
Merkel Cell Polyomavirus: Oncogenesis in a Stable Genome


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Two Types of Merkel Cell Carcinoma
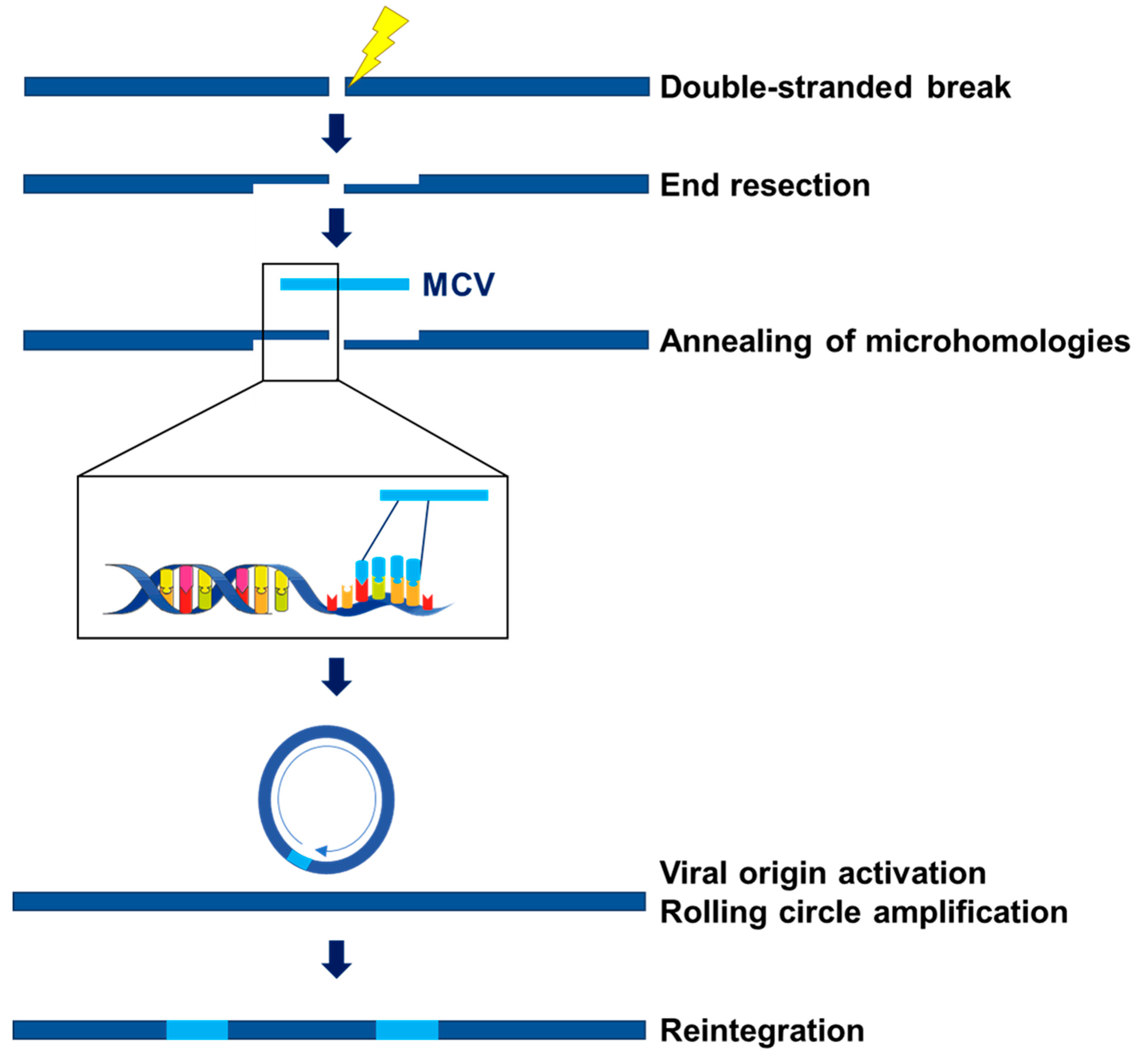
3. Origins of MCC
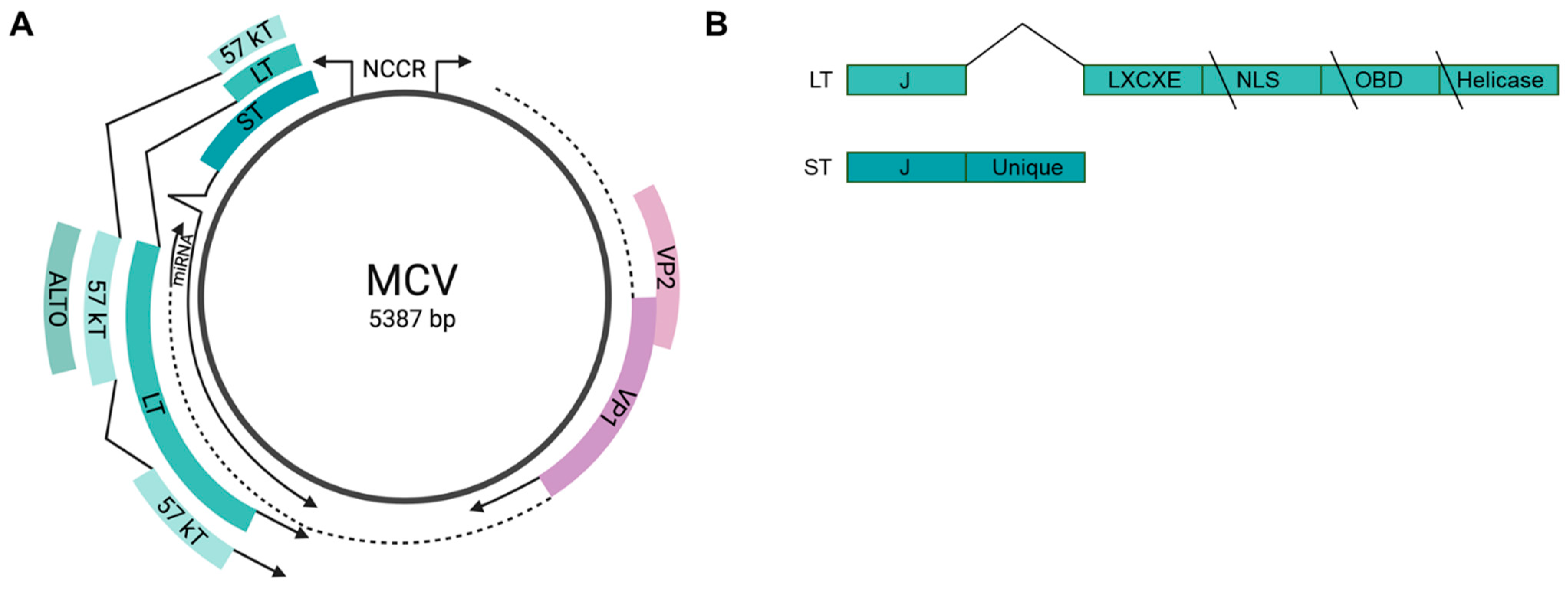
4. Merkel Cell Polyomavirus
5. LT Inhibits RB
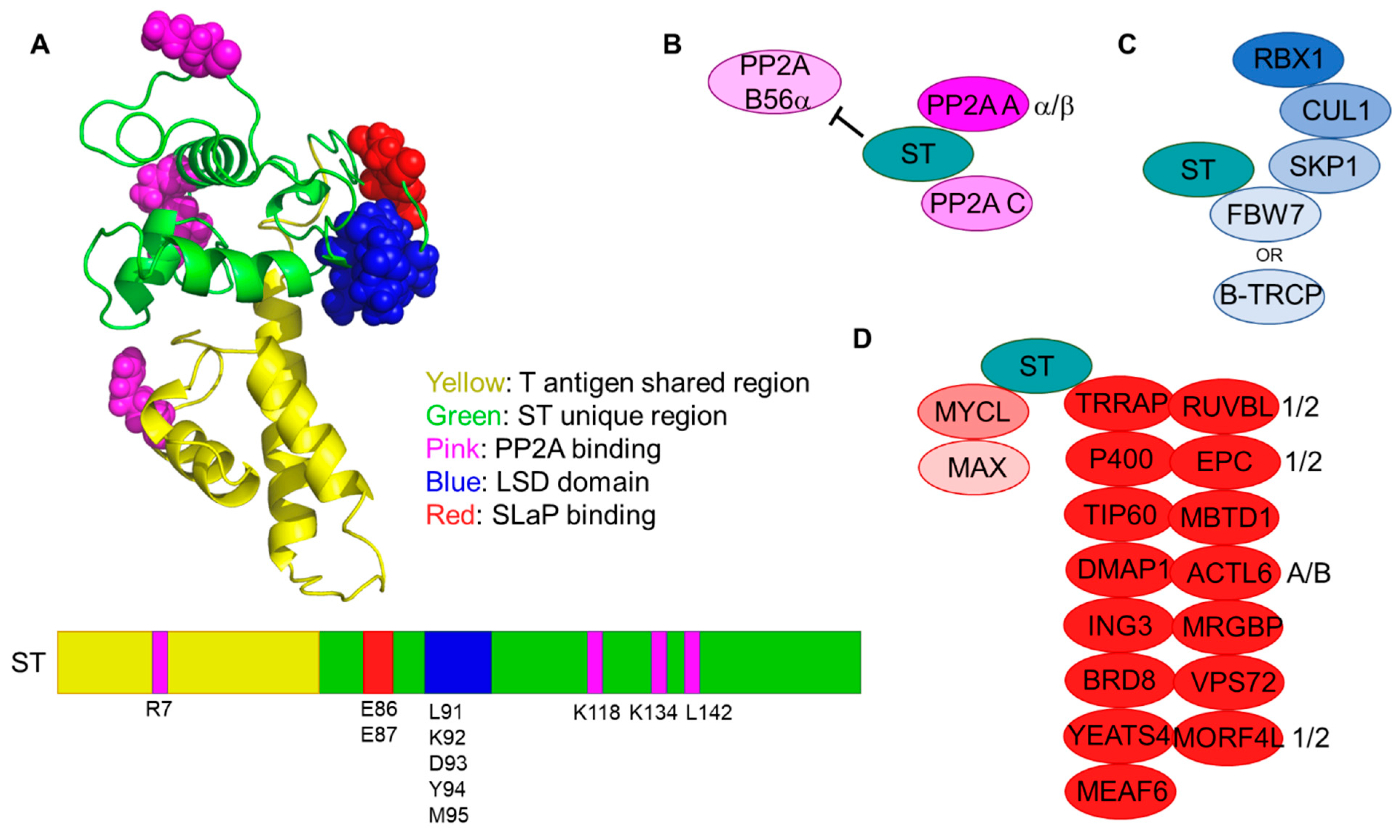
6. MCV ST Interactions
6.1. Protein Phosphatase 2A (PP2A)
6.2. SKP1-CUL1-FBOX (SCF) Complexes
6.3. ST, MYCL, and P400: The SLaP Complex
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef]
- Hahn, W.C.; Dessain, S.K.; Brooks, M.W.; King, J.E.; Elenbaas, B.; Sabatini, D.M.; DeCaprio, J.A.; Weinberg, R.A. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol. Cell. Biol. 2002, 22, 2111–2123. [Google Scholar] [CrossRef] [Green Version]
- Voorhoeve, P.M.; Agami, R. The tumor-suppressive functions of the human INK4A locus. Cancer Cell 2003, 4, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- Whitman, M.; Kaplan, D.R.; Schaffhausen, B.; Cantley, L.; Roberts, T.M. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 1985, 315, 239–242. [Google Scholar] [CrossRef]
- Carbone, M.; Gazdar, A.; Butel, J.S. SV40 and human mesothelioma. Transl. Lung Cancer Res. 2020, 9, S47–S59. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096. [Google Scholar] [CrossRef] [Green Version]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [Green Version]
- Houben, R.; Adam, C.; Baeurle, A.; Hesbacher, S.; Grimm, J.; Angermeyer, S.; Henzel, K.; Hauser, S.; Elling, R.; Bröcker, E.B.; et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int. J. Cancer 2012, 130, 847–856. [Google Scholar] [CrossRef]
- Starrett, G.J.; Marcelus, C.; Cantalupo, P.G.; Katz, J.P.; Cheng, J.; Akagi, K.; Thakuria, M.; Rabinowits, G.; Wang, L.C.; Symer, D.E.; et al. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. mBio 2017, 8, e02079-16. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2016, 7, 3403–3415. [Google Scholar] [CrossRef] [Green Version]
- DeCaprio, J.A. Molecular Pathogenesis of Merkel Cell Carcinoma. Annu. Rev. Pathol. 2021, 16, 69–91. [Google Scholar] [CrossRef]
- Paulson, K.G.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463.e452. [Google Scholar] [CrossRef]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17077. [Google Scholar] [CrossRef]
- Lilo, M.T.; Chen, Y.; LeBlanc, R.E. INSM1 Is More Sensitive and Interpretable than Conventional Immunohistochemical Stains Used to Diagnose Merkel Cell Carcinoma. Am. J. Surg. Pathol. 2018, 42, 1541–1548. [Google Scholar] [CrossRef]
- Jaeger, T.; Ring, J.; Andres, C. Histological, Immunohistological, and Clinical Features of Merkel Cell Carcinoma in Correlation to Merkel Cell Polyomavirus Status. J. Skin Cancer 2012, 2012, 983421. [Google Scholar] [CrossRef] [Green Version]
- Kervarrec, T.; Tallet, A.; Miquelestorena-Standley, E.; Houben, R.; Schrama, D.; Gambichler, T.; Berthon, P.; Le Corre, Y.; Hainaut-Wierzbicka, E.; Aubin, F.; et al. Morphologic and immunophenotypical features distinguishing Merkel cell polyomavirus-positive and negative Merkel cell carcinoma. Mod. Pathol. 2019, 32, 1605–1616. [Google Scholar] [CrossRef]
- Iwasaki, T.; Matsushita, M.; Kuwamoto, S.; Kato, M.; Murakami, I.; Higaki-Mori, H.; Nakajima, H.; Sano, S.; Hayashi, K. Usefulness of significant morphologic characteristics in distinguishing between Merkel cell polyomavirus-positive and Merkel cell polyomavirus-negative Merkel cell carcinomas. Hum. Pathol. 2013, 44, 1912–1917. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, S.; Carter, M.D.; Ly, T.Y.; Doucette, S.; Walsh, N.M. Immunohistochemical profiles of different subsets of Merkel cell carcinoma. Hum. Pathol. 2018, 82, 232–238. [Google Scholar] [CrossRef]
- Moshiri, A.S.; Doumani, R.; Yelistratova, L.; Blom, A.; Lachance, K.; Shinohara, M.M.; Delaney, M.; Chang, O.; McArdle, S.; Thomas, H.; et al. Polyomavirus-Negative Merkel Cell Carcinoma: A More Aggressive Subtype Based on Analysis of 282 Cases Using Multimodal Tumor Virus Detection. J. Investig. Dermatol. 2017, 137, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I.; et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5961. [Google Scholar] [CrossRef] [Green Version]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A.; et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLOS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef]
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toker, C. Trabecular Carcinoma of the Skin. Arch. Dermatol. 1972, 105, 107–110. [Google Scholar] [CrossRef]
- Tang, C.K.; Toker, C. Trabecular carcinoma of the skin: An ultrastructural study. Cancer 1978, 42, 2311–2321. [Google Scholar] [CrossRef]
- Sibley, R.K.; Rosai, J.; Foucar, E.; Dehner, L.P.; Bosl, G. Neuroendocrine (Merkel cell) carcinoma of the skin. A histologic and ultrastructural study of two cases. Am. J. Surg. Pathol. 1980, 4, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Moll, I.; Kuhn, C.; Moll, R. Cytokeratin 20 is a general marker of cutaneous Merkel cells while certain neuronal proteins are absent. J. Investig. Dermatol. 1995, 104, 910–915. [Google Scholar] [CrossRef] [Green Version]
- Moll, R.; Löwe, A.; Laufer, J.; Franke, W.W. Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am. J. Pathol. 1992, 140, 427–447. [Google Scholar]
- Eispert, A.C.; Fuchs, F.; Brandner, J.M.; Houdek, P.; Wladykowski, E.; Moll, I. Evidence for distinct populations of human Merkel cells. Histochem. Cell Biol. 2009, 132, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Gallego, R.; García-Caballero, T.; Fraga, M.; Beiras, A.; Forteza, J. Neural cell adhesion molecule immunoreactivity in Merkel cells and Merkel cell tumours. Virchows Arch. 1995, 426, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Nabeshima, K.; Akiyama, Y.; Maeda, S.; Nishida, T.; Nakayama, F.; Amano, M.; Ogata, K.; Setoyama, M. CD56: A useful marker for diagnosing Merkel cell carcinoma. J. Dermatol. Sci. 2003, 31, 219–224. [Google Scholar] [CrossRef]
- García-Caballero, T.; Cuevas, J.; Gallego, R.; Rosón, E.; Forteza, J.; Beiras, A. Synaptophysinlike immunoreactivity in the Merkel cells of pig-snout skin. Ultrastruct. Pathol. 1989, 13, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Llombart, B.; Monteagudo, C.; López-Guerrero, J.A.; Carda, C.; Jorda, E.; Sanmartín, O.; Almenar, S.; Molina, I.; Martín, J.M.; Llombart-Bosch, A. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology 2005, 46, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Acebo, E.; Vidaurrazaga, N.; Varas, C.; Burgos-Bretones, J.J.; Díaz-Pérez, J.L. Merkel cell carcinoma: A clinicopathological study of 11 cases. J. Eur. Acad Dermatol. Venereol. 2005, 19, 546–551. [Google Scholar] [CrossRef]
- Shah, I.A.; Netto, D.; Schlageter, M.O.; Muth, C.; Fox, I.; Manne, R.K. Neurofilament immunoreactivity in Merkel-cell tumors: A differentiating feature from small-cell carcinoma. Mod. Pathol. 1993, 6, 3–9. [Google Scholar]
- Moll, I.; Roessler, M.; Brandner, J.M.; Eispert, A.C.; Houdek, P.; Moll, R. Human Merkel cells--aspects of cell biology, distribution and functions. Eur. J. Cell Biol. 2005, 84, 259–271. [Google Scholar] [CrossRef]
- Tilling, T.; Moll, I. Which are the cells of origin in merkel cell carcinoma? J. Skin Cancer 2012, 2012, 680410. [Google Scholar] [CrossRef] [Green Version]
- Lemasson, G.; Coquart, N.; Lebonvallet, N.; Boulais, N.; Galibert, M.D.; Marcorelles, P.; Misery, L. Presence of putative stem cells in Merkel cell carcinomas. J. Eur. Acad Dermatol. Venereol. 2012, 26, 789–795. [Google Scholar] [CrossRef]
- Visscher, D.; Cooper, P.H.; Zarbo, R.J.; Crissman, J.D. Cutaneous neuroendocrine (Merkel cell) carcinoma: An immunophenotypic, clinicopathologic, and flow cytometric study. Mod. Pathol. 1989, 2, 331–338. [Google Scholar]
- Zur Hausen, A.; Rennspiess, D.; Winnepenninckx, V.; Speel, E.J.; Kurz, A.K. Early B-cell differentiation in Merkel cell carcinomas: Clues to cellular ancestry. Cancer Res. 2013, 73, 4982–4987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.H.; Stumpfova, M.; Jensen, U.B.; Lumpkin, E.A.; Owens, D.M. Identification of epidermal progenitors for the Merkel cell lineage. Development 2010, 137, 3965–3971. [Google Scholar] [CrossRef] [Green Version]
- Vieites, B.; Suárez-Peñaranda, J.M.; Delgado, V.; Vázquez-Veiga, H.; Varela, J.; Forteza, J. Merkel cell carcinoma associated with in situ and invasive squamous cell carcinoma. Acta Derm. Venereol. 2009, 89, 184–186. [Google Scholar] [CrossRef]
- Morrison, K.M.; Miesegaes, G.R.; Lumpkin, E.A.; Maricich, S.M. Mammalian Merkel cells are descended from the epidermal lineage. Dev. Biol. 2009, 336, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Van Keymeulen, A.; Mascre, G.; Youseff, K.K.; Harel, I.; Michaux, C.; De Geest, N.; Szpalski, C.; Achouri, Y.; Bloch, W.; Hassan, B.A.; et al. Epidermal progenitors give rise to Merkel cells during embryonic development and adult homeostasis. J. Cell Biol. 2009, 187, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Kervarrec, T.; Aljundi, M.; Appenzeller, S.; Samimi, M.; Maubec, E.; Cribier, B.; Deschamps, L.; Sarma, B.; Sarosi, E.M.; Berthon, P.; et al. Polyomavirus-Positive Merkel Cell Carcinoma Derived from a Trichoblastoma Suggests an Epithelial Origin of this Merkel Cell Carcinoma. J. Investig. Dermatol. 2020, 140, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Kervarrec, T.; Appenzeller, S.; Samimi, M.; Sarma, B.; Sarosi, E.M.; Berthon, P.; Le Corre, Y.; Hainaut-Wierzbicka, E.; Blom, A.; Benethon, N.; et al. Merkel Cell Polyomavirus-Negative Merkel Cell Carcinoma Originating from In Situ Squamous Cell Carcinoma: A Keratinocytic Tumor with Neuroendocrine Differentiation. J. Investig. Dermatol. 2021. [Google Scholar] [CrossRef]
- Harms, P.W.; Verhaegen, M.E.; Hu, K.; Hrycaj, S.M.; Chan, M.P.; Liu, C.J.; Grachtchouk, M.; Patel, R.M.; Udager, A.M.; Dlugosz, A.A. Genomic evidence suggests that cutaneous neuroendocrine carcinomas can arise from squamous dysplastic precursors. Mod. Pathol. 2021. [Google Scholar] [CrossRef]
- Metallo, C.M.; Azarin, S.M.; Moses, L.E.; Ji, L.; de Pablo, J.J.; Palecek, S.P. Human embryonic stem cell-derived keratinocytes exhibit an epidermal transcription program and undergo epithelial morphogenesis in engineered tissue constructs. Tissue Eng. Part A 2010, 16, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Larouche, D.; Lavoie, A.; Paquet, C.; Simard-Bisson, C.; Germain, L. Identification of epithelial stem cells in vivo and in vitro using keratin 19 and BrdU. Methods Mol. Biol. 2010, 585, 383–400. [Google Scholar] [CrossRef]
- Calder, K.B.; Smoller, B.R. New insights into merkel cell carcinoma. Adv. Anat. Pathol. 2010, 17, 155–161. [Google Scholar] [CrossRef]
- Sommer, L. Generation of melanocytes from neural crest cells. Pigment Cell Melanoma Res. 2011, 24, 411–421. [Google Scholar] [CrossRef]
- Zabierowski, S.E.; Fukunaga-Kalabis, M.; Li, L.; Herlyn, M. Dermis-derived stem cells: A source of epidermal melanocytes and melanoma? Pigment Cell Melanoma Res. 2011, 24, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Clewes, O.; Narytnyk, A.; Gillinder, K.R.; Loughney, A.D.; Murdoch, A.P.; Sieber-Blum, M. Human Epidermal Neural Crest Stem Cells (hEPI-NCSC)—Characterization and Directed Differentiation into Osteocytes and Melanocytes. Stem Cell Rev. Rep. 2011, 7, 799–814. [Google Scholar] [CrossRef] [Green Version]
- Laga, A.C.; Lai, C.Y.; Zhan, Q.; Huang, S.J.; Velazquez, E.F.; Yang, Q.; Hsu, M.Y.; Murphy, G.F. Expression of the embryonic stem cell transcription factor SOX2 in human skin: Relevance to melanocyte and merkel cell biology. Am. J. Pathol. 2010, 176, 903–913. [Google Scholar] [CrossRef] [Green Version]
- Kolhe, R.; Reid, M.D.; Lee, J.R.; Cohen, C.; Ramalingam, P. Immunohistochemical expression of PAX5 and TdT by Merkel cell carcinoma and pulmonary small cell carcinoma: A potential diagnostic pitfall but useful discriminatory marker. Int. J. Clin. Exp. Pathol. 2013, 6, 142–147. [Google Scholar] [PubMed]
- Dong, H.Y.; Liu, W.; Cohen, P.; Mahle, C.E.; Zhang, W. B-cell specific activation protein encoded by the PAX-5 gene is commonly expressed in merkel cell carcinoma and small cell carcinomas. Am. J. Surg. Pathol. 2005, 29, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Mhawech-Fauceglia, P.; Saxena, R.; Zhang, S.; Terracciano, L.; Sauter, G.; Chadhuri, A.; Herrmann, F.R.; Penetrante, R. Pax-5 immunoexpression in various types of benign and malignant tumours: A high-throughput tissue microarray analysis. J. Clin. Pathol. 2007, 60, 709. [Google Scholar] [CrossRef] [Green Version]
- Sur, M.; AlArdati, H.; Ross, C.; Alowami, S. TdT expression in Merkel cell carcinoma: Potential diagnostic pitfall with blastic hematological malignancies and expanded immunohistochemical analysis. Mod. Pathol. 2007, 20, 1113–1120. [Google Scholar] [CrossRef] [Green Version]
- Buresh, C.J.; Oliai, B.R.; Miller, R.T. Reactivity with TdT in Merkel cell carcinoma: A potential diagnostic pitfall. Am. J. Clin. Pathol. 2008, 129, 894–898. [Google Scholar] [CrossRef] [Green Version]
- Sidiropoulos, M.; Hanna, W.; Raphael, S.J.; Ghorab, Z. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am. J. Clin. Pathol. 2011, 135, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Sauer, C.M.; Haugg, A.M.; Chteinberg, E.; Rennspiess, D.; Winnepenninckx, V.; Speel, E.J.; Becker, J.C.; Kurz, A.K.; zur Hausen, A. Reviewing the current evidence supporting early B-cells as the cellular origin of Merkel cell carcinoma. Crit. Rev. Oncol. Hematol. 2017, 116, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.C.; Jahchan, N.S.; Sage, J.; Choi, J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene 2018, 37, 1409–1416. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Lambert, P.F. Merkel cell polyomavirus: A newly discovered human virus with oncogenic potential. Virology 2013, 435, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.J.; Chen, C.J.; Sullivan, C.S. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology 2009, 383, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yang, R.; Payne, A.S.; Schowalter, R.M.; Spurgeon, M.E.; Lambert, P.F.; Xu, X.; Buck, C.B.; You, J. Identifying the Target Cells and Mechanisms of Merkel Cell Polyomavirus Infection. Cell Host Microbe 2016, 19, 775–787. [Google Scholar] [CrossRef] [Green Version]
- DeCaprio, J.A. Merkel cell polyomavirus and Merkel cell carcinoma. Philos. Trans. R. Soc. Lond B Biol. Sci. 2017, 372, 20160276. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Paulson, K.G.; Murchison, E.P.; Afanasiev, O.K.; Alkan, C.; Leonard, J.H.; Byrd, D.R.; Hannon, G.J.; Nghiem, P. Identification and validation of a novel mature microRNA encoded by the Merkel cell polyomavirus in human Merkel cell carcinomas. J. Clin. Virol. 2011, 52, 272–275. [Google Scholar] [CrossRef] [Green Version]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, K.G.; Lewis, C.W.; Redman, M.W.; Simonson, W.T.; Lisberg, A.; Ritter, D.; Morishima, C.; Hutchinson, K.; Mudgistratova, L.; Blom, A.; et al. Viral oncoprotein antibodies as a marker for recurrence of Merkel cell carcinoma: A prospective validation study. Cancer 2017, 123, 1464–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef] [Green Version]
- Schrama, D.; Sarosi, E.M.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; König, E.M.; Utikal, J.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef]
- Cobrinik, D. Pocket proteins and cell cycle control. Oncogene 2005, 24, 2796–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech-Sioli, M.; Günther, T.; Therre, M.; Spohn, M.; Indenbirken, D.; Theiss, J.; Riethdorf, S.; Qi, M.; Alawi, M.; Wülbeck, C.; et al. High-resolution analysis of Merkel Cell Polyomavirus in Merkel Cell Carcinoma reveals distinct integration patterns and suggests NHEJ and MMBIR as underlying mechanisms. PLOS Pathog. 2020, 16, e1008562. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef]
- Pardee, A.B. A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 1974, 71, 1286–1290. [Google Scholar] [CrossRef] [Green Version]
- Pardee, A.B. G1 events and regulation of cell proliferation. Science 1989, 246, 603–608. [Google Scholar] [CrossRef]
- Sullivan, C.S.; Gilbert, S.P.; Pipas, J.M. ATP-dependent simian virus 40 T-antigen-Hsc70 complex formation. J. Virol. 2001, 75, 1601–1610. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, A.; McClellan, A.J.; Vartikar, J.; Marks, I.; Cantalupo, P.; Li, Y.; Whyte, P.; Rundell, K.; Brodsky, J.L.; Pipas, J.M. The amino-terminal transforming region of simian virus 40 large T and small t antigens functions as a J domain. Mol. Cell. Biol. 1997, 17, 4761–4773. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, D.; Sáenz-Robles, M.T.; Pipas, J.M. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005, 24, 7729–7745. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, C.S.; Cantalupo, P.; Pipas, J.M. The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol. Cell. Biol. 2000, 20, 6233–6243. [Google Scholar] [CrossRef]
- Stubdal, H.; Zalvide, J.; Campbell, K.S.; Schweitzer, C.; Roberts, T.M.; DeCaprio, J.A. Inactivation of pRB-related proteins p130 and p107 mediated by the J domain of simian virus 40 large T antigen. Mol. Cell. Biol. 1997, 17, 4979–4990. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Q.; Denis, D.; Ratnofsky, M.; Roberts, T.M.; DeCaprio, J.A.; Schaffhausen, B. The DnaJ domain of polyomavirus large T antigen is required to regulate Rb family tumor suppressor function. J. Virol. 1997, 71, 9410–9416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houben, R.; Angermeyer, S.; Haferkamp, S.; Aue, A.; Goebeler, M.; Schrama, D.; Hesbacher, S. Characterization of functional domains in the Merkel cell polyoma virus Large T antigen. Int. J. Cancer 2015, 136, E290–E300. [Google Scholar] [CrossRef] [PubMed]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [Green Version]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.S.; et al. Merkel Cell Polyomavirus Small T Antigen Induces Cancer and Embryonic Merkel Cell Proliferation in a Transgenic Mouse Model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef] [Green Version]
- Seshacharyulu, P.; Pandey, P.; Datta, K.; Batra, S.K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013, 335, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Guergnon, J.; Godet, A.N.; Galioot, A.; Falanga, P.B.; Colle, J.H.; Cayla, X.; Garcia, A. PP2A targeting by viral proteins: A widespread biological strategy from DNA/RNA tumor viruses to HIV-1. Biochim. Biophys. Acta 2011, 1812, 1498–1507. [Google Scholar] [CrossRef]
- Moens, U.; Macdonald, A. Effect of the Large and Small T-Antigens of Human Polyomaviruses on Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 3914. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Berrios, C.; Kim, M.; Schade, A.E.; Adelmant, G.; Yeerna, H.; Damato, E.; Iniguez, A.B.; Florens, L.; Washburn, M.P.; et al. STRIPAK directs PP2A activity toward MAP4K4 to promote oncogenic transformation of human cells. Elife 2020, 9, e53003. [Google Scholar] [CrossRef]
- Kwun, H.J.; Shuda, M.; Camacho, C.J.; Gamper, A.M.; Thant, M.; Chang, Y.; Moore, P.S. Restricted protein phosphatase 2A targeting by Merkel cell polyomavirus small T antigen. J. Virol. 2015, 89, 4191–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dye, K.N.; Welcker, M.; Clurman, B.E.; Roman, A.; Galloway, D.A. Merkel cell polyomavirus Tumor antigens expressed in Merkel cell carcinoma function independently of the ubiquitin ligases Fbw7 and β-TrCP. PLoS Pathog. 2019, 15, e1007543. [Google Scholar] [CrossRef]
- Yumimoto, K.; Nakayama, K.I. Recent insight into the role of FBXW7 as a tumor suppressor. Semin. Cancer Biol. 2020, 67, 1–15. [Google Scholar] [CrossRef]
- Zhao, J.; Jia, Y.; Shen, S.; Kim, J.; Wang, X.; Lee, E.; Brownell, I.; Cho-Vega, J.H.; Lewis, C.; Homsi, J.; et al. Merkel Cell Polyomavirus Small T Antigen Activates Noncanonical NF-κB Signaling to Promote Tumorigenesis. Mol. Cancer Res. 2020, 18, 1623–1637. [Google Scholar] [CrossRef]
- Nwogu, N.; Ortiz, L.E.; Whitehouse, A.; Kwun, H.J. Merkel Cell Polyomavirus Small Tumor Antigen Activates Matrix Metallopeptidase-9 Gene Expression for Cell Migration and Invasion. J. Virol. 2020, 94, e00786-20. [Google Scholar] [CrossRef]
- Kwun, H.J.; Wendzicki, J.A.; Shuda, Y.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen induces genome instability by E3 ubiquitin ligase targeting. Oncogene 2017, 36, 6784–6792. [Google Scholar] [CrossRef]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortés-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; DeCaprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2019, 116, 1027–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.E.; Cheng, J.; McGrath, J.P.; Lim, M.Y.; Cushman, C.; Swanson, S.K.; Tillgren, M.L.; Paulo, J.A.; Gokhale, P.C.; Florens, L.; et al. Merkel cell polyomavirus activates LSD1-mediated blockade of non-canonical BAF to regulate transformation and tumorigenesis. Nat. Cell Biol. 2020, 22, 603–615. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, M.M.; Cushman, C.H.; DeCaprio, J.A. Merkel Cell Polyomavirus: Oncogenesis in a Stable Genome. Viruses 2022, 14, 58. https://doi.org/10.3390/v14010058
Ahmed MM, Cushman CH, DeCaprio JA. Merkel Cell Polyomavirus: Oncogenesis in a Stable Genome. Viruses. 2022; 14(1):58. https://doi.org/10.3390/v14010058
Chicago/Turabian StyleAhmed, Mona M., Camille H. Cushman, and James A. DeCaprio. 2022. "Merkel Cell Polyomavirus: Oncogenesis in a Stable Genome" Viruses 14, no. 1: 58. https://doi.org/10.3390/v14010058
APA StyleAhmed, M. M., Cushman, C. H., & DeCaprio, J. A. (2022). Merkel Cell Polyomavirus: Oncogenesis in a Stable Genome. Viruses, 14(1), 58. https://doi.org/10.3390/v14010058