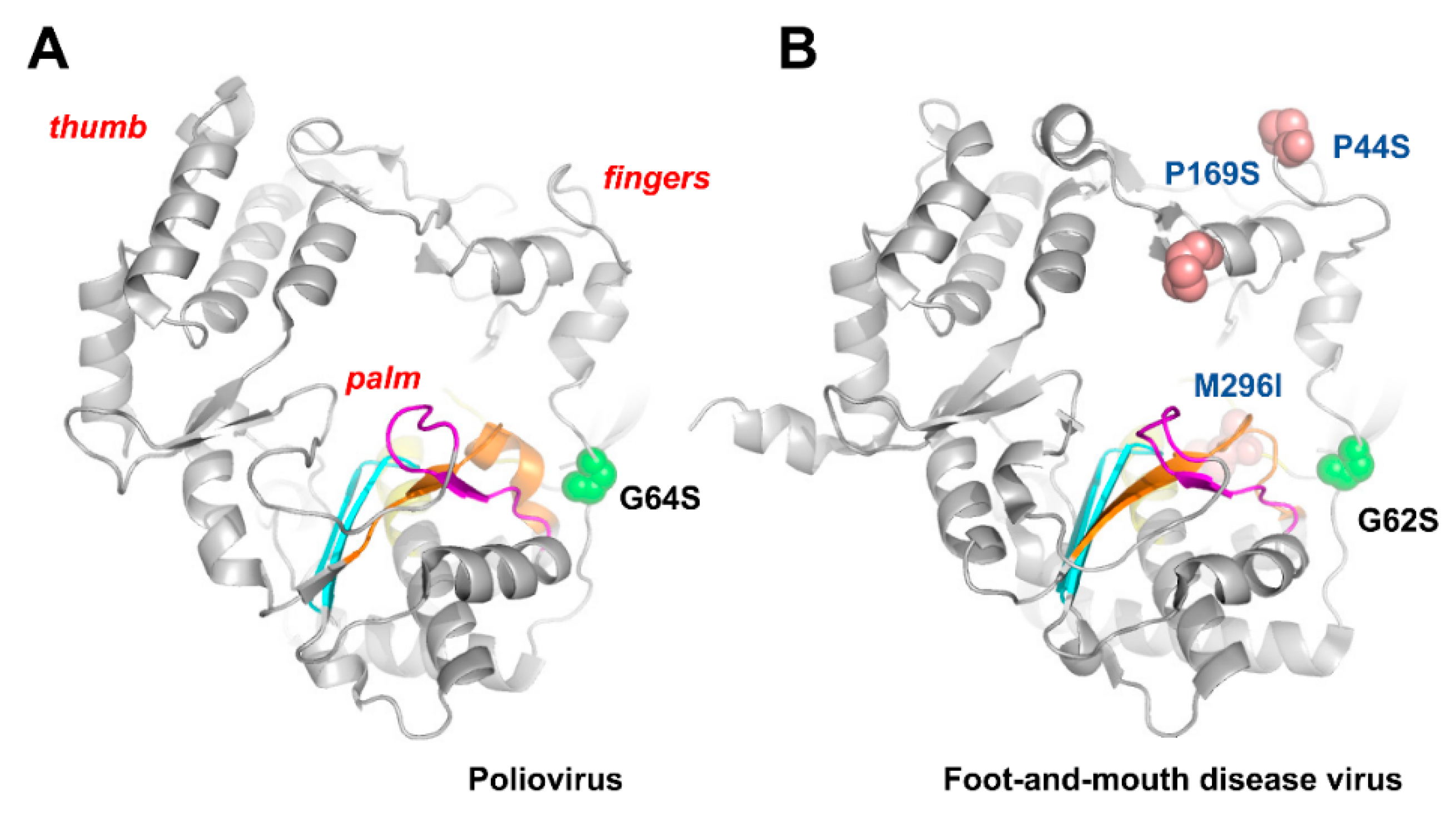
2. Mutation Rates and Fidelity of Viral Polymerases
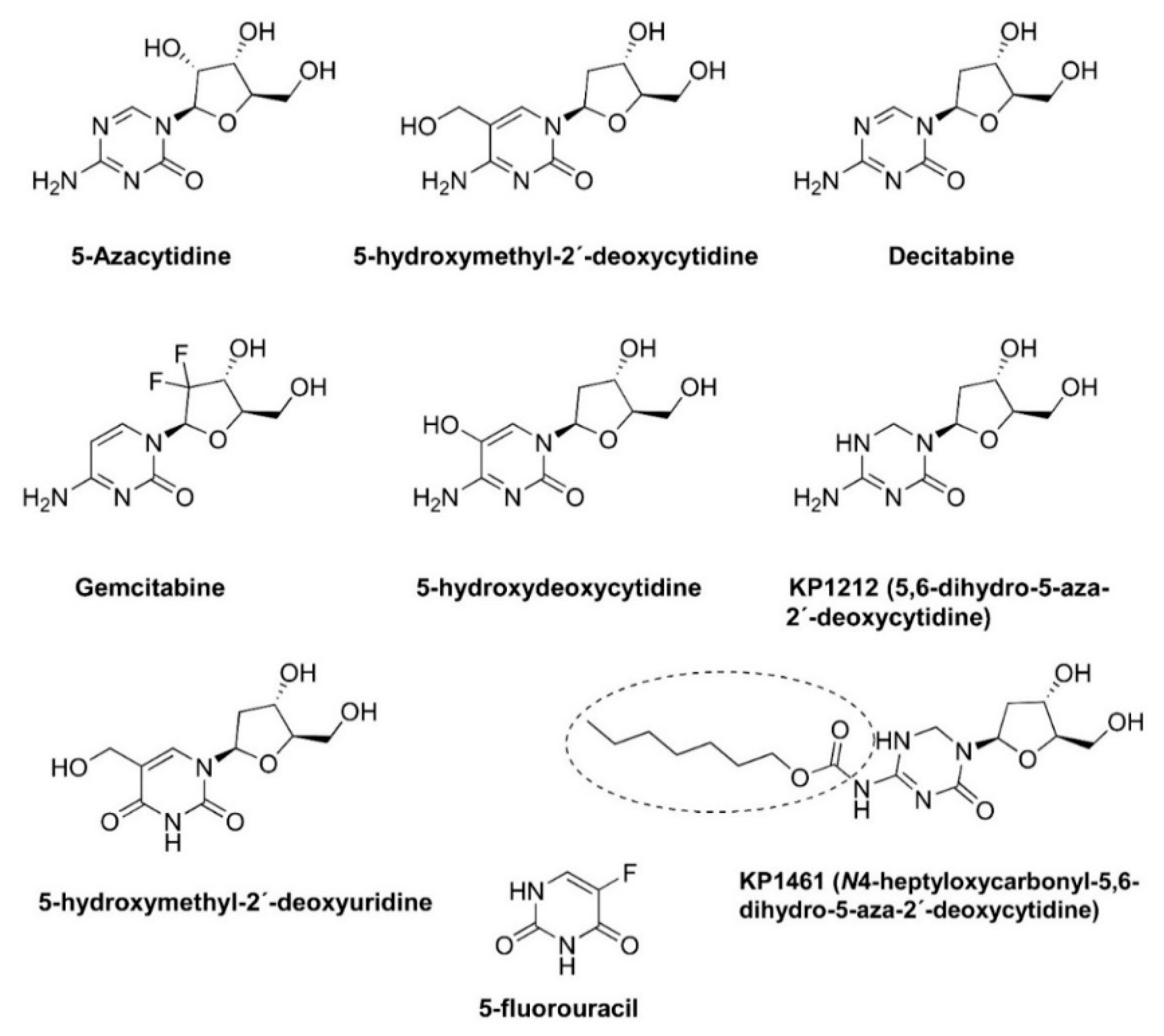
3. Driving HIV into Error Catastrophe and Preliminary Clinical Development of KP1212/KP1461 as an Antiretroviral Agent Causing Lethal Mutagenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutagenic Nucleoside | Increase in Mutation Frequency | Mutational Preference | References |
|---|---|---|---|
| 5-azacytidine | 2.3-fold | G/C transversions | [45,47] |
| 5-fluorouracil | <1.5-fold | A/G, U/C transitions | [48] |
| 5-hydroxymethyl-2′-deoxycytidine | 3.4-fold | G→A, G→T | [49] |
| 5-hydroxymethyl-2′-deoxyuridine | 3.1-fold | A→G, G→C | [49] |
| Decitabine (5-aza-2′-deoxycytidine) | 4.1-fold | G/C transversions | [50] |
| Gemcitabine (2,2(′)-difluoro-2(′)-deoxycytidine) | <1.5-fold | [48] |
4. Lethal Mutagenesis in Non-Retroviral RNA Viruses: An Overview of Studies Showing the Effects of Base and Nucleoside Analogs
| Virus Names | Family/Genus | Mutagenic Base and Nucleoside Analogs | Refs. | |
|---|---|---|---|---|
| Positive-strand RNA viruses | ||||
| - | Poliovirus | Picornaviridae/Enterovirus | Ribavirin, 5-nitrocytidine, 6-(β-d-ribofuranosyl)-3,4-dihydro-8H-pyrimido [4,5-c][1,2]oxazin-7-one, and N-6-substituted purine analogs (JA28 and JA30) | [69,70,71,72] |
| Coxsackievirus | Picornaviridae/Enterovirus | Ribavirin, and N-6-substituted purine analogs | [72,73] | |
| Encephalomyocarditis virus | Picornaviridae/Cardiovirus | 5-Fluorouracil | [74] | |
| Foot-and-mouth disease virus | Picornaviridae/Aphthovirus | 5-Fluorouracil, ribavirin, and favipiravir | [74,75,76,77,78,79] | |
| Murine norovirus | Caliciviridae/Norovirus | Favipiravir | [80] | |
| Dengue virus | Flaviviridae/Flavivirus | 3-Hydroxy-2-pyrazinecarboxamide (T-1105), and its ribose derivative (T-1106) | [81] | |
| Usutu virus | Flaviviridae/Flavivirus | 5-Fluorouracil and favipiravir, while ribavirin effects are less pronounced. | [82] | |
| West Nile virus | Flaviviridae/Flavivirus | Ribavirin and favipiravir | [83,84] | |
| Zika virus | Flaviviridae/Flavivirus | Ribavirin and favipiravir | [82] | |
| GB virus B | Flaviviridae/Hepacivirus | Ribavirin | [85] | |
| Hepatitis C virus | Flaviviridae/Hepacivirus | Ribavirin and favipiravir | [86,87,88,89,90,91,92] | |
| Hepatitis E virus | Hepeviridae/Orthohepevirus | Ribavirin | [93] | |
| Venezuelan equine encephalitis virus | Togaviridae/Alphavirus | β-d-N4-hydroxycytidine (molnupiravir) | [94] | |
| SARS-CoV-2 | Coronaviridae/Betacoronavirus | Favipiravir and β-d-N4-hydroxycytidine (molnupiravir) | [68,95,96] | |
| Tobacco mosaic virus | Virgaviridae/Tobamovirus | 5-Fluorouracil | [97] | |
| Negative-strand RNA viruses | ||||
| Influenza A virus | Orthomyxoviridae/ Alphainfluenzavirus | Ribavirin, 5-azacytidine, 5-fluorouracil, and β-d-N4-hydroxycytidine (molnupiravir) | [98,99] | |
| Vesicular stomatitis virus | Rhabdoviridae/Vesiculovirus | 5-Fluorouracil | [74,100] | |
| Hantaan virus | Hantaviridae/Orthohantaviridae | Ribavirin | [101,102] | |
| Rift Valley fever virus | Phenuiviridae/Phlebovirus | Favipiravir | [103] | |
| Lymphocytic choriomeningitis virus | Arenaviridae/Mammarenavirus | 5-Fluorouracil | [74,104] | |
| Ebola virus | Filoviridae/Ebolavirus | Favipiravir | [105] | |
| Marburg virus | Filoviridae/Marburgvirus | Favipiravir | [105] | |
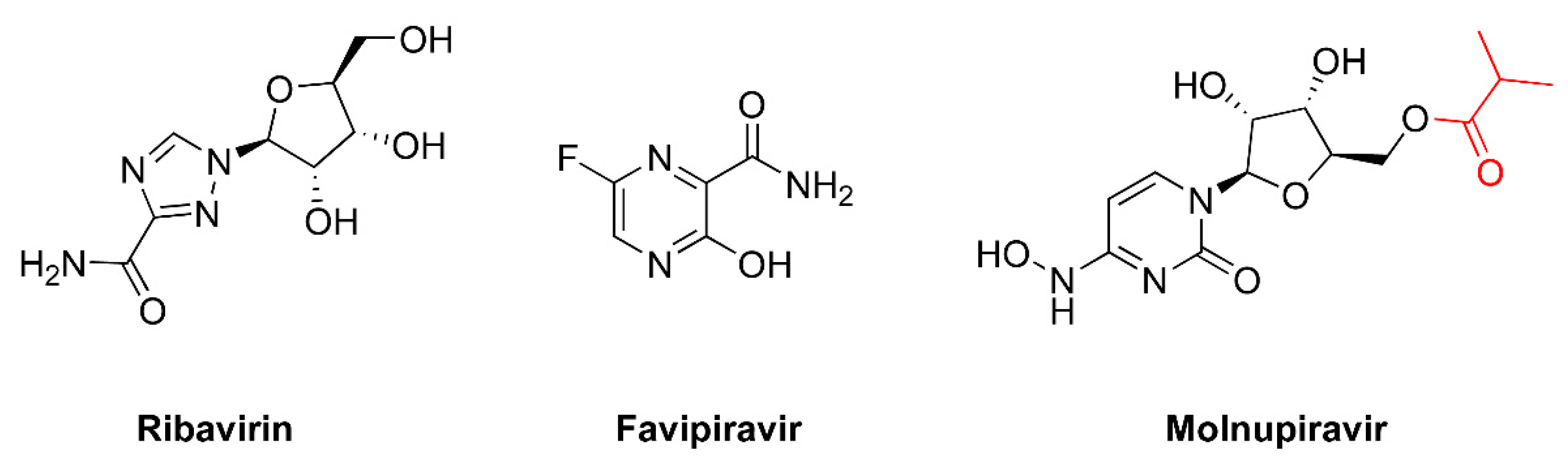
5. Mutagenic Effects of Ribavirin

6. Favipiravir as a Lethal Mutagenesis Agent
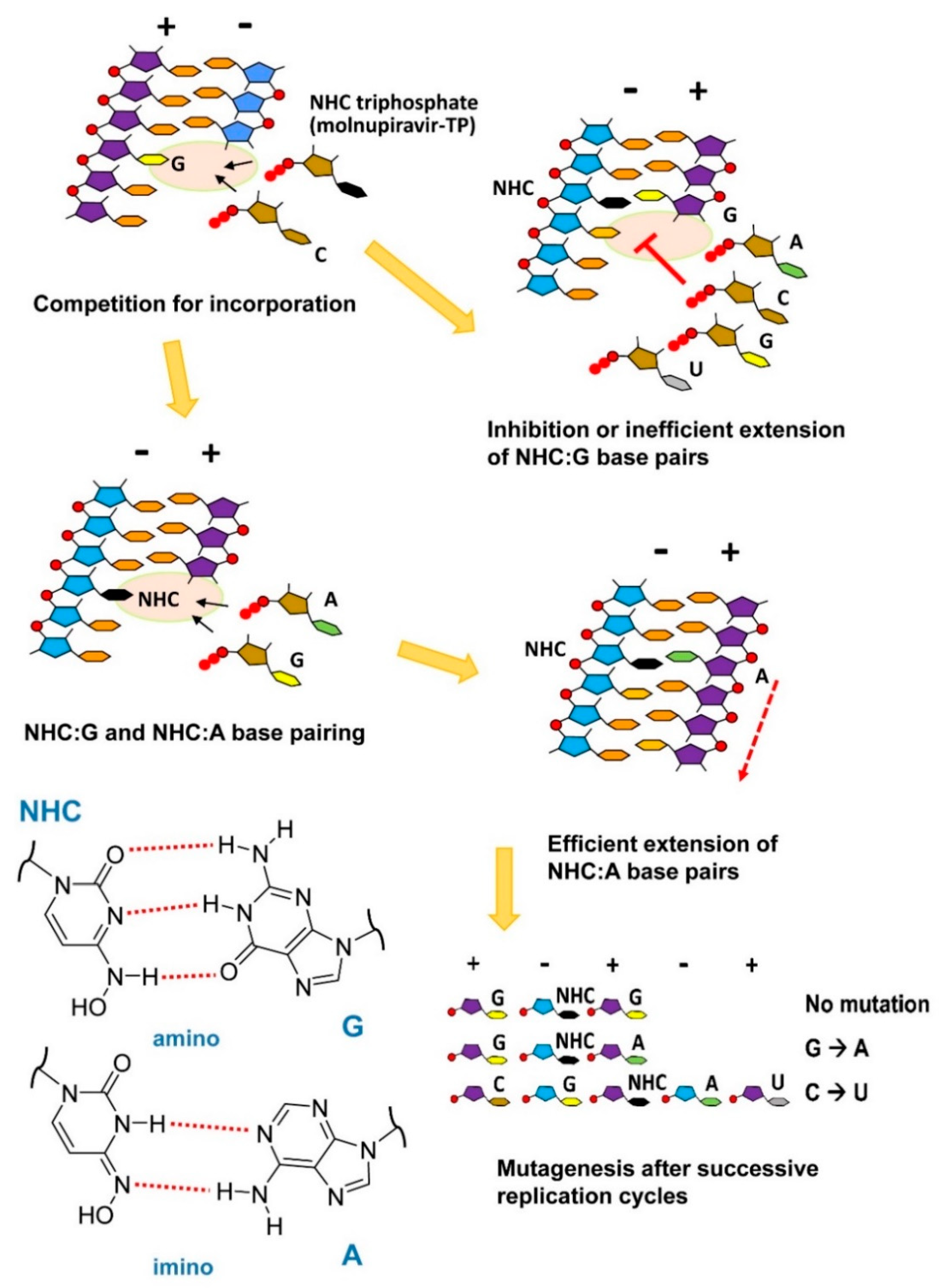
7. Molnupiravir as a Broad-Spectrum Antiviral Drug Effective against SARS-CoV-2

8. Future Directions and Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; García-Crespo, C.; Perales, C. Historical perspective on the discovery of the quasispecies concept. Annu. Rev. Virol. 2021, 8, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. USA 2002, 99, 13374–13376. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. From Strange Simplicity to Complex Familiarity: A Treatise on Matter, Information, Life and Thought; Oxford University Press: Cary, NC, USA, 2013. [Google Scholar]
- Weissmann, C.; Billeter, M.A.; Goodman, H.M.; Hindley, J.; Weber, H. Structure and function of phage RNA. Annu. Rev. Biochem. 1973, 42, 303–328. [Google Scholar] [CrossRef] [PubMed]
- Haruna, I.; Spiegelman, S. Recognition of size and sequence by an RNA replicase. Proc. Natl. Acad. Sci. USA 1965, 54, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Haruna, I.; Spiegelman, S. Specific template requirments of RNA replicases. Proc. Natl. Acad. Sci. USA 1965, 54, 579–587. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef]
- Perales, C.; Domingo, E. Antiviral strategies based on lethal mutagenesis and error threshold. Curr. Top. Microbiol. Immunol. 2016, 392, 323–339. [Google Scholar] [CrossRef]
- Holland, J.J.; Domingo, E.; de la Torre, J.C.; Steinhauer, D.A. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol. 1990, 64, 3960–3962. [Google Scholar] [CrossRef]
- Anderson, J.P.; Daifuku, R.; Loeb, L.A. Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 2004, 58, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Essigmann, J.M.; Kazazi, F.; Zhang, J.; Rose, K.D.; Mullins, J.I. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. USA 1999, 96, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Combe, M.; Sanjuán, R. Variation in RNA virus mutation rates across host cells. PLoS Pathog. 2014, 10, e1003855. [Google Scholar] [CrossRef]
- Peck, K.M.; Lauring, A.S. Complexities of viral mutation rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef]
- Coffin, J.M. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [CrossRef]
- Menéndez-Arias, L.; Sebastián-Martín, A.; Álvarez, M. Viral reverse transcriptases. Virus Res. 2017, 234, 153–176. [Google Scholar] [CrossRef]
- Menéndez-Arias, L. Mutation rates and intrinsic fidelity of retroviral reverse transcriptases. Viruses 2009, 1, 1137–1165. [Google Scholar] [CrossRef]
- O’Neil, P.K.; Sun, G.; Yu, H.; Ron, Y.; Dougherty, J.P.; Preston, B.D. Mutational analysis of HIV-1 long terminal repeats to explore the relative contribution of reverse transcriptase and RNA polymerase II to viral mutagenesis. J. Biol. Chem. 2002, 277, 38053–38061. [Google Scholar] [CrossRef]
- Menéndez-Arias, L. Molecular basis of fidelity of DNA synthesis and nucleotide specificity of retroviral reverse transcriptases. Prog. Nucleic Acid Res. Mol. Biol. 2002, 71, 91–147. [Google Scholar] [CrossRef]
- Elder, J.H.; Lerner, D.L.; Hasselkus-Light, C.S.; Fontenot, D.J.; Hunter, E.; Luciw, P.A.; Montelaro, R.C.; Phillips, T.R. Distinct subsets of retroviruses encode dUTPase. J. Virol. 1992, 66, 1791–1794. [Google Scholar] [CrossRef] [PubMed]
- Köppe, B.; Menéndez-Arias, L.; Oroszlan, S. Expression and purification of the mouse mammary tumor virus gag-pro transframe protein p30 and characterization of its dUTPase activity. J. Virol. 1994, 68, 2313–2319. [Google Scholar] [CrossRef] [PubMed]
- Lerner, D.L.; Wagaman, P.C.; Phillips, T.R.; Prospero-García, O.; Henriksen, S.J.; Fox, H.S.; Bloom, F.E.; Elder, J.H. Increased mutation frequency of feline immunodeficiency virus lacking a functional deoxyuridine-triphosphatase. Proc. Natl. Acad. Sci. USA 1995, 92, 7480–7484. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M. The mutation rate of human immunodeficiency virus type 1 is influenced by the vpr gene. Virology 1996, 222, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Takaori-Kondo, A.; Shindo, K. HIV-1 Vif: A guardian of the virus that opens up a new era in the research field of restriction factors. Front Microbiol. 2013, 4, 34. [Google Scholar] [CrossRef]
- Shah, F.S.; Curr, K.A.; Hamburgh, M.E.; Parniak, M.; Mitsuya, H.; Arnez, J.G.; Prasad, V.R. Differential influence of nucleoside analog-resistance mutations K65R and L74V on the overall mutation rate and error specificity of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 2000, 275, 27037–27044. [Google Scholar] [CrossRef]
- Barrioluengo, V.; Alvarez, M.; Barbieri, D.; Menéndez-Arias, L. Thermostable HIV-1 group O reverse transcriptase variants with the same fidelity as murine leukaemia virus reverse transcriptase. Biochem. J. 2011, 436, 599–607. [Google Scholar] [CrossRef]
- Martín-Hernández, A.M.; Domingo, E.; Menéndez-Arias, L. Human immunodeficiency virus type 1 reverse transcriptase: Role of Tyr115 in deoxynucleotide binding and misinsertion fidelity of DNA synthesis. EMBO J. 1996, 15, 4434–4442. [Google Scholar] [CrossRef]
- Cases-González, C.E.; Gutiérrez-Rivas, M.; Menéndez-Arias, L. Coupling ribose selection to fidelity of DNA synthesis: The role of Tyr-115 of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 2000, 275, 19759–19767. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Le Rouzic, E.; Benichou, S.; Gajary, L.C. Influence of reverse transcriptase variants, drugs, and Vpr on human immunodeficiency virus type 1 mutant frequencies. J. Virol. 2003, 77, 2071–2080. [Google Scholar] [CrossRef] [PubMed]
- Peersen, O.B. Picornaviral polymerase structure, function, and fidelity modulation. Virus Res. 2017, 234, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Gong, P. A structural view of the RNA-dependent RNA polymerases from the Flavivirus genus. Virus Res. 2017, 234, 34–43. [Google Scholar] [CrossRef]
- Hillen, H.S. Structure and function of SARS-CoV-2 polymerase. Curr. Opin. Virol. 2021, 48, 82–90. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.-L. Coronavirus RNA proofreading: Molecular basis and therapeutic targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of SARS-CoV nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010, 6, e1000896. [Google Scholar] [CrossRef]
- Pathak, V.K.; Temin, H.M. 5-Azacytidine and RNA secondary structure increase the retrovirus mutation rate. J. Virol. 1992, 66, 3093–3100. [Google Scholar] [CrossRef]
- LaCasse, R.A.; Remington, K.M.; North, T.W. The mutation frequency of feline immunodeficiency virus enhanced by 3′-azido-3′-deoxythymidine. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 12, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Julias, J.G.; Kim, T.; Arnold, G.; Pathak, V.K. The antiretrovirus drug 3′-azido-3′-deoxythymidine increases the retrovirus mutation rate. J. Virol. 1997, 71, 4254–4263. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Bernard, L.C. 3′-Azido-3′-deoxythymidine (AZT) and AZT-resistant reverse transcriptase can increase the in vivo mutation rate of human immunodeficiency virus type 1. J. Virol. 2000, 74, 9532–9539. [Google Scholar] [CrossRef]
- Dapp, M.J.; Clouser, C.L.; Patterson, S.; Mansky, L.M. 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J. Virol. 2009, 83, 11950–11958. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; McDaniel, Y.Z.; Daly, M.B.; Talledge, N.; Greggs, W.M., 3rd; Patterson, S.E.; Kim, B.; Mansky, L.M. Distinct antiretroviral mechanisms elicited by a viral mutagen. J. Mol. Biol. 2021, 433, 167111. [Google Scholar] [CrossRef] [PubMed]
- Rawson, J.M.O.; Daly, M.B.; Xie, J.; Clouser, C.L.; Landman, S.R.; Reilly, C.S.; Bonnac, L.; Kim, B.; Patterson, S.E.; Mansky, L.M. 5-Azacytidine enhances the mutagenesis of HIV-1 by reduction to 5-aza-2′-deoxycytidine. Antimicrob. Agents Chemother. 2016, 60, 2318–2325. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, C.; Olivares, I.; de Ávila Lucas, A.I.; Toledano, V.; Gutiérrez-Rivas, M.; Lorenzo-Redondo, R.; Grande-Pérez, A.; Domingo, E.; López-Galíndez, C. Mutagen-mediated enhancement of HIV-1 replication in persistently infected cells. Virology 2012, 424, 147–153. [Google Scholar] [CrossRef]
- Vivet-Boudou, V.; Isel, C.; El Safadi, Y.; Smyth, R.P.; Laumond, G.; Moog, C.; Paillart, J.-C.; Marquet, R. Evaluation of anti-HIV-1 mutagenic nucleoside analogues. J. Biol. Chem. 2015, 290, 371–383. [Google Scholar] [CrossRef]
- Rawson, J.M.O.; Landman, S.R.; Reilly, C.S.; Bonnac, L.; Patterson, S.E.; Mansky, L.M. Lack of mutational hot spots during decitabine-mediated HIV-1 mutagenesis. Antimicrob. Agents Chemother. 2015, 59, 6834–6843. [Google Scholar] [CrossRef][Green Version]
- Menéndez-Arias, L. Targeting HIV: Antiretroviral therapy and development of drug resistance. Trends Pharmacol. Sci. 2002, 23, 381–388. [Google Scholar] [CrossRef]
- Menéndez-Arias, L.; Delgado, R. Update and latest advances in antiretroviral therapy. Trends Pharmacol. Sci. 2022, 43, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Tapia, N.; Fernàndez, G.; Parera, M.; Gómez-Mariano, G.; Clotet, B.; Quiñones-Mateu, M.; Domingo, E.; Martínez, M.A. Combination of a mutagenic agent with a reverse transcriptase inhibitor results in systematic inhibition of HIV-1 infection. Virology 2005, 338, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Seier, T.; Zilberberg, G.; Zeiger, D.M.; Lovett, S.T. Azidothymidine and other chain terminators are mutagenic for template-switch-generated genetic mutations. Proc. Natl. Acad. Sci. USA 2012, 109, 6171–6174. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Brabant, W.; Styrchak, S.; Gall, A.; Daifuku, R. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antivir. Res. 2005, 67, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Fedeles, B.I.; Singh, V.; Peng, C.S.; Silvestre, K.J.; Simi, A.K.; Simpson, J.H.; Tokmakoff, A.; Essigmann, J.M. Tautomerism provides a molecular explanation for the mutagenic properties of the anti-HIV nucleoside 5-aza-5,6-dihydro-2′-deoxycytidine. Proc. Natl. Acad. Sci. USA 2014, 111, E3252–E3259. [Google Scholar] [CrossRef]
- Mullins, J.I.; Heath, L.; Hughes, J.P.; Kicha, J.; Styrchak, S.; Wong, K.G.; Rao, U.; Hansen, A.; Harris, K.S.; Laurent, J.-P.; et al. Mutation of HIV-1 genomes in a clinical population treated with the mutagenic nucleoside KP1461. PLoS ONE 2011, 6, e15135. [Google Scholar] [CrossRef]
- Fontecave, M.; Lepoivre, M.; Elleingand, E.; Gerez, C.; Guittet, O. Resveratrol, a remarkable inhibitor of ribonucleotide reductase. FEBS Lett. 1998, 421, 277–279. [Google Scholar] [CrossRef]
- Rawson, J.M.; Heineman, R.H.; Beach, L.B.; Martin, J.L.; Schnettler, E.K.; Dapp, M.J.; Patterson, S.E.; Mansky, L.M. 5,6-Dihydro-5-aza-2′-deoxycytidine potentiates the anti-HIV-1 activity of ribonucleotide reductase inhibitors. Bioorg. Med. Chem. 2013, 21, 7222–7228. [Google Scholar] [CrossRef]
- Guarino, E.; Salguero, I.; Kearsey, S.E. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin. Cell Dev. Biol. 2014, 30, 97–103. [Google Scholar] [CrossRef]
- Musiałek, M.W.; Rybaczek, D. Hydroxyurea-The good, the bad and the ugly. Genes 2021, 12, 1096. [Google Scholar] [CrossRef]
- Clouser, C.L.; Patterson, S.E.; Mansky, L.M. Exploiting drug repositioning for discovery of a novel HIV combination therapy. J. Virol. 2010, 84, 9301–9309. [Google Scholar] [CrossRef] [PubMed]
- Rawson, J.M.O.; Roth, M.E.; Xie, J.; Daly, M.B.; Clouser, C.L.; Landman, S.R.; Reilly, C.S.; Bonnac, L.; Kim, B.; Patterson, S.E.; et al. Synergistic reduction of HIV-1 infectivity by 5-azacytidine and inhibitors of ribonucleotide reductase. Bioorg. Med. Chem. 2016, 24, 2410–2422. [Google Scholar] [CrossRef] [PubMed]
- Beach, L.B.; Rawson, J.M.; Kim, B.; Patterson, S.E.; Mansky, L.M. Novel inhibitors of human immunodeficiency virus type 2 infectivity. J. Gen. Virol. 2014, 95 Pt 12, 2778–2783. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, M.; Sebastián-Martín, A.; García-Marquina, G.; Menéndez-Arias, L. Fidelity of classwide-resistant HIV-2 reverse transcriptase and differential contribution of K65R to the accuracy of HIV-1 and HIV-2 reverse transcriptases. Sci. Rep. 2017, 7, 44834. [Google Scholar] [CrossRef]
- McDaniel, Y.Z.; Patterson, S.E.; Mansky, L.M. Distinct dual antiviral mechanism that enhances hepatitis B virus mutagenesis and reduces viral DNA synthesis. Antiviral. Res. 2019, 170, 104540. [Google Scholar] [CrossRef]
- Holland, J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; VandePol, S. Rapid evolution of RNA genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef]
- Zhou, S.; Hill, C.S.; Sarkar, S.; Tse, L.V.; Woodburn, B.M.D.; Schinazi, R.F.; Sheahan, T.P.; Baric, R.S.; Heise, M.T.; Swanstrom, R. β-D-N4-hydroxycytidine inhibits SARS-CoV-2 through lethal mutagenesis but is also mutagenic to mammalian cells. J. Infect. Dis. 2021, 224, 415–419. [Google Scholar] [CrossRef]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [CrossRef]
- Harki, D.A.; Graci, J.D.; Galarraga, J.E.; Chain, W.J.; Cameron, C.E.; Peterson, B.R. Synthesis and antiviral activity of 5-substituted cytidine analogues: Identification of a potent inhibitor of viral RNA-dependent RNA polymerases. J. Med. Chem. 2006, 49, 6166–6169. [Google Scholar] [CrossRef]
- Graci, J.D.; Harki, D.A.; Korneeva, V.S.; Edathil, J.P.; Too, K.; Franco, D.; Smidansky, E.D.; Paul, A.V.; Peterson, B.R.; Brown, D.M.; et al. Lethal mutagenesis of poliovirus mediated by a mutagenic pyrimidine analogue. J. Virol. 2007, 81, 11256–11266. [Google Scholar] [CrossRef]
- Graci, J.D.; Too, K.; Smidansky, E.D.; Edathil, J.P.; Barr, E.W.; Harki, D.A.; Galarraga, J.E.; Bollinger, J.M., Jr.; Peterson, B.R.; Loakes, D.; et al. Lethal mutagenesis of picornaviruses with N-6-modified purine nucleoside analogues. Antimicrob. Agents Chemother. 2008, 52, 971–979. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Graci, J.D.; Gnädig, N.F.; Galarraga, J.E.; Castro, C.; Vignuzzi, M.; Cameron, C.E. Mutational robustness of an RNA virus influences sensitivity to lethal mutagenesis. J. Virol. 2012, 86, 2869–2873. [Google Scholar] [CrossRef] [PubMed]
- Moreno, H.; Tejero, H.; de la Torre, J.C.; Domingo, E.; Martín, V. Mutagenesis-mediated virus extinction: Virus-dependent effect of viral load on sensitivity to lethal defection. PLoS ONE 2012, 7, e32550. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, A.; Pariente, N.; Menéndez-Arias, L.; Domingo, E. Curing of foot-and-mouth disease virus from persistently infected cells by ribavirin involves enhanced mutagenesis. Virology 2003, 311, 339–349. [Google Scholar] [CrossRef]
- Pariente, N.; Airaksinen, A.; Domingo, E. Mutagenesis versus inhibition in the efficiency of extinction of foot-and-mouth disease virus. J. Virol. 2003, 77, 7131–7138. [Google Scholar] [CrossRef][Green Version]
- Sierra, M.; Airaksinen, A.; González-López, C.; Agudo, R.; Arias, A.; Domingo, E. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: Implications for error catastrophe. J. Virol. 2007, 81, 2012–2024. [Google Scholar] [CrossRef]
- Agudo, R.; Ferrer-Orta, C.; Arias, A.; de la Higuera, I.; Perales, C.; Pérez-Luque, R.; Verdaguer, N.; Domingo, E. A multi-step process of viral adaptation to a mutagenic nucleoside analogue by modulation of transition types leads to extinction-escape. PLoS Pathog. 2010, 6, e1001072. [Google Scholar] [CrossRef]
- de Avila, A.I.; Moreno, E.; Perales, C.; Domingo, E. Favipiravir can evoke lethal mutagenesis and extinction of foot-and-mouth disease virus. Virus Res. 2017, 233, 105–112. [Google Scholar] [CrossRef]
- Arias, A.; Thorne, L.; Goodfellow, I. Favipiravir elicits antiviral mutagenesis during virus replication in vivo. Elife 2014, 3, e03679. [Google Scholar] [CrossRef]
- Qiu, L.; Patterson, S.E.; Bonnac, L.F.; Geraghty, R.J. Nucleobases and corresponding nucleosides display potent antiviral activities against dengue virus possibly through viral lethal mutagenesis. PLoS Negl. Trop. Dis. 2018, 12, e0006421. [Google Scholar] [CrossRef]
- Bassi, M.R.; Sempere, R.N.; Meyn, P.; Polacek, C.; Arias, A. Extinction of Zika virus and Usutu virus by lethal mutagenesis reveals different patterns of sensitivity to three mutagenic drugs. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Day, C.W.; Smee, D.F.; Julander, J.G.; Yamshchikov, V.F.; Sidwell, R.W.; Morrey, J.D. Error-prone replication of West Nile virus caused by ribavirin. Antiviral. Res. 2005, 67, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Romero, E.; Jiménez de Oya, N.; Domingo, E.; Saiz, J.C. Extinction of West Nile virus by favipiravir through lethal mutagenesis. Antimicrob. Agents Chemother. 2017, 61, e01400-17. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Chavez, D.; Guerra, B.; Lau, J.Y.; Hong, Z.; Brasky, K.M.; Beames, B. Ribavirin induces error-prone replication of GB virus B in primary tamarin hepatocytes. J. Virol. 2001, 75, 8074–8081. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.M.; Hiasa, Y.; He, W.; Terella, A.; Schmidt, E.V.; Chung, R.T. Viral RNA mutations are region specific and increased by ribavirin in a full-length hepatitis C virus replication system. J. Virol. 2002, 76, 8505–8517. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, R.; Baroudy, B.M.; Malcolm, B.A.; Reyes, G.R. The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology 2003, 310, 333–342. [Google Scholar] [CrossRef]
- Cuevas, J.M.; González-Candelas, F.; Moya, A.; Sanjuán, R. Effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. J. Virol. 2009, 83, 5760–5764. [Google Scholar] [CrossRef]
- Ortega-Prieto, A.M.; Sheldon, J.; Grande-Pérez, A.; Tejero, H.; Gregori, J.; Quer, J.; Esteban, J.I.; Domingo, E.; Perales, C. Extinction of hepatitis C virus by ribavirin in hepatoma cells involves lethal mutagenesis. PLoS ONE 2013, 8, e71039. [Google Scholar] [CrossRef]
- Dietz, J.; Schelhorn, S.-E.; Fitting, D.; Mihm, U.; Susser, S.; Welker, M.-W.; Füller, C.; Däumer, M.; Teuber, G.; Wedemeyer, H.; et al. Deep sequencing reveals mutagenic effects of ribavirin during monotherapy of hepatitis C virus genotype 1-infected patients. J. Virol. 2013, 87, 6172–6181. [Google Scholar] [CrossRef]
- Gallego, I.; Soria, M.E.; Gregori, J.; de Ávila, A.I.; García-Crespo, C.; Moreno, E.; Gadea, I.; Esteban, J.; Fernández-Roblas, R.; Esteban, J.I.; et al. Synergistic lethal mutagenesis of hepatitis C virus. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- de Ávila, A.I.; Gallego, I.; Soria, M.E.; Gregori, J.; Quer, J.; Esteban, J.I.; Rice, C.M.; Domingo, E.; Perales, C. Lethal mutagenesis of hepatitis C virus induced by favipiravir. PLoS ONE 2016, 11, e0164691. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; Walter, S.; Brown, R.; Steinmann, E. Mutagenic effects of ribavirin on hepatitis E virus—Viral extinction versus selection of fitness-enhancing mutations. Viruses 2016, 8, 283. [Google Scholar] [CrossRef]
- Urakova, N.; Kuznetsova, V.; Crossman, D.K.; Sokratian, A.; Guthrie, D.B.; Kolykhalov, A.A.; Lockwood, M.A.; Natchus, M.G.; Crowley, M.R.; Painter, G.R.; et al. β-D-N4-hydroxycytidine is a potent anti-alphavirus compound that induces a high level of mutations in the viral genome. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Shannon, A.; Selisko, B.; Le, N.-T.-T.; Huchting, J.; Touret, F.; Piorkowski, G.; Fattorini, V.; Ferron, F.; Decroly, E.; Meier, C.; et al. Rapid incorporation of favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat. Commun. 2020, 11, 4682. [Google Scholar] [CrossRef]
- Agostini, M.L.; Pruijssers, A.J.; Chappell, J.D.; Gribble, J.; Lu, X.; Andres, E.L.; Bluemling, G.R.; Lockwood, M.A.; Sheahan, T.P.; Sims, A.C.; et al. Small-molecule antiviral β-D-N4-hydroxycytidine inhibits a proofreading-intact coronavirus with a high genetic barrier to resistance. J. Virol. 2019, 93, e01348-19. [Google Scholar] [CrossRef]
- Díaz-Martínez, L.; Brichette-Mieg, I.; Pineño-Ramos, A.; Domínguez-Huerta, G.; Grande-Pérez, A. Lethal mutagenesis of an RNA plant virus via lethal defection. Sci. Rep. 2018, 8, 1444. [Google Scholar] [CrossRef]
- Pauly, M.D.; Lauring, A.S. Effective lethal mutagenesis of influenza virus by three nucleoside analogs. J. Virol. 2015, 89, 3584–3597. [Google Scholar] [CrossRef]
- Toots, M.; Yoon, J.-J.; Cox, R.M.; Hart, M.; Sticher, Z.M.; Makhsous, N.; Plesker, R.; Barrena, A.H.; Reddy, P.G.; Mitchell, D.G.; et al. Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia. Sci. Transl. Med. 2019, 11, eaax5866. [Google Scholar] [CrossRef]
- Grande-Pérez, A.; Lázaro, E.; Lowenstein, P.; Domingo, E.; Manrubia, S.C. Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. USA 2005, 102, 4448–4452. [Google Scholar] [CrossRef]
- Severson, W.E.; Schmaljohn, C.S.; Javadian, A.; Jonsson, C.B. Ribavirin causes error catastrophe during Hantaan virus replication. J. Virol. 2003, 77, 481–488. [Google Scholar] [CrossRef]
- Chung, D.-H.; Sun, Y.; Parker, W.B.; Arterburn, J.B.; Bartolucci, A.; Jonsson, C.B. Ribavirin reveals a lethal threshold of allowable mutation frequency for Hantaan virus. J. Virol. 2007, 81, 11722–11729. [Google Scholar] [CrossRef] [PubMed]
- Borrego, B.; de Ávila, A.I.; Domingo, E.; Brun, A. Lethal mutagenesis of Rift Valley fever virus induced by favipiravir. Antimicrob. Agents Chemother. 2019, 63, e00669-19. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Jarabo, C.M.; Ly, C.; Domingo, E.; de la Torre, J.C. Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV). Virology 2003, 308, 37–47. [Google Scholar] [CrossRef]
- Espy, N.; Nagle, E.; Pfeffer, B.; Garcia, K.; Chitty, A.J.; Wiley, M.; Sanchez-Lockhart, M.; Bavari, S.; Warren, T.; Palacios, G. T-705 induces lethal mutagenesis in Ebola and Marburg populations in macaques. Antiviral. Res. 2019, 170, 104529. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Andino, R. Ribavirin and lethal mutagenesis of poliovirus: Molecular mechanisms, resistance and biological implications. Virus Res. 2005, 107, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Olivencia, G.; Estébanez, M.; Membrillo, F.J.; Ybarra, M.D.C. Uso de ribavirina en virus distintos de la hepatitis C. una revisión de la evidencia. Enferm. Infecc. Microbiol. Clin. (Engl.) 2019, 37, 602–608. [Google Scholar] [CrossRef]
- Graci, J.D.; Cameron, C.E. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 2006, 16, 37–48. [Google Scholar] [CrossRef]
- Paeshuyse, J.; Dallmeier, K.; Neyts, J. Ribavirin for the treatment of chronic hepatitis C virus infection: A review of the proposed mechanisms of action. Curr. Opin. Virol. 2011, 1, 590–598. [Google Scholar] [CrossRef]
- Kentsis, A.; Topisirovic, I.; Culjkovic, B.; Shao, L.; Borden, K.L.B. Ribavirin suppresses EIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc. Natl. Acad. Sci. USA 2004, 101, 18105–18110. [Google Scholar] [CrossRef]
- Sidwell, R.W.; Huffman, J.H.; Khare Lois, G.P.; Allen, B.; Witkowski Roland, J.T.; Robins, K. Broad-spectrum antiviral activity of virazole: 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 1972, 177, 705–706. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Aliota, M.T.; Bonnac, L.F. Broad-spectrum antiviral strategies and nucleoside analogues. Viruses 2021, 13, 667. [Google Scholar] [CrossRef]
- Arnold, J.J.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3Dpol) is sufficient for template switching in vitro. J. Biol. Chem. 1999, 274, 2706–2716. [Google Scholar] [CrossRef] [PubMed]
- Gohara, D.W.; Crotty, S.; Arnold, J.J.; Yoder, J.D.; Andino, R.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3Dpol): Structural, biochemical, and biological analysis of conserved structural motifs a and b. J. Biol. Chem. 2000, 275, 25523–25532. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Orta, C.; Sierra, M.; Agudo, R.; de la Higuera, I.; Arias, A.; Pérez-Luque, R.; Escarmís, C.; Domingo, E.; Verdaguer, N. Structure of foot-and-mouth disease virus mutant polymerases with reduced sensitivity to ribavirin. J. Virol. 2010, 84, 6188–6199. [Google Scholar] [CrossRef] [PubMed]
- Young, K.-C.; Lindsay, K.L.; Lee, K.-J.; Liu, W.-C.; He, J.-W.; Milstein, S.L.; Lai, M.M.C. Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology 2003, 38, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Lutchman, G.; Danehower, S.; Song, B.-C.; Liang, T.J.; Hoofnagle, J.H.; Thomson, M.; Ghany, M.G. Mutation rate of the hepatitis C virus NS5B in patients undergoing treatment with ribavirin monotherapy. Gastroenterology 2007, 132, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Mejer, N.; Galli, A.; Ramirez, S.; Fahnøe, U.; Benfield, T.; Bukh, J. Ribavirin inhibition of cell-culture infectious hepatitis C genotype 1-3 viruses is strain-dependent. Virology 2020, 540, 132–140. [Google Scholar] [CrossRef]
- Mejer, N.; Fahnøe, U.; Galli, A.; Ramirez, S.; Weiland, O.; Benfield, T.; Bukh, J. Mutations identified in the hepatitis C virus (HCV) polymerase of patients with chronic HCV treated with ribavirin cause resistance and affect viral replication fidelity. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Gong, P.; Peersen, O.B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2010, 107, 22505–22510. [Google Scholar] [CrossRef]
- Gallego, I.; Gregori, J.; Soria, M.E.; García-Crespo, C.; García-Álvarez, M.; Gómez-González, A.; Valiergue, R.; Gómez, J.; Esteban, J.I.; Quer, J.; et al. Resistance of high fitness hepatitis C virus to lethal mutagenesis. Virology 2018, 523, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Delang, L.; Abdelnabi, R.; Neyts, J. Favipiravir as a potential countermeasure against neglected and emerging RNA viruses. Antiviral. Res. 2018, 153, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Jordan, P.C.; Stevens, S.K.; Deval, J. Nucleosides for the treatment of respiratory RNA virus infections. Antivir. Chem. Chemother. 2018, 26, 2040206618764483. [Google Scholar] [CrossRef] [PubMed]
- Sangawa, H.; Komeno, T.; Nishikawa, H.; Yoshida, A.; Takahashi, K.; Nomura, N.; Furuta, Y. Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob. Agents Chemother. 2013, 57, 5202–5208. [Google Scholar] [CrossRef]
- Jin, Z.; Smith, L.K.; Rajwanshi, V.K.; Kim, B.; Deval, J. The ambiguous base-pairing and high substrate efficiency of T-705 (favipiravir) ribofuranosyl 5’-triphosphate towards influenza A virus polymerase. PLoS ONE 2013, 8, e68347. [Google Scholar] [CrossRef]
- Baranovich, T.; Wong, S.-S.; Armstrong, J.; Marjuki, H.; Webby, R.J.; Webster, R.G.; Govorkova, E.A. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 2013, 87, 3741–3751. [Google Scholar] [CrossRef]
- Goldhill, D.H.; Te Velthuis, A.J.W.; Fletcher, R.A.; Langat, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA 2018, 115, 11613–11618. [Google Scholar] [CrossRef]
- Goldhill, D.H.; Yan, A.; Frise, R.; Zhou, J.; Shelley, J.; Gallego Cortés, A.; Miah, S.; Akinbami, O.; Galiano, M.; Zambon, M.; et al. Favipiravir-resistant influenza A virus shows potential for transmission. PLoS Pathog. 2021, 17, e1008937. [Google Scholar] [CrossRef]
- Delang, L.; Segura Guerrero, N.; Tas, A.; Quérat, G.; Pastorino, B.; Froeyen, M.; Dallmeier, K.; Jochmans, D.; Herdewijn, P.; Bello, F.; et al. Mutations in the Chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. J. Antimicrob. Chemother. 2014, 69, 2770–2784. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.; Yuan, S.; Gao, Q.; Lan, K.; Altmeyer, R.; Zou, G. In vitro assessment of combinations of enterovirus inhibitors against enterovirus 71. Antimicrob. Agents Chemother. 2016, 60, 5357–5367. [Google Scholar] [CrossRef]
- Naydenova, K.; Muir, K.W.; Wu, L.-F.; Zhang, Z.; Coscia, F.; Peet, M.J.; Castro-Hartmann, P.; Qian, P.; Sader, K.; Dent, K.; et al. Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc. Natl. Acad. Sci. USA 2021, 118, e2021946118. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Dandapat, J.; Saudagar, P.; Uversky, V.N.; Tripathi, T. Interface-based design of the favipiravir-binding site in SARS-CoV-2 RNA-dependent RNA polymerase reveals mutations conferring resistance to chain termination. FEBS Lett. 2021, 595, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Guo, S.; Yi, D.; Li, Q.; Ma, L.; Zhang, Y.; Wang, J.; Li, X.; Guo, F.; Lin, R.; et al. A cell-based assay to discover inhibitors of SARS-CoV-2 RNA dependent RNA polymerase. Antiviral. Res. 2021, 190, 105078. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H., 3rd; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Götte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef] [PubMed]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Jena, N.R. Role of different tautomers in the base-pairing abilities of some of the vital antiviral drugs used against COVID-19. Phys. Chem. Chem. Phys. 2020, 22, 28115–28122. [Google Scholar] [CrossRef]
- Rosenke, K.; Hansen, F.; Schwarz, B.; Feldmann, F.; Haddock, E.; Rosenke, R.; Barbian, K.; Meade-White, K.; Okumura, A.; Leventhal, S.; et al. Orally delivered MK-4482 inhibits SARS-CoV-2 replication in the Syrian hamster model. Nat. Commun. 2021, 12, 2295. [Google Scholar] [CrossRef]
- Cox, R.M.; Wolf, J.D.; Plemper, R.K. Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat. Microbiol. 2021, 6, 11–18. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; De los Reyes, V.; Martín-Quirós, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for oral treatment of COVID-19 in nonhospitalized patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef]
- Fischer, W.A., 2nd; Eron, J.J., Jr.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.J.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. A phase 2a clinical trial of molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci. Transl. Med. 2022, 14, eabl7430. [Google Scholar] [CrossRef] [PubMed]
- Abdelnabi, R.; Foo, C.S.; De Jonghe, S.; Maes, P.; Weynand, B.; Neyts, J. Molnupiravir inhibits replication of the emerging SARS-CoV-2 variants of concern in a hamster infection model. J. Infect. Dis. 2021, 224, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, Y.; Lavrijsen, M.; Lamers, M.M.; de Vries, A.C.; Rottier, R.J.; Bruno, M.J.; Peppelenbosch, M.P.; Haagmans, B.L.; Pan, Q. SARS-CoV-2 omicron variant is highly sensitive to molnupiravir, nirmatrelvir, and the combination. Cell Res. 2022, 32, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Vangeel, L.; Chiu, W.; De Jonghe, S.; Maes, P.; Slechten, B.; Raymenants, J.; André, E.; Leyssen, P.; Neyts, J.; Jochmans, D. Remdesivir, molnupiravir and nirmatrelvir remain active against SARS-CoV-2 omicron and other variants of concern. Antiviral. Res. 2022, 198, 105252. [Google Scholar] [CrossRef]
- Menéndez-Arias, L. Decoding molnupiravir-induced mutagenesis in SARS-CoV-2. J. Biol. Chem. 2021, 297, 100867. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Foo, C.S.; Kaptein, S.J.F.; Zhang, X.; Do, T.N.D.; Langendries, L.; Vangeel, L.; Breuer, J.; Pang, J.; Williams, R.; et al. The combined treatment of molnupiravir and favipiravir results in a potentiation of antiviral efficacy in a SARS-CoV-2 hamster infection model. EBioMedicine 2021, 72, 103595. [Google Scholar] [CrossRef]
- Schultz, D.C.; Johnson, R.M.; Ayyanathan, K.; Miller, J.; Whig, K.; Kamalia, B.; Dittmar, M.; Weston, S.; Hammond, H.L.; Dillen, C.; et al. Pyrimidine inhibitors synergize with nucleoside analogues to block SARS-CoV-2. Nature 2022, 604, 134–140. [Google Scholar] [CrossRef]
- Painter, G.R.; Natchus, M.G.; Cohen, O.; Holman, W.; Painter, W.P. Developing a direct acting, orally available antiviral agent in a pandemic: The evolution of molnupiravir as a potential treatment for COVID-19. Curr. Opin. Virol. 2021, 50, 17–22. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, T.; Xi, J.; Zhang, G.; Wang, T.; Liu, W.; You, X.; Zhang, X.; Xia, Z.; Luan, Y. PIG-A gene mutation as a genotoxicity biomarker in human population studies: An investigation in lead-exposed workers. Environ. Mol. Mutagen. 2020, 61, 611–621. [Google Scholar] [CrossRef]
- Waters, M.D.; Warren, S.; Hughes, C.; Lewis, P.; Zhang, F. Human genetic risk of treatment with antiviral nucleoside analog drugs that induce lethal mutagenesis: The special case of molnupiravir. Environ. Mol. Mutagen. 2022, 63, 37–63. [Google Scholar] [CrossRef]
- Ma, Y.; Frutos-Beltrán, E.; Kang, D.; Pannecouque, C.; De Clercq, E.; Menéndez-Arias, L.; Liu, X.; Zhan, P. Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chem. Soc. Rev. 2021, 50, 4514–4540. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadj Hassine, I.; Ben M’hadheb, M.; Menéndez-Arias, L. Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity. Viruses 2022, 14, 841. https://doi.org/10.3390/v14040841
Hadj Hassine I, Ben M’hadheb M, Menéndez-Arias L. Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity. Viruses. 2022; 14(4):841. https://doi.org/10.3390/v14040841
Chicago/Turabian StyleHadj Hassine, Ikbel, Manel Ben M’hadheb, and Luis Menéndez-Arias. 2022. "Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity" Viruses 14, no. 4: 841. https://doi.org/10.3390/v14040841
APA StyleHadj Hassine, I., Ben M’hadheb, M., & Menéndez-Arias, L. (2022). Lethal Mutagenesis of RNA Viruses and Approved Drugs with Antiviral Mutagenic Activity. Viruses, 14(4), 841. https://doi.org/10.3390/v14040841