
So Pathogenic or So What?—A Brief Overview of SIV Pathogenesis with an Emphasis on Cure Research


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
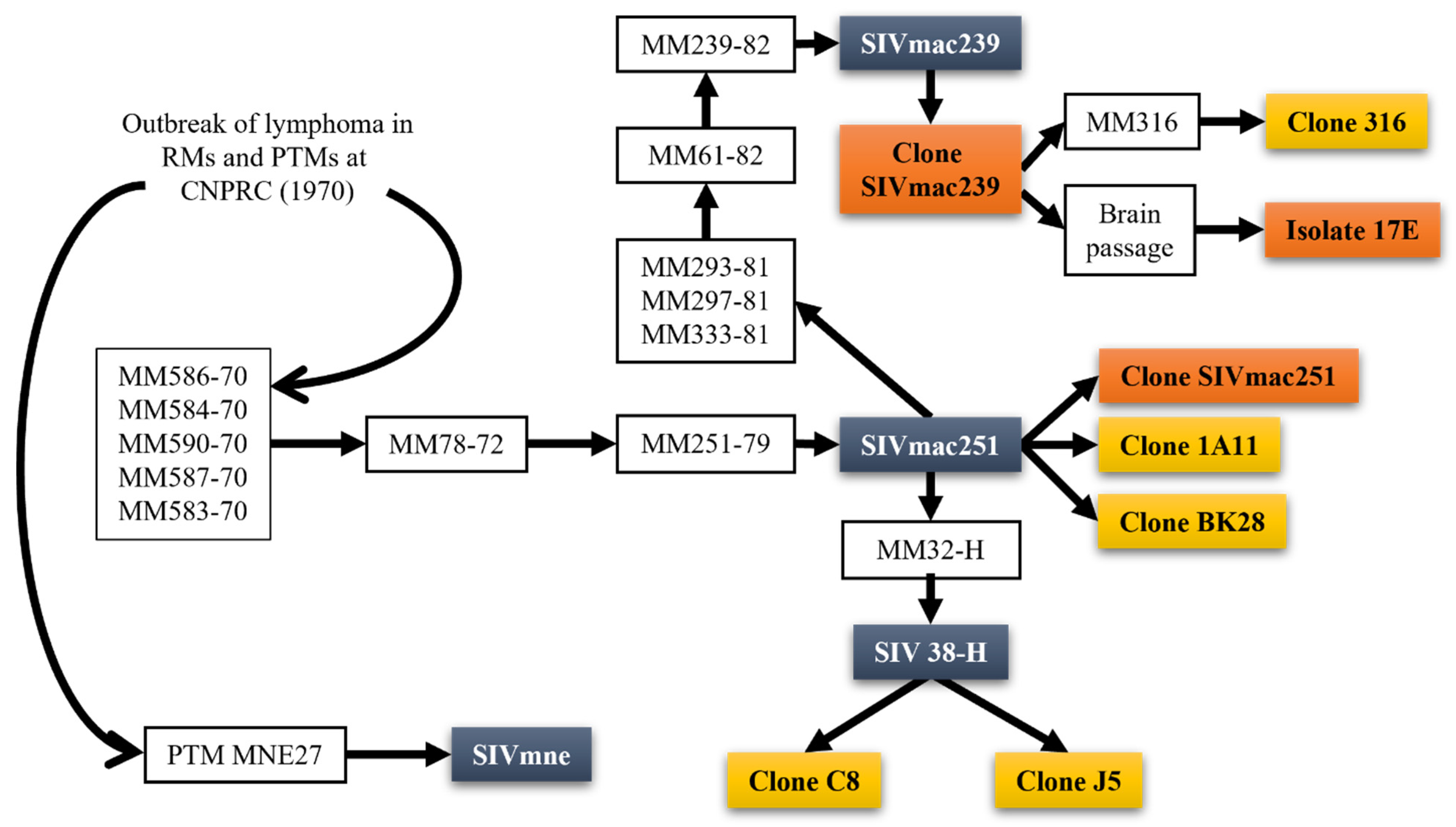
1. You Can Call Me SIV: Introduction and the Origin of SIVmac
2. Why Don’t You Infect Me? The Animal Model for AIDS Research
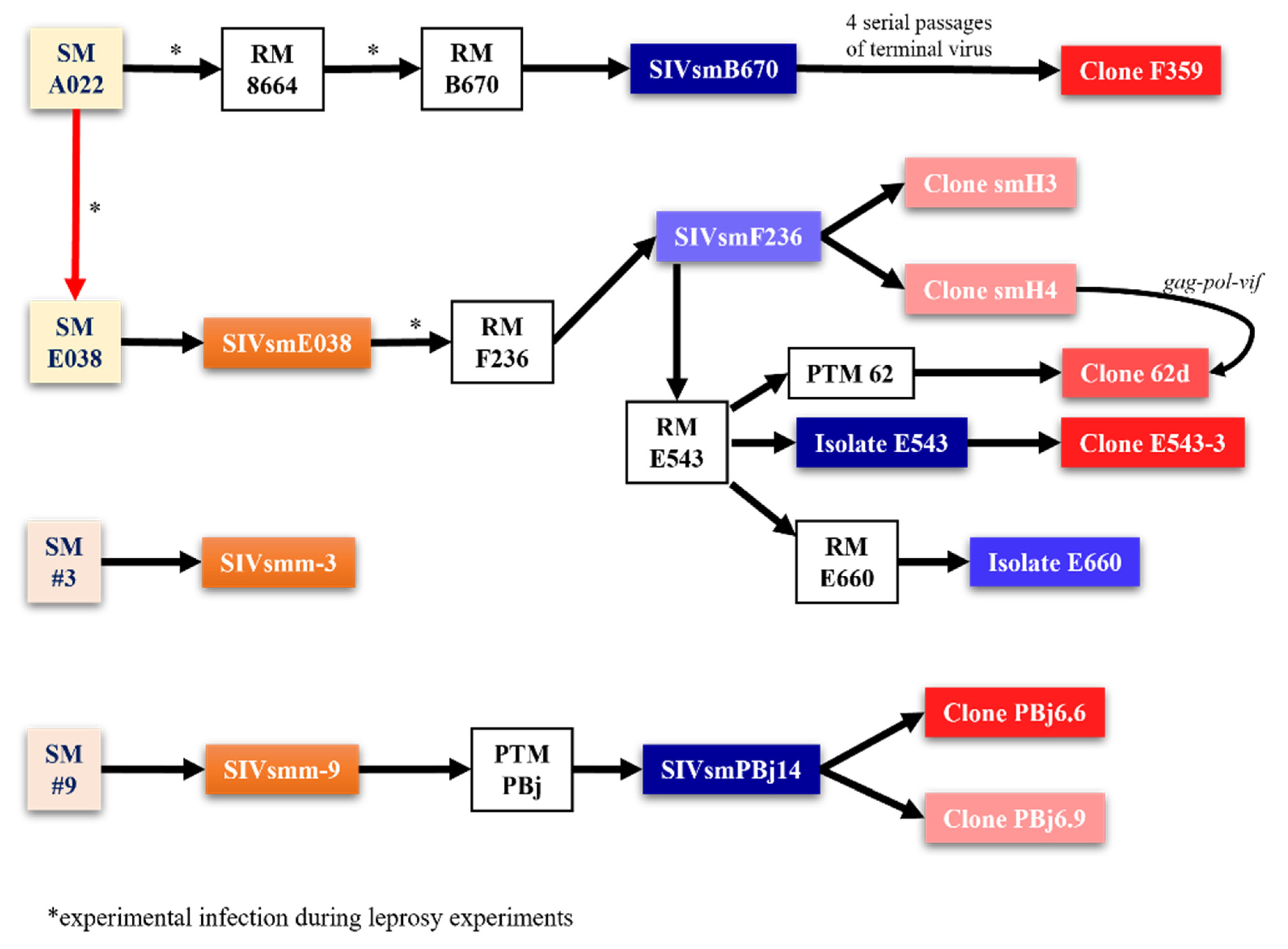
Fifty Ways to Infect a Monkey—SIV/SHIV Strains for Use in Nonhuman Primates
3. Everything Put Together Falls Apart. A Brief Introduction to HIV/SIV Pathogenesis
3.1. HIV/SIV Reservoir Is Rapidly Seeded after Transmission
3.2. Immune Response during Acute Infection Drives Viral Set-Point
3.3. Control at Last—Antiretroviral Therapy for HIV
4. At the Zoo. Nonhuman Primate Models for HIV Pathogenesis
4.1. Similarities and Recapitulation of Specific Pathogenicities
4.2. Everything about It Is Inflammation and Immune Activation
4.3. Gut Dysfunction and Microbial Translocation Potentiate Immune Activation and Inflammation
4.4. Under African Skies—Study of Natural Hosts Demonstrates Important Differences between Pathogenic and Nonpathogenic Infections
4.5. How the Heart Approaches What It Yearns—SIV as Models for the Study of HIV-Related Comorbidities
5. For the Cure, Whenever We May Find Her
5.1. The Need for an HIV Cure
5.2. The Latent Reservoir Currently Prevents HIV Cure
5.3. Multi-Trick Pony—Mechanisms of HIV Latency Establishment
5.4. Reservoir Decay Is Not Curative
5.5. Somewhere Researchers Cannot Find Me: Technical Obstacles towards Reservoir Quantification
5.6. SIVmac-Infected RMs as a Model for Cure Research
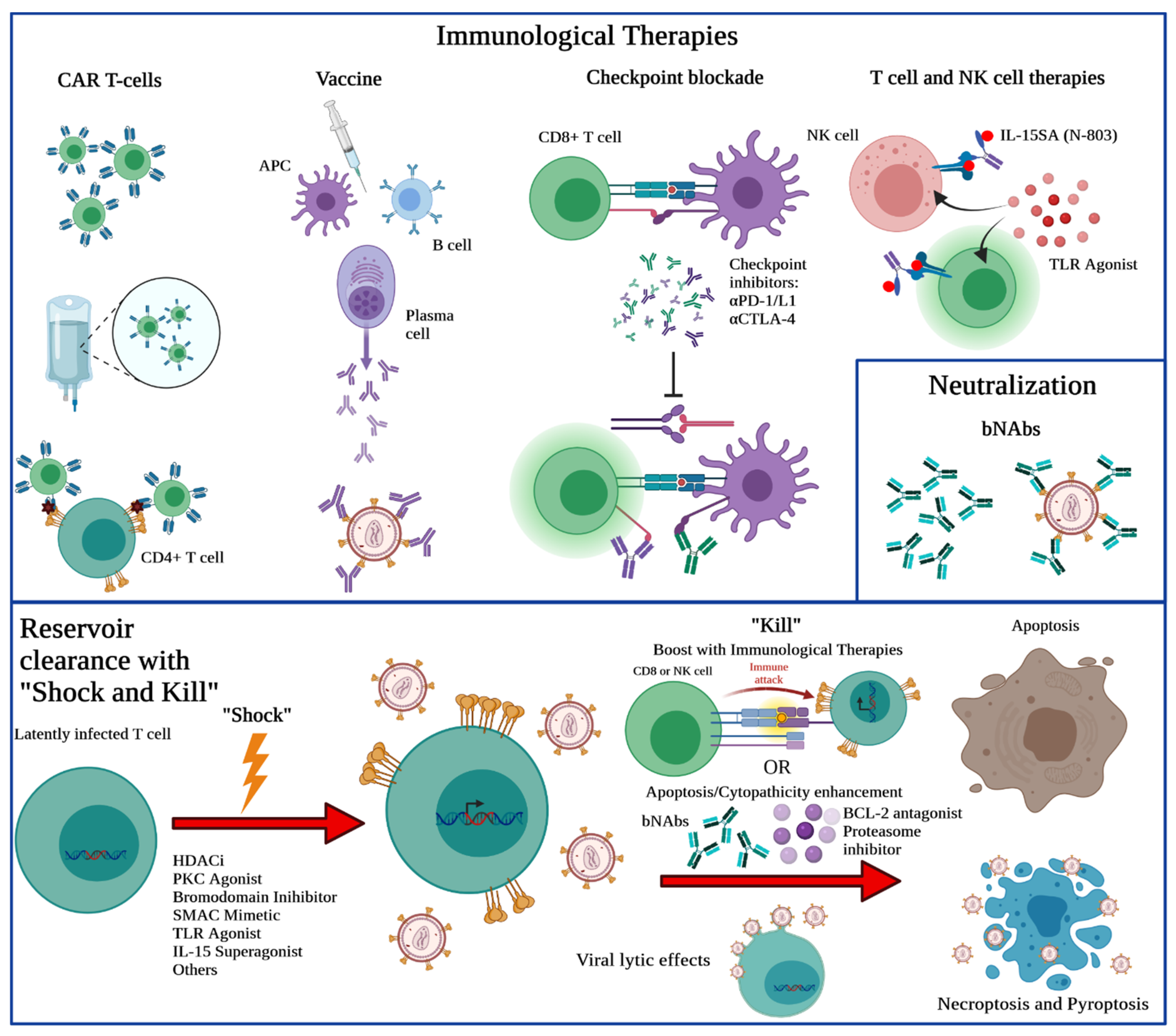
6. Still Searching after All These Years: Strategies towards an HIV/SIV Cure
6.1. ART Intensification as a Cure Strategy
6.2. “Block and Lock”—Transcriptional Silencing for HIV Functional Cure
6.3. Gene Therapy and Engineered CAR T Cells for HIV Cure
6.4. Enhancing Apoptosis and Cytopathic Effects as a Cure Strategy
6.5. Bone Marrow Transplant for HIV Cure
6.6. Broadly Neutralizing Antibodies for the HIV Cure
6.7. HIV Vaccines for HIV Prevention or Therapeutics
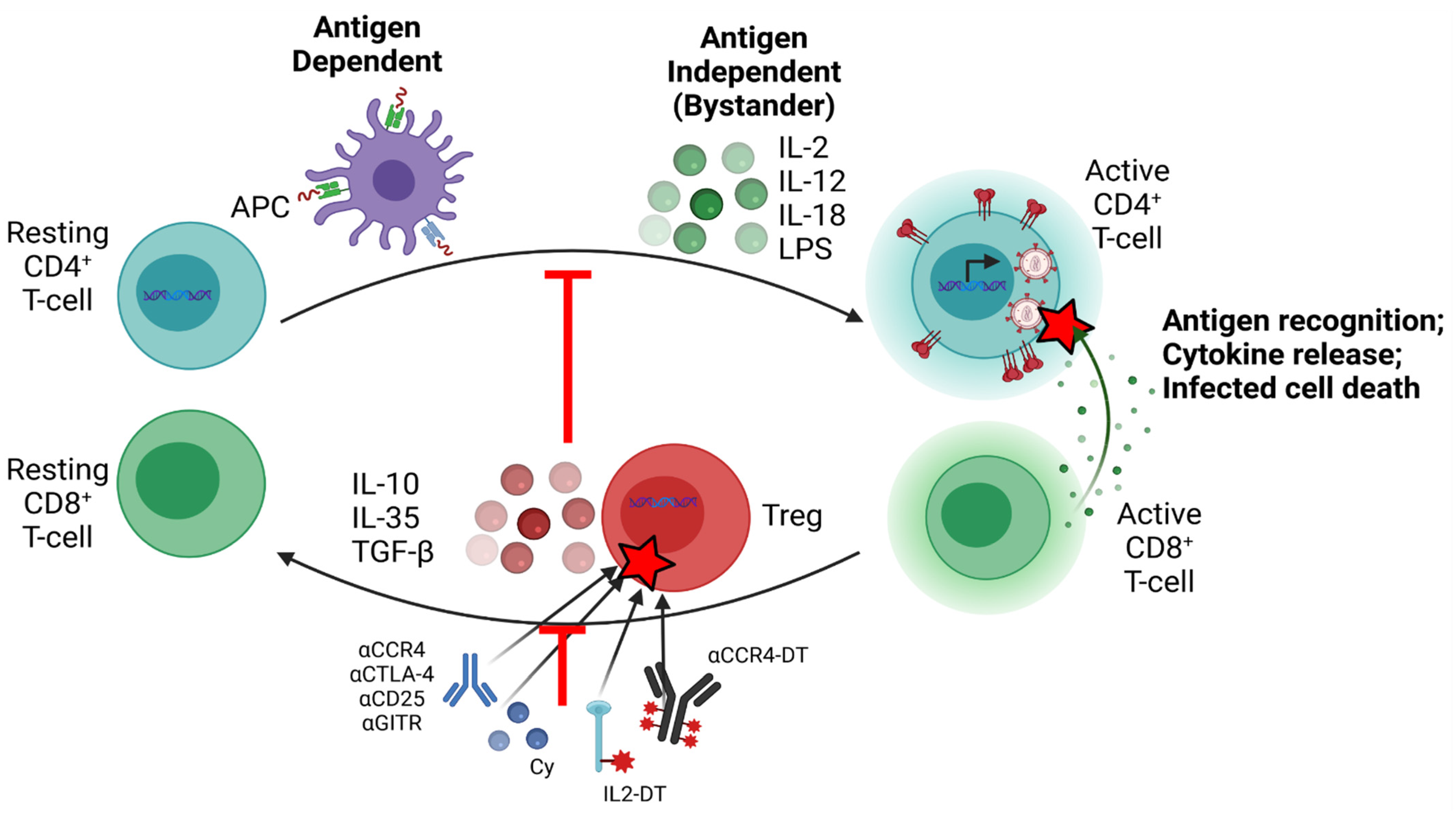
6.8. Targeting Tregs as a Strategy for Cure Research
6.8.1. Targeting CD25
6.8.2. Targeting CCR4
6.8.3. Targeting GITR
6.8.4. Cyclophosphamide (Cy) for Treg Depletion
6.9. T-Cell Exhaustion and Targeted Therapies
6.9.1. CTLA-4 Blockade in HIV Cure
6.9.2. PD-1 Blockade in HIV Cure
6.9.3. IL-15 for HIV Cure
6.9.4. IL-21 in HIV Cure
6.10. “Shock and Kill”—Latency Reactivation for HIV Cure
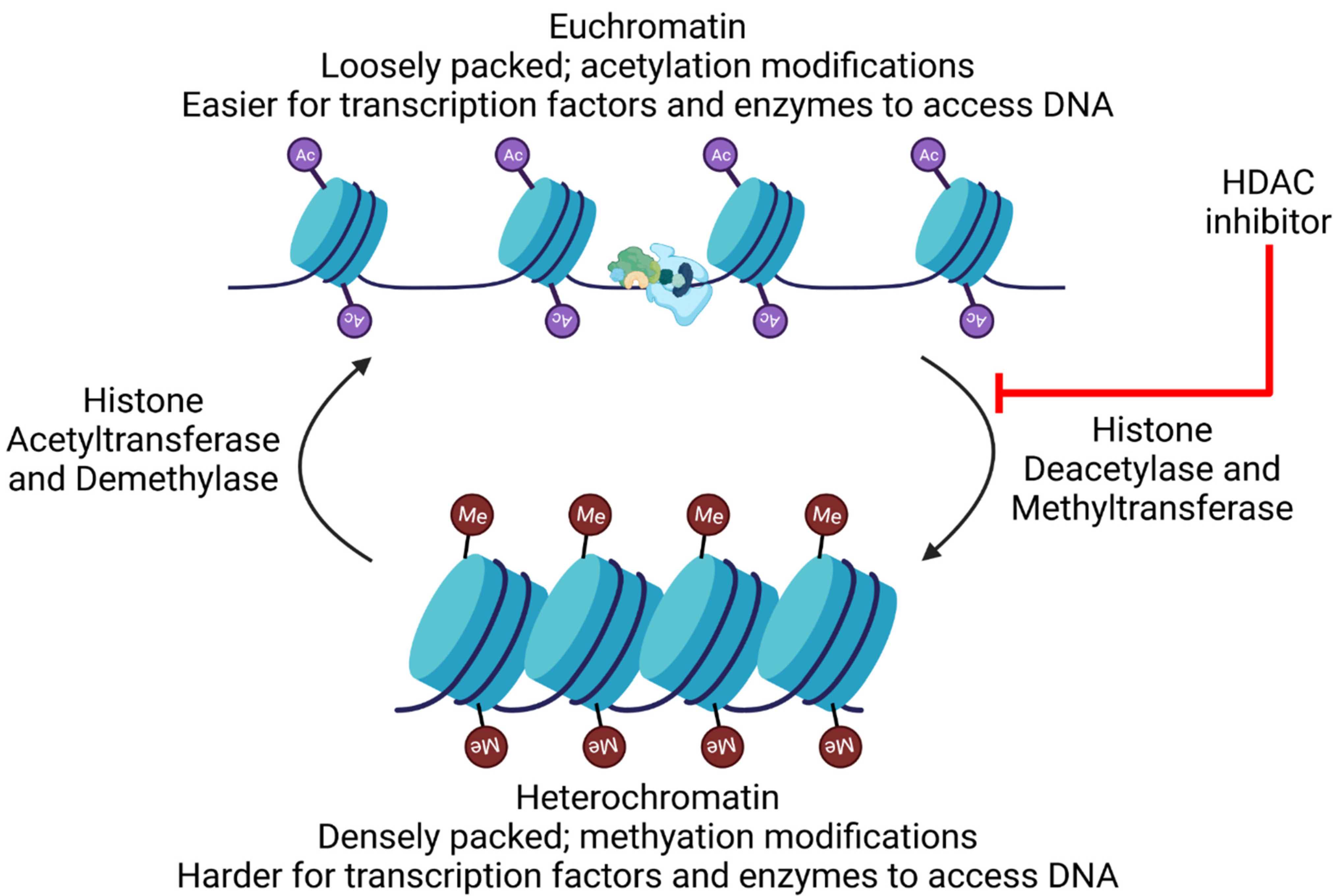
6.10.1. HDAC Inhibitors for “Shock and Kill”
6.10.2. Protein Kinase C (PKC) Agonists for “Shock and Kill”
6.10.3. Ingenol Derivatives for “Shock and Kill”
6.10.4. Bromodomain Inhibitors for “Shock and Kill”
6.10.5. Second Mitochondrial Activator of Caspases (SMAC) Mimetics for “Shock and Kill”
6.10.6. Stimulator of Interferon Genes (STING) Agonists for “Shock and Kill”
6.10.7. Toll-like Receptor (TLR) Agonists for “Shock and Kill”
6.11. When Calories Get Serious—Role of Immunometabolism in HIV Pathogenesis and Cure
6.11.1. Immunometabolism and HIV Pathogenesis
6.11.2. Modulation of Immunometabolic Programming for HIV Cure/Therapeutics
6.12. True or False—Macrophages and the HIV/SIV Reservoir
6.12.1. Macrophages Contribution to the HIV/SIV reservoir
6.12.2. Crosstalk between Macrophages and Exhausted T Cells
6.12.3. Targeting Macrophages with “Shock and Kill”
7. Further to Fly: Future Perspectives for HIV Cure and Avenues to Explore
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Barré-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.; Letvin, N.; King, N.; Kannagi, M.; Sehgal, P.; Hunt, R.; Kanki, P.; Essex, M.; Desrosiers, R. Isolation of T-cell tropic HTLV-III-like retrovirus from macaques. Science 1985, 228, 1201–1204. [Google Scholar] [CrossRef]
- Johnson, L.F.; Mossong, J.; Dorrington, R.E.; Schomaker, M.; Hoffmann, C.J.; Keiser, O.; Fox, M.P.; Wood, R.; Prozesky, H.; Giddy, J.; et al. Life Expectancies of South African Adults Starting Antiretroviral Treatment: Collaborative Analysis of Cohort Studies. PLoS Med. 2013, 10, e1001418. [Google Scholar] [CrossRef]
- Guaraldi, G.; Orlando, G.; Zona, S.; Menozzi, M.; Carli, F.; Garlassi, E.; Berti, A.; Rossi, E.; Roverato, A.; Palella, F. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2011, 53, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Zona, S.; Brothers, T.D.; Carli, F.; Stentarelli, C.; Dolci, G.; Santoro, A.; Beghetto, B.; Menozzi, M.; Mussini, C.; et al. Aging with HIV vs. HIV seroconversion at older age: A diverse population with distinct comorbidity profiles. PLoS ONE 2015, 10, e0118531. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.; Hsue, P.; Budd, D.; Meyer, N. Healthcare utilization and direct costs of non-infectious comorbidities in HIV-infected patients in the USA. Curr. Med. Res. Opin. 2018, 34, 13–23. [Google Scholar] [CrossRef]
- Allers, K.; Hütter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Stowell, R.E.; Smith, E.K.; España, C.; Nelson, V.G. Outbreak of malignant lymphoma in rhesus monkeys. Lab. Investig. A J. Tech. Methods Pathol. 1971, 25, 476–479. [Google Scholar]
- Terrell, T.G.; Gribble, D.H.; Osburn, B.I. Malignant lymphoma in macaques: A clinicopathologic study of 45 cases. J. Natl. Cancer Inst. 1980, 64, 561–568. [Google Scholar]
- Gardner, M.B. The history of simian AIDS. J. Med. Primatol. 1996, 25, 148–157. [Google Scholar] [CrossRef]
- Letvin, N.L.; Daniel, M.D.; Sehgal, P.K.; Desrosiers, R.C.; Hunt, R.D.; Waldron, L.M.; MacKey, J.J.; Schmidt, D.K.; Chalifoux, L.V.; King, N.W. Induction of AIDS-like disease in macaque monkeys with T-cell tropic retrovirus STLV-III. Science 1985, 230, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.A.; Silvestri, G. Natural Hosts of SIV: Implication in AIDS; Newnes: Oxford, UK, 2014. [Google Scholar]
- Lowenstine, L.J.; Pedersen, N.C.; Higgins, J.; Pallis, K.C.; Uyeda, A.; Marx, P.; Lerche, N.W.; Munn, R.J.; Gardner, M.B. Seroepidemiologic survey of captive Old-World primates for antibodies to human and simian retroviruses, and isolation of a lentivirus from sooty mangabeys (Cercocebus atys). Int. J. Cancer 1986, 38, 563–574. [Google Scholar] [CrossRef]
- Ohta, Y.; Masuda, T.; Tsujimoto, H.; Ishikawa, K.; Kodama, T.; Morikawa, S.; Nakai, M.; Honjo, S.; Hayami, M. Isolation of simian immunodeficiency virus from African green monkeys and seroepidemiologic survey of the virus in various non-human primates. Int. J. Cancer 1988, 41, 115–122. [Google Scholar] [CrossRef]
- Ma, D.; Jasinska, A.; Kristoff, J.; Grobler, J.P.; Turner, T.; Jung, Y.; Schmitt, C.; Raehtz, K.; Feyertag, F.; Martinez Sosa, N.; et al. SIVagm infection in wild African green monkeys from South Africa: Epidemiology, natural history, and evolutionary considerations. PLoS Pathog. 2013, 9, e1003011. [Google Scholar] [CrossRef]
- Raehtz, K.; Pandrea, I.; Apetrei, C. The well-tempered SIV infection: Pathogenesis of SIV infection in natural hosts in the wild, with emphasis on virus transmission and early events post-infection that may contribute to protection from disease progression. Infect. Genet. Evol. 2016, 46, 308–323. [Google Scholar] [CrossRef] [PubMed]
- VandeWoude, S.; Apetrei, C. Going wild: Lessons from naturally occurring T-lymphotropic lentiviruses. Clin. Microbiol. Rev. 2006, 19, 728–762. [Google Scholar] [CrossRef] [PubMed]
- Apetrei, C.; Lerche, N.W.; Pandrea, I.; Gormus, B.; Silvestri, G.; Kaur, A.; Robertson, D.L.; Hardcastle, J.; Lackner, A.A.; Marx, P.A. Kuru experiments triggered the emergence of pathogenic SIVmac. AIDS (Lond. Engl.) 2006, 20, 317–321. [Google Scholar] [CrossRef]
- Corbet, S.; Müller-Trutwin, M.C.; Versmisse, P.; Delarue, S.; Ayouba, A.; Lewis, J.; Brunak, S.; Martin, P.; Brun-Vezinet, F.; Simon, F.; et al. env sequences of simian immunodeficiency viruses from chimpanzees in Cameroon are strongly related to those of human immunodeficiency virus group N from the same geographic area. J. Virol. 2000, 74, 529–534. [Google Scholar] [CrossRef]
- Chen, Z.; Telfier, P.; Gettie, A.; Reed, P.; Zhang, L.; Ho, D.D.; Marx, P.A. Genetic characterization of new West African simian immunodeficiency virus SIVsm: Geographic clustering of household-derived SIV strains with human immunodeficiency virus type 2 subtypes and genetically diverse viruses from a single feral sooty mangabey troop. J. Virol. 1996, 70, 3617–3627. [Google Scholar] [CrossRef]
- Apetrei, C.; Kaur, A.; Lerche, N.W.; Metzger, M.; Pandrea, I.; Hardcastle, J.; Falkenstein, S.; Bohm, R.; Koehler, J.; Traina-Dorge, V.; et al. Molecular epidemiology of simian immunodeficiency virus SIVsm in U.S. primate centers unravels the origin of SIVmac and SIVstm. J. Virol. 2005, 79, 8991–9005. [Google Scholar] [CrossRef]
- Mansfield, K.G.; Lerch, N.W.; Gardner, M.B.; Lackner, A.A. Origins of simian immunodeficiency virus infection in macaques at the New England Regional Primate Research Center. J. Med. Primatol. 1995, 24, 116–122. [Google Scholar] [CrossRef]
- Benveniste, R.E.; Morton, W.R.; Clark, E.A.; Tsai, C.C.; Ochs, H.D.; Ward, J.M.; Kuller, L.; Knott, W.B.; Hill, R.W.; Gale, M.J. Inoculation of baboons and macaques with simian immunodeficiency virus/Mne, a primate lentivirus closely related to human immunodeficiency virus type 2. J. Virol. 1988, 62, 2091–2101. [Google Scholar] [CrossRef]
- Khan, A.S.; Galvin, T.A.; Jennings, M.B.; Gardner, M.B.; Lowenstine, L.J. SIV of stump-tailed macaque (SIVstm) is a divergent Asian isolate. J. Med. Primatol. 1991, 20, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Novembre, F.J.; Hirsch, V.M.; McClure, H.M.; Fultz, P.N.; Johnson, P.R. SIV from stump-tailed macaques: Molecular characterization of a highly transmissible primate lentivirus. Virology 1992, 186, 783–787. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Johnson, W.E.; Kirmaier, A. Phylogeny and History of the Lost SIV from Crab-Eating Macaques: SIVmfa. PLoS ONE 2016, 11, e0159281. [Google Scholar] [CrossRef]
- Whitney, J.B.; Hill, A.L.; Sanisetty, S.; Penaloza-MacMaster, P.; Liu, J.; Shetty, M.; Parenteau, L.; Cabral, C.; Shields, J.; Blackmore, S.; et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 2014, 512, 74–77. [Google Scholar] [CrossRef]
- Henrich, T.J.; Hatano, H.; Bacon, O.; Hogan, L.E.; Rutishauser, R.; Hill, A.; Kearney, M.F.; Anderson, E.M.; Buchbinder, S.P.; Cohen, S.E.; et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PLOS Med. 2017, 14, e1002417. [Google Scholar] [CrossRef] [PubMed]
- Okoye, A.A.; Hansen, S.G.; Vaidya, M.; Fukazawa, Y.; Park, H.; Duell, D.M.; Lum, R.; Hughes, C.M.; Ventura, A.B.; Ainslie, E.; et al. Early antiretroviral therapy limits SIV reservoir establishment to delay or prevent post-treatment viral rebound. Nat. Med. 2018, 24, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Policicchio, B.B.; Pandrea, I.; Apetrei, C. Animal Models for HIV Cure Research. Front. Immunol. 2016, 7, 12. [Google Scholar] [CrossRef]
- Deleage, C.; Wietgrefe, S.W.; Del Prete, G.; Morcock, D.R.; Hao, X.P.; Piatak, M., Jr.; Bess, J.; Anderson, J.L.; Perkey, K.E.; Reilly, C.; et al. Defining HIV and SIV Reservoirs in Lymphoid Tissues. Pathog. Immun. 2016, 1, 68–106. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Marshall, E.E.; Malouli, D.; Ventura, A.B.; Hughes, C.M.; Ainslie, E.; Ford, J.C.; Morrow, D.; Gilbride, R.M.; Bae, J.Y.; et al. A live-attenuated RhCMV/SIV vaccine shows long-term efficacy against heterologous SIV challenge. Sci. Transl. Med. 2019, 11, eaaw2607. [Google Scholar] [CrossRef]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-Y.; Osuna, C.E.; Hraber, P.T.; Hesselgesser, J.; Gerold, J.M.; Barnes, T.L.; Sanisetty, S.; Seaman, M.S.; Lewis, M.G.; Geleziunas, R.; et al. TLR7 agonists induce transient viremia and reduce the viral reservoir in SIV-infected rhesus macaques on antiretroviral therapy. Sci. Transl. Med. 2018, 10, eaao4521. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Perez, S.; Gao, Y.; Doyle-Meyers, L.; Foley, B.T.; Li, Q.; Ling, B. Persistent Viral Reservoirs in Lymphoid Tissues in SIV-Infected Rhesus Macaques of Chinese-Origin on Suppressive Antiretroviral Therapy. Viruses 2019, 11, 105. [Google Scholar] [CrossRef]
- Veazey, R.S.; DeMaria, M.; Chalifoux, L.V.; Shvetz, D.E.; Pauley, D.R.; Knight, H.L.; Rosenzweig, M.; Johnson, R.P.; Desrosiers, R.C.; Lackner, A.A. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 1998, 280, 427–431. [Google Scholar] [CrossRef]
- Smit-McBride, Z.; Mattapallil, J.J.; McChesney, M.; Ferrick, D.; Dandekar, S. Gastrointestinal T lymphocytes retain high potential for cytokine responses but have severe CD4(+) T-cell depletion at all stages of simian immunodeficiency virus infection compared to peripheral lymphocytes. J. Virol. 1998, 72, 6646–6656. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Schacker, T.W.; Ruff, L.E.; Price, D.A.; Taylor, J.H.; Beilman, G.J.; Nguyen, P.L.; Khoruts, A.; Larson, M.; Haase, A.T.; et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 2004, 200, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Mehandru, S.; Poles, M.A.; Tenner-Racz, K.; Horowitz, A.; Hurley, A.; Hogan, C.; Boden, D.; Racz, P.; Markowitz, M. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 2004, 200, 761–770. [Google Scholar] [CrossRef]
- He, T.; Xu, C.; Krampe, N.; Dillon, S.M.; Sette, P.; Falwell, E.; Haret-Richter, G.S.; Butterfield, T.; Dunsmore, T.L.; McFadden, W.M., Jr.; et al. High-fat diet exacerbates SIV pathogenesis and accelerates disease progression. J. Clin. Investig. 2019, 129, 5474–5488. [Google Scholar] [CrossRef]
- Miller, C.J.; Hu, J. T Cell—Tropic Simian Immunodeficiency Virus (SIV) and Simian-Human Immunodeficiency Viruses Are Readily Transmitted by Vaginal Inoculation of Rhesus Macaques, and Langerhans’ Cells of the Female Genital Tract Are Infected with SIV. J. Infect. Dis. 1999, 179, S413–S417. [Google Scholar] [CrossRef] [PubMed]
- Heeney, J.L.; Rutjens, E.; Verschoor, E.J.; Niphuis, H.; ten Haaft, P.; Rouse, S.; McClure, H.; Balla-Jhagjhoorsingh, S.; Bogers, W.; Salas, M.; et al. Transmission of simian immunodeficiency virus SIVcpz and the evolution of infection in the presence and absence of concurrent human immunodeficiency virus type 1 infection in chimpanzees. J. Virol. 2006, 80, 7208–7218. [Google Scholar] [CrossRef][Green Version]
- Stone, M.; Keele, B.F.; Ma, Z.M.; Bailes, E.; Dutra, J.; Hahn, B.H.; Shaw, G.M.; Miller, C.J. A limited number of simian immunodeficiency virus (SIV) env variants are transmitted to rhesus macaques vaginally inoculated with SIVmac251. J. Virol. 2010, 84, 7083–7095. [Google Scholar] [CrossRef]
- Deleage, C.; Immonen, T.T.; Fennessey, C.M.; Reynaldi, A.; Reid, C.; Newman, L.; Lipkey, L.; Schlub, T.E.; Camus, C.; O’Brien, S.; et al. Defining early SIV replication and dissemination dynamics following vaginal transmission. Sci. Adv. 2019, 5, eaav7116. [Google Scholar] [CrossRef]
- Barrenas, F.; Raehtz, K.; Xu, C.; Law, L.; Green, R.R.; Silvestri, G.; Bosinger, S.E.; Nishida, A.; Li, Q.; Lu, W.; et al. Macrophage-associated wound healing contributes to African green monkey SIV pathogenesis control. Nat. Commun. 2019, 10, 5101. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, C.; Ploquin, M.J.; Pandrea, I.; Faye, A.; Onanga, R.; Apetrei, C.; Poaty-Mavoungou, V.; Rouquet, P.; Estaquier, J.; Mortara, L.; et al. Antiinflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J. Clin. Investig. 2005, 115, 1082–1091. [Google Scholar] [CrossRef]
- Raehtz, K.D.; Barrenäs, F.; Xu, C.; Busman-Sahay, K.; Valentine, A.; Law, L.; Ma, D.; Policicchio, B.B.; Wijewardana, V.; Brocca-Cofano, E.; et al. African green monkeys avoid SIV disease progression by preventing intestinal dysfunction and maintaining mucosal barrier integrity. PLoS Pathog. 2020, 16, e1008333. [Google Scholar] [CrossRef] [PubMed]
- Kuller, L.H.; Tracy, R.; Belloso, W.; De Wit, S.; Drummond, F.; Lane, H.C.; Ledergerber, B.; Lundgren, J.; Neuhaus, J.; Nixon, D.; et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008, 5, e203. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Apetrei, C. Where the wild things are: Pathogenesis of SIV infection in African nonhuman primate hosts. Curr. HIV/AIDS Rep. 2010, 7, 28–36. [Google Scholar] [CrossRef]
- Pandrea, I.; Cornell, E.; Wilson, C.; Ribeiro, R.M.; Ma, D.; Kristoff, J.; Xu, C.; Haret-Richter, G.S.; Trichel, A.; Apetrei, C.; et al. Coagulation biomarkers predict disease progression in SIV-infected nonhuman primates. Blood 2012, 120, 1357–1366. [Google Scholar] [CrossRef]
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Landay, A.; Wilson, C.; Stock, J.; Tracy, R.; Apetrei, C. Using the pathogenic and nonpathogenic nonhuman primate model for studying non-AIDS comorbidities. Curr. HIV/AIDS Rep. 2015, 12, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Murphey-Corb, M.; Martin, L.N.; Rangan, S.R.; Baskin, G.B.; Gormus, B.J.; Wolf, R.H.; Andes, W.A.; West, M.; Montelaro, R.C. Isolation of an HTLV-III-related retrovirus from macaques with simian AIDS and its possible origin in asymptomatic mangabeys. Nature 1986, 321, 435–437. [Google Scholar] [CrossRef]
- Hirsch, V.M.; Dapolito, G.; McGann, C.; Olmsted, R.A.; Purcell, R.H.; Johnson, P.R. Molecular Cloning of SIV From Sooty Mangabey Monkeys. J. Med. Primatol. 1989, 18, 279–285. [Google Scholar] [CrossRef]
- Hirsch, V.M.; Olmsted, R.A.; Murphey-Corb, M.; Purcell, R.H.; Johnson, P.R. An African primate lentivirus (SIVsm) closely related to HIV-2. Nature 1989, 339, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, V.; Adger-Johnson, D.; Campbell, B.; Goldstein, S.; Brown, C.; Elkins, W.R.; Montefiori, D.C. A molecularly cloned, pathogenic, neutralization-resistant simian immunodeficiency virus, SIVsmE543-3. J. Virol. 1997, 71, 1608–1620. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, V.M.; Zack, P.M.; Johnson, P.R. Molecular characterization of SIV in tissues from experimentally infected macaques. J. Med. Primatol. 1990, 19, 287–294. [Google Scholar] [CrossRef]
- Hirsch, V.M.; Johnson, P.R. Pathogenic diversity of simian immunodeficiency viruses. Virus Res. 1994, 32, 183–203. [Google Scholar] [CrossRef]
- Letvin, N.L.; Rao, S.S.; Montefiori, D.C.; Seaman, M.S.; Sun, Y.; Lim, S.Y.; Yeh, W.W.; Asmal, M.; Gelman, R.S.; Shen, L.; et al. Immune and Genetic Correlates of Vaccine Protection Against Mucosal Infection by SIV in Monkeys. Sci. Transl. Med. 2011, 3, 81ra36. [Google Scholar] [CrossRef]
- Wu, F.; Ourmanov, I.; Kuwata, T.; Goeken, R.; Brown Charles, R.; Buckler-White, A.; Iyengar, R.; Plishka, R.; Aoki Scott, T.; Hirsch Vanessa, M. Sequential Evolution and Escape from Neutralization of Simian Immunodeficiency Virus SIVsmE660 Clones in Rhesus Macaques. J. Virol. 2012, 86, 8835–8847. [Google Scholar] [CrossRef] [PubMed]
- Kirmaier, A.; Wu, F.; Newman, R.M.; Hall, L.R.; Morgan, J.S.; O’Connor, S.; Marx, P.A.; Meythaler, M.; Goldstein, S.; Buckler-White, A.; et al. TRIM5 Suppresses Cross-Species Transmission of a Primate Immunodeficiency Virus and Selects for Emergence of Resistant Variants in the New Species. PLoS Biol. 2010, 8, e1000462. [Google Scholar] [CrossRef]
- Lopker, M.J.; Del Prete, G.Q.; Estes, J.D.; Li, H.; Reid, C.; Newman, L.; Lipkey, L.; Camus, C.; Easlick, J.L.; Wang, S.; et al. Derivation and Characterization of Pathogenic Transmitted/Founder Molecular Clones from Simian Immunodeficiency Virus SIVsmE660 and SIVmac251 following Mucosal Infection. J. Virol. 2016, 90, 8435–8453. [Google Scholar] [CrossRef] [PubMed]
- Brocca-Cofano, E.; Xu, C.; Wetzel, K.S.; Cottrell, M.L.; Policicchio, B.B.; Raehtz, K.D.; Ma, D.; Dunsmore, T.; Haret-Richter, G.S.; Musaitif, K.; et al. Marginal Effects of Systemic CCR5 Blockade with Maraviroc on Oral Simian Immunodeficiency Virus Transmission to Infant Macaques. J. Virol. 2018, 92, e00576-00518. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, G.Q.; Park, H.; Fennessey, C.M.; Reid, C.; Lipkey, L.; Newman, L.; Oswald, K.; Kahl, C.; Piatak, M., Jr.; Quinones, O.A.; et al. Molecularly tagged simian immunodeficiency virus SIVmac239 synthetic swarm for tracking independent infection events. J. Virol. 2014, 88, 8077–8090. [Google Scholar] [CrossRef]
- Fennessey, C.M.; Pinkevych, M.; Immonen, T.T.; Reynaldi, A.; Venturi, V.; Nadella, P.; Reid, C.; Newman, L.; Lipkey, L.; Oswald, K.; et al. Genetically-barcoded SIV facilitates enumeration of rebound variants and estimation of reactivation rates in nonhuman primates following interruption of suppressive antiretroviral therapy. PLoS Pathog. 2017, 13, e1006359. [Google Scholar] [CrossRef]
- Khanal, S.; Fennessey, C.M.; O’Brien, S.P.; Thorpe, A.; Reid, C.; Immonen, T.T.; Smith, R.; Bess, J.W., Jr.; Swanstrom, A.E.; Del Prete, G.Q.; et al. In Vivo Validation of the Viral Barcoding of Simian Immunodeficiency Virus SIVmac239 and the Development of New Barcoded SIV and Subtype B and C Simian-Human Immunodeficiency Viruses. J. Virol. 2019, 94, e01420-01419. [Google Scholar] [CrossRef]
- Diop, O.M.; Gueye, A.; Dias-Tavares, M.; Kornfeld, C.; Faye, A.; Ave, P.; Huerre, M.; Corbet, S.; Barre-Sinoussi, F.; Müller-Trutwin, M.C. High levels of viral replication during primary simian immunodeficiency virus SIVagm infection are rapidly and strongly controlled in African green monkeys. J. Virol. 2000, 74, 7538–7547. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gnanadurai, C.W.; Pandrea, I.; Parrish, N.F.; Kraus, M.H.; Learn, G.H.; Salazar, M.G.; Sauermann, U.; Töpfer, K.; Gautam, R.; Münch, J.; et al. Genetic identity and biological phenotype of a transmitted/founder virus representative of nonpathogenic simian immunodeficiency virus infection in African green monkeys. J. Virol. 2010, 84, 12245–12254. [Google Scholar] [CrossRef][Green Version]
- Mandell, D.T.; Kristoff, J.; Gaufin, T.; Gautam, R.; Ma, D.; Sandler, N.; Haret-Richter, G.; Xu, C.; Aamer, H.; Dufour, J.; et al. Pathogenic features associated with increased virulence upon Simian immunodeficiency virus cross-species transmission from natural hosts. J. Virol. 2014, 88, 6778–6792. [Google Scholar] [CrossRef]
- Pandrea, I.; Gaufin, T.; Gautam, R.; Kristoff, J.; Mandell, D.; Montefiori, D.; Keele, B.F.; Ribeiro, R.M.; Veazey, R.S.; Apetrei, C. Functional Cure of SIVagm Infection in Rhesus Macaques Results in Complete Recovery of CD4+ T Cells and Is Reverted by CD8+ Cell Depletion. PLoS Pathog. 2011, 7, e1002170. [Google Scholar] [CrossRef]
- Malikov, V.; da Silva, E.S.; Jovasevic, V.; Bennett, G.; de Souza Aranha Vieira, D.A.; Schulte, B.; Diaz-Griffero, F.; Walsh, D.; Naghavi, M.H. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun. 2015, 6, 6660. [Google Scholar] [CrossRef] [PubMed]
- Hatziioannou, T.; Bieniasz, P.D. Antiretroviral restriction factors. Curr. Opin. Virol. 2011, 1, 526–532. [Google Scholar] [CrossRef][Green Version]
- Li, J.; Lord, C.I.; Haseltine, W.; Letvin, N.L.; Sodroski, J. Infection of cynomolgus monkeys with a chimeric HIV-1/SIVmac virus that expresses the HIV-1 envelope glycoproteins. J. Acquir. Immune Defic. Syndr. 1992, 5, 639–646. [Google Scholar] [PubMed]
- Reimann, K.A.; Li, J.T.; Voss, G.; Lekutis, C.; Tenner-Racz, K.; Racz, P.; Lin, W.; Montefiori, D.C.; Lee-Parritz, D.E.; Lu, Y.; et al. An env gene derived from a primary human immunodeficiency virus type 1 isolate confers high in vivo replicative capacity to a chimeric simian/human immunodeficiency virus in rhesus monkeys. J. Virol. 1996, 70, 3198–3206. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, G.B.; Halloran, M.; Li, J.; Park, I.W.; Gomila, R.; Reimann, K.A.; Axthelm, M.K.; Iliff, S.A.; Letvin, N.L.; Sodroski, J. Characterization of molecularly cloned simian-human immunodeficiency viruses causing rapid CD4+ lymphocyte depletion in rhesus monkeys. J. Virol. 1997, 71, 4218–4225. [Google Scholar] [CrossRef]
- Nishimura, Y.; Igarashi, T.; Donau, O.K.; Buckler-White, A.; Buckler, C.; Lafont, B.A.; Goeken, R.M.; Goldstein, S.; Hirsch, V.M.; Martin, M.A. Highly pathogenic SHIVs and SIVs target different CD4+ T cell subsets in rhesus monkeys, explaining their divergent clinical courses. Proc. Natl. Acad. Sci. USA 2004, 101, 12324–12329. [Google Scholar] [CrossRef]
- Del Prete, G.Q.; Lifson, J.D.; Keele, B.F. Nonhuman primate models for the evaluation of HIV-1 preventive vaccine strategies: Model parameter considerations and consequences. Curr. Opin. HIV AIDS 2016, 11. [Google Scholar] [CrossRef]
- Sharma, A.; Boyd, D.F.; Overbaugh, J. Development of SHIVs with circulating, transmitted HIV-1 variants. J. Med. Primatol. 2015, 44, 296–300. [Google Scholar] [CrossRef]
- Ndung’u, T.; Lu, Y.; Renjifo, B.; Touzjian, N.; Kushner, N.; Pena-Cruz, V.; Novitsky, V.A.; Lee, T.H.; Essex, M. Infectious simian/human immunodeficiency virus with human immunodeficiency virus type 1 subtype C from an African isolate: Rhesus macaque model. J. Virol. 2001, 75, 11417–11425. [Google Scholar] [CrossRef]
- Pal, R.; Taylor, B.; Foulke, J.S.; Woodward, R.; Merges, M.; Praschunus, R.; Gibson, A.; Reitz, M. Characterization of a simian human immunodeficiency virus encoding the envelope gene from the CCR5-tropic HIV-1 Ba-L. J. Acquir. Immune Defic. Syndr. (1999) 2003, 33, 300–307. [Google Scholar] [CrossRef]
- Harouse, J.M.; Gettie, A.; Eshetu, T.; Tan, R.C.; Bohm, R.; Blanchard, J.; Baskin, G.; Cheng-Mayer, C. Mucosal transmission and induction of simian AIDS by CCR5-specific simian/human immunodeficiency virus SHIV(SF162P3). J. Virol. 2001, 75, 1990–1995. [Google Scholar] [CrossRef]
- Chen, Z.; Huang, Y.; Zhao, X.; Skulsky, E.; Lin, D.; Ip, J.; Gettie, A.; Ho, D.D. Enhanced infectivity of an R5-tropic simian/human immunodeficiency virus carrying human immunodeficiency virus type 1 subtype C envelope after serial passages in pig-tailed macaques (Macaca nemestrina). J. Virol. 2000, 74, 6501–6510. [Google Scholar] [CrossRef]
- Nishimura, Y.; Shingai, M.; Willey, R.; Sadjadpour, R.; Lee, W.R.; Brown, C.R.; Brenchley, J.M.; Buckler-White, A.; Petros, R.; Eckhaus, M.; et al. Generation of the pathogenic R5-tropic simian/human immunodeficiency virus SHIVAD8 by serial passaging in rhesus macaques. J. Virol. 2010, 84, 4769–4781. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.J.; Gupte, S.; Pastores, K.C.; Trott, S.; Abbink, P.; Mercado, N.B.; Li, Z.; Liu, P.T.; Borducchi, E.N.; Chandrashekar, A.; et al. Differential Outcomes following Optimization of Simian-Human Immunodeficiency Viruses from Clades AE, B, and C. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.M.; Bar, K.J. Advances in simian--human immunodeficiency viruses for nonhuman primate studies of HIV prevention and cure. Curr. Opin. HIV AIDS 2020, 15, 275–281. [Google Scholar] [CrossRef]
- Gautam, R.; Nishimura, Y.; Lee, W.R.; Donau, O.; Buckler-White, A.; Shingai, M.; Sadjadpour, R.; Schmidt, S.D.; LaBranche, C.C.; Keele, B.F.; et al. Pathogenicity and mucosal transmissibility of the R5-tropic simian/human immunodeficiency virus SHIV(AD8) in rhesus macaques: Implications for use in vaccine studies. J. Virol. 2012, 86, 8516–8526. [Google Scholar] [CrossRef] [PubMed]
- Shingai, M.; Donau, O.K.; Schmidt, S.D.; Gautam, R.; Plishka, R.J.; Buckler-White, A.; Sadjadpour, R.; Lee, W.R.; LaBranche, C.C.; Montefiori, D.C.; et al. Most rhesus macaques infected with the CCR5-tropic SHIV(AD8) generate cross-reactive antibodies that neutralize multiple HIV-1 strains. Proc. Natl. Acad. Sci. USA 2012, 109, 19769–19774. [Google Scholar] [CrossRef]
- Nishimura, Y.; Francis, J.N.; Donau, O.K.; Jesteadt, E.; Sadjadpour, R.; Smith, A.R.; Seaman, M.S.; Welch, B.D.; Martin, M.A.; Kay, M.S. Prevention and treatment of SHIVAD8 infection in rhesus macaques by a potent d-peptide HIV entry inhibitor. Proc. Natl. Acad. Sci. USA 2020, 117, 22436–22442. [Google Scholar] [CrossRef]
- Song, R.J.; Chenine, A.L.; Rasmussen, R.A.; Ruprecht, C.R.; Mirshahidi, S.; Grisson, R.D.; Xu, W.; Whitney, J.B.; Goins, L.M.; Ong, H.; et al. Molecularly cloned SHIV-1157ipd3N4: A highly replication- competent, mucosally transmissible R5 simian-human immunodeficiency virus encoding HIV clade C Env. J. Virol. 2006, 80, 8729–8738. [Google Scholar] [CrossRef]
- Keele, B.F.; Derdeyn, C.A. Genetic and antigenic features of the transmitted virus. Curr. Opin. HIV AIDS 2009, 4, 352–357. [Google Scholar] [CrossRef]
- Del Prete, G.Q.; Ailers, B.; Moldt, B.; Keele, B.F.; Estes, J.D.; Rodriguez, A.; Sampias, M.; Oswald, K.; Fast, R.; Trubey, C.M.; et al. Selection of unadapted, pathogenic SHIVs encoding newly transmitted HIV-1 envelope proteins. Cell Host Microbe 2014, 16, 412–418. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; Kong, R.; Ding, W.; Lee, F.H.; Parker, Z.; Kim, E.; Learn, G.H.; Hahn, P.; Policicchio, B.; et al. Envelope residue 375 substitutions in simian-human immunodeficiency viruses enhance CD4 binding and replication in rhesus macaques. Proc. Natl. Acad. Sci. USA 2016, 113, E3413–E3422. [Google Scholar] [CrossRef]
- O’Brien, S.P.; Swanstrom, A.E.; Pegu, A.; Ko, S.-Y.; Immonen, T.T.; Del Prete, G.Q.; Fennessey, C.M.; Gorman, J.; Foulds, K.E.; Schmidt, S.D.; et al. Rational design and in vivo selection of SHIVs encoding transmitted/founder subtype C HIV-1 envelopes. PLoS Pathog. 2019, 15, e1007632. [Google Scholar] [CrossRef]
- Bauer, A.M.; Ziani, W.; Lindemuth, E.; Kuri-Cervantes, L.; Li, H.; Lee, F.H.; Watkins, M.; Ding, W.; Xu, H.; Veazey, R.; et al. Novel Transmitted/Founder Simian-Human Immunodeficiency Viruses for Human Immunodeficiency Virus Latency and Cure Research. J. Virol. 2020, 94, e01659-19. [Google Scholar] [CrossRef]
- Bar, K.J.; Coronado, E.; Hensley-McBain, T.; O’Connor, M.A.; Osborn, J.M.; Miller, C.; Gott, T.M.; Wangari, S.; Iwayama, N.; Ahrens, C.Y.; et al. Simian-Human Immunodeficiency Virus SHIV.CH505 Infection of Rhesus Macaques Results in Persistent Viral Replication and Induces Intestinal Immunopathology. J. Virol. 2019, 93, e00372-19. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.J.; Li, Q.; Abel, K.; Kim, E.Y.; Ma, Z.M.; Wietgrefe, S.; La Franco-Scheuch, L.; Compton, L.; Duan, L.; Shore, M.D.; et al. Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. J. Virol. 2005, 79, 9217–9227. [Google Scholar] [CrossRef] [PubMed]
- Sagar, M. HIV-1 transmission biology: Selection and characteristics of infecting viruses. J. Infect. Dis. 2010, 202 (Suppl. 2), S289–S296. [Google Scholar] [CrossRef]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef]
- Haaland, R.E.; Sullivan, S.T.; Evans-Strickfaden, T.; Lennox, J.L.; Hart, C.E. Female genital tract shedding of CXCR4-tropic HIV Type 1 is associated with a majority population of CXCR4-tropic HIV Type 1 in blood and declining CD4(+) cell counts. AIDS Res. Hum. Retrovir. 2012, 28, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Gianella, S.; Mehta, S.R.; Young, J.A.; Vargas, M.V.; Little, S.J.; Richman, D.D.; Kosakovsky Pond, S.L.; Smith, D.M. Sexual transmission of predicted CXCR4-tropic HIV-1 likely originating from the source partner’s seminal cells. Virology 2012, 434, 2–4. [Google Scholar] [CrossRef][Green Version]
- Muciaccia, B.; Padula, F.; Gandini, L.; Lenzi, A.; Stefanini, M. HIV-1 chemokine co-receptor CCR5 is expressed on the surface of human spermatozoa. AIDS (Lond. Engl.) 2005, 19, 1424–1426. [Google Scholar] [CrossRef]
- Chohan, B.; Lang, D.; Sagar, M.; Korber, B.; Lavreys, L.; Richardson, B.; Overbaugh, J. Selection for human immunodeficiency virus type 1 envelope glycosylation variants with shorter V1-V2 loop sequences occurs during transmission of certain genetic subtypes and may impact viral RNA levels. J. Virol. 2005, 79, 6528–6531. [Google Scholar] [CrossRef] [PubMed]
- Gnanakaran, S.; Bhattacharya, T.; Daniels, M.; Keele, B.F.; Hraber, P.T.; Lapedes, A.S.; Shen, T.; Gaschen, B.; Krishnamoorthy, M.; Li, H.; et al. Recurrent signature patterns in HIV-1 B clade envelope glycoproteins associated with either early or chronic infections. PLoS Pathog. 2011, 7, e1002209. [Google Scholar] [CrossRef]
- Parrish, N.F.; Gao, F.; Li, H.; Giorgi, E.E.; Barbian, H.J.; Parrish, E.H.; Zajic, L.; Iyer, S.S.; Decker, J.M.; Kumar, A.; et al. Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. USA 2013, 110, 6626–6633. [Google Scholar] [CrossRef] [PubMed]
- Fenton-May, A.E.; Dibben, O.; Emmerich, T.; Ding, H.; Pfafferott, K.; Aasa-Chapman, M.M.; Pellegrino, P.; Williams, I.; Cohen, M.S.; Gao, F.; et al. Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology 2013, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Amerongen, H.M.; Weltzin, R.; Farnet, C.M.; Michetti, P.; Haseltine, W.A.; Neutra, M.R. Transepithelial transport of HIV-1 by intestinal M cells: A mechanism for transmission of AIDS. J. Acquir. Immune Defic. Syndr. 1991, 4, 760–765. [Google Scholar] [PubMed]
- Fotopoulos, G.; Harari, A.; Michetti, P.; Trono, D.; Pantaleo, G.; Kraehenbuhl, J.-P. Transepithelial transport of HIV-1 by M cells is receptor-mediated. Proc. Natl. Acad. Sci. USA 2002, 99, 9410–9414. [Google Scholar] [CrossRef]
- Spira, A.I.; Marx, P.A.; Patterson, B.K.; Mahoney, J.; Koup, R.A.; Wolinsky, S.M.; Ho, D.D. Cellular targets of infection and route of viral dissemination after an intravaginal inoculation of simian immunodeficiency virus into rhesus macaques. J. Exp. Med. 1996, 183, 215–225. [Google Scholar] [CrossRef]
- Hu, J.; Gardner, M.B.; Miller, C.J. Simian immunodeficiency virus rapidly penetrates the cervicovaginal mucosa after intravaginal inoculation and infects intraepithelial dendritic cells. J. Virol. 2000, 74, 6087–6095. [Google Scholar] [CrossRef]
- Kreiss, J.K.; Hopkins, S.G. The association between circumcision status and human immunodeficiency virus infection among homosexual men. J. Infect. Dis. 1993, 168, 1404–1408. [Google Scholar] [CrossRef] [PubMed]
- Veazey, R.S.; Tham, I.C.; Mansfield, K.G.; DeMaria, M.; Forand, A.E.; Shvetz, D.E.; Chalifoux, L.V.; Sehgal, P.K.; Lackner, A.A. Identifying the target cell in primary simian immunodeficiency virus (SIV) infection: Highly activated memory CD4(+) T cells are rapidly eliminated in early SIV infection in vivo. J. Virol. 2000, 74, 57–64. [Google Scholar] [CrossRef]
- Mattapallil, J.J.; Douek, D.C.; Hill, B.; Nishimura, Y.; Martin, M.; Roederer, M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 2005, 434, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Dos Santos, P.; Rancez, M.; Prétet, J.-L.; Michel-Salzat, A.; Messent, V.; Bogdanova, A.; Couëdel-Courteille, A.; Souil, E.; Cheynier, R.; Butor, C. Rapid dissemination of SIV follows multisite entry after rectal inoculation. PLoS ONE 2011, 6, e19493. [Google Scholar] [CrossRef]
- Miyake, A.; Ibuki, K.; Enose, Y.; Suzuki, H.; Horiuchi, R.; Motohara, M.; Saito, N.; Nakasone, T.; Honda, M.; Watanabe, T.; et al. Rapid dissemination of a pathogenic simian/human immunodeficiency virus to systemic organs and active replication in lymphoid tissues following intrarectal infection. J. Gen. Virol. 2006, 87, 1311–1320. [Google Scholar] [CrossRef]
- Haase, A.T. Early events in sexual transmission of HIV and SIV and opportunities for interventions. Annu. Rev. Med. 2011, 62, 127–139. [Google Scholar] [CrossRef]
- Li, Q.; Duan, L.; Estes, J.D.; Ma, Z.M.; Rourke, T.; Wang, Y.; Reilly, C.; Carlis, J.; Miller, C.J.; Haase, A.T. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 2005, 434, 1148–1152. [Google Scholar] [CrossRef]
- Huang, X.; Liu, X.; Meyers, K.; Liu, L.; Su, B.; Wang, P.; Li, Z.; Li, L.; Zhang, T.; Li, N.; et al. Cytokine cascade and networks among MSM HIV seroconverters: Implications for early immunotherapy. Sci. Rep. 2016, 6, 36234. [Google Scholar] [CrossRef] [PubMed]
- Muema, D.M.; Akilimali, N.A.; Ndumnego, O.C.; Rasehlo, S.S.; Durgiah, R.; Ojwach, D.B.A.; Ismail, N.; Dong, M.; Moodley, A.; Dong, K.L.; et al. Association between the cytokine storm, immune cell dynamics, and viral replicative capacity in hyperacute HIV infection. BMC Med. 2020, 18, 81. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.; Passmore, J.-A.S.; Williamson, C.; Little, F.; Bebell, L.M.; Mlisana, K.; Burgers, W.A.; van Loggerenberg, F.; Walzl, G.; Djoba Siawaya, J.F.; et al. Plasma cytokine levels during acute HIV-1 infection predict HIV disease progression. AIDS (Lond. Engl.) 2010, 24, 819–831. [Google Scholar] [CrossRef]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef]
- Sabado, R.L.; Babcock, E.; Kavanagh, D.G.; Tjomsland, V.; Walker, B.D.; Lifson, J.D.; Bhardwaj, N.; Larsson, M. Pathways utilized by dendritic cells for binding, uptake, processing and presentation of antigens derived from HIV-1. Eur. J. Immunol. 2007, 37, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, C.; Della Chiesa, M.; Kohler, S.; Moewes, B.; Radbruch, A.; Moretta, L.; Moretta, A.; Thiel, A. Activation of human NK cells by plasmacytoid dendritic cells and its modulation by CD4+ T helper cells and CD4+ CD25hi T regulatory cells. Eur. J. Immunol. 2005, 35, 2452–2458. [Google Scholar] [CrossRef]
- Florez-Alvarez, L.; Hernandez, J.C.; Zapata, W. NK Cells in HIV-1 Infection: From Basic Science to Vaccine Strategies. Front. Immunol 2018, 9, 2290. [Google Scholar] [CrossRef] [PubMed]
- Pardo, J.; Balkow, S.; Anel, A.; Simon, M.M. Granzymes are essential for natural killer cell-mediated and perf-facilitated tumor control. Eur. J. Immunol. 2002, 32, 2881–2886. [Google Scholar] [CrossRef]
- Scott-Algara, D.; Truong, L.X.; Versmisse, P.; David, A.; Luong, T.T.; Nguyen, N.V.; Theodorou, I.; Barré-Sinoussi, F.; Pancino, G. Cutting Edge: Increased NK Cell Activity in HIV-1-Exposed but Uninfected Vietnamese Intravascular Drug Users. J. Immunol. 2003, 171, 5663–5667. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.; Kinter, A.L.; Vaccarezza, M.; Rubbert, A.; Catanzaro, A.; Moir, S.; Monaco, J.; Ehler, L.; Mizell, S.; Jackson, R.; et al. Natural killer cells from human immunodeficiency virus (HIV)-infected individuals are an important source of CC-chemokines and suppress HIV-1 entry and replication in vitro. J. Clin. Investig. 1998, 102, 223–231. [Google Scholar] [CrossRef]
- Demers, K.R.; Reuter, M.A.; Betts, M.R. CD8(+) T-cell effector function and transcriptional regulation during HIV pathogenesis. Immunol Rev. 2013, 254, 190–206. [Google Scholar] [CrossRef]
- Ndhlovu, Z.M.; Kamya, P.; Mewalal, N.; Kløverpris, H.N.; Nkosi, T.; Pretorius, K.; Laher, F.; Ogunshola, F.; Chopera, D.; Shekhar, K.; et al. Magnitude and Kinetics of CD8+ T Cell Activation during Hyperacute HIV Infection Impact Viral Set Point. Immunity 2015, 43, 591–604. [Google Scholar] [CrossRef]
- Bernardin, F.; Kong, D.; Peddada, L.; Baxter-Lowe, L.A.; Delwart, E. Human immunodeficiency virus mutations during the first month of infection are preferentially found in known cytotoxic T-lymphocyte epitopes. J. Virol. 2005, 79, 11523–11528. [Google Scholar] [CrossRef][Green Version]
- Goonetilleke, N.; Liu, M.K.P.; Salazar-Gonzalez, J.F.; Ferrari, G.; Giorgi, E.; Ganusov, V.V.; Keele, B.F.; Learn, G.H.; Turnbull, E.L.; Salazar, M.G.; et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 2009, 206, 1253–1272. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, G.D.; Yates, N.L.; Liu, P.; Qin, L.; Fouda, G.G.; Chavez, L.L.; Decamp, A.C.; Parks, R.J.; Ashley, V.C.; Lucas, J.T.; et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: Virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J. Virol. 2008, 82, 12449–12463. [Google Scholar] [CrossRef]
- Levesque, M.C.; Moody, M.A.; Hwang, K.K.; Marshall, D.J.; Whitesides, J.F.; Amos, J.D.; Gurley, T.C.; Allgood, S.; Haynes, B.B.; Vandergrift, N.A.; et al. Polyclonal B cell differentiation and loss of gastrointestinal tract germinal centers in the earliest stages of HIV-1 infection. PLoS Med. 2009, 6, e1000107. [Google Scholar] [CrossRef]
- Le Hingrat, Q.; Sereti, I.; Landay, A.L.; Pandrea, I.; Apetrei, C. The Hitchhiker Guide to CD4(+) T-Cell Depletion in Lentiviral Infection. A Critical Review of the Dynamics of the CD4(+) T Cells in SIV and HIV Infection. Front. Immunol 2021, 12, 695674. [Google Scholar] [CrossRef]
- Lavreys, L.; Baeten, J.M.; Chohan, V.; McClelland, R.S.; Hassan, W.M.; Richardson, B.A.; Mandaliya, K.; Achola, J.O.N.; Overbaugh, J. Higher Set Point Plasma Viral Load and More-Severe Acute HIV Type 1 (HIV-1) Illness Predict Mortality among High-Risk HIV-1–Infected African Women. Clin. Infect. Dis. 2006, 42, 1333–1339. [Google Scholar] [CrossRef]
- Mellors, J.W.; Rinaldo, C.R.; Gupta, P.; White, R.M.; Todd, J.A.; Kingsley, L.A. Prognosis in HIV-1 Infection Predicted by the Quantity of Virus in Plasma. Science 1996, 272, 1167–1170. [Google Scholar] [CrossRef]
- Deeks, S.G.; Kitchen, C.M.R.; Liu, L.; Guo, H.; Gascon, R.; Narváez, A.B.; Hunt, P.; Martin, J.N.; Kahn, J.O.; Levy, J.; et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 2004, 104, 942–947. [Google Scholar] [CrossRef]
- Zaunders, J.J.; Cunningham, P.H.; Kelleher, A.D.; Kaufmann, G.R.; Jaramillo, A.B.; Wright, R.; Smith, D.; Grey, P.; Vizzard, J.; Carr, A.; et al. Potent antiretroviral therapy of primary human immunodeficiency virus type 1 (HIV-1) infection: Partial normalization of T lymphocyte subsets and limited reduction of HIV-1 DNA despite clearance of plasma viremia. J. Infect. Dis. 1999, 180, 320–329. [Google Scholar] [CrossRef][Green Version]
- Tilling, R.; Kinloch, S.; Goh, L.E.; Cooper, D.; Perrin, L.; Lampe, F.; Zaunders, J.; Hoen, B.; Tsoukas, C.; Andersson, J.; et al. Parallel decline of CD8+/CD38++ T cells and viraemia in response to quadruple highly active antiretroviral therapy in primary HIV infection. AIDS (Lond. Engl.) 2002, 16, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Mohri, H.; Perelson, A.S.; Tung, K.; Ribeiro, R.M.; Ramratnam, B.; Markowitz, M.; Kost, R.; Hurley, A.; Weinberger, L.; Cesar, D.; et al. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J. Exp. Med. 2001, 194, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Saravolatz, L.D.; Winslow, D.L.; Collins, G.; Hodges, J.S.; Pettinelli, C.; Stein, D.S.; Markowitz, N.; Reves, R.; Loveless, M.O.; Crane, L.; et al. Zidovudine alone or in combination with didanosine or zalcitabine in HIV-infected patients with the acquired immunodeficiency syndrome or fewer than 200 CD4 cells per cubic millimeter. Investigators for the Terry Beirn Community Programs for Clinical Research on AIDS. N. Engl. J. Med. 1996, 335, 1099–1106. [Google Scholar] [CrossRef]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef] [PubMed]
- DHHS. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Available online: https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/AdultandAdolescentGL.pdf (accessed on 9 September 2021).
- Eisinger, R.W.; Dieffenbach, C.W.; Fauci, A.S. HIV Viral Load and Transmissibility of HIV Infection: Undetectable Equals Untransmittable. JAMA 2019, 321, 451–452. [Google Scholar] [CrossRef]
- Guadalupe, M.; Reay, E.; Sankaran, S.; Prindiville, T.; Flamm, J.; McNeil, A.; Dandekar, S. Severe CD4+ T-Cell Depletion in Gut Lymphoid Tissue during Primary Human Immunodeficiency Virus Type 1 Infection and Substantial Delay in Restoration following Highly Active Antiretroviral Therapy. J. Virol. 2003, 77, 11708–11717. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, D.; Sankaran, S.; Silvey, M.; Dandekar, S. Antiviral Therapy during Primary Simian Immunodeficiency Virus Infection Fails To Prevent Acute Loss of CD4+ T Cells in Gut Mucosa but Enhances Their Rapid Restoration through Central Memory T Cells. J. Virol. 2008, 82, 4016–4027. [Google Scholar] [CrossRef] [PubMed]
- Macal, M.; Sankaran, S.; Chun, T.W.; Reay, E.; Flamm, J.; Prindiville, T.J.; Dandekar, S. Effective CD4+ T-cell restoration in gut-associated lymphoid tissue of HIV-infected patients is associated with enhanced Th17 cells and polyfunctional HIV-specific T-cell responses. Mucosal Immunol. 2008, 1, 475–488. [Google Scholar] [CrossRef]
- Deleage, C.; Schuetz, A.; Alvord, W.G.; Johnston, L.; Hao, X.-P.; Morcock, D.R.; Rerknimitr, R.; Fletcher, J.L.K.; Puttamaswin, S.; Phanuphak, N.; et al. Impact of early cART in the gut during acute HIV infection. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Kelly, C.; Gaskell, K.M.; Richardson, M.; Klein, N.; Garner, P.; MacPherson, P. Discordant Immune Response with Antiretroviral Therapy in HIV-1: A Systematic Review of Clinical Outcomes. PLoS ONE 2016, 11, e0156099. [Google Scholar] [CrossRef]
- Schuetz, A.; Deleage, C.; Sereti, I.; Rerknimitr, R.; Phanuphak, N.; Phuang-Ngern, Y.; Estes, J.D.; Sandler, N.G.; Sukhumvittaya, S.; Marovich, M.; et al. Initiation of ART during Early Acute HIV Infection Preserves Mucosal Th17 Function and Reverses HIV-Related Immune Activation. PLoS Pathog. 2014, 10, e1004543. [Google Scholar] [CrossRef]
- de Paula, H.H.S.; Ferreira, A.C.G.; Caetano, D.G.; Delatorre, E.; Teixeira, S.L.M.; Coelho, L.E.; João, E.G.; de Andrade, M.M.; Cardoso, S.W.; Grinsztejn, B.; et al. Reduction of inflammation and T cell activation after 6 months of cART initiation during acute, but not in early chronic HIV-1 infection. Retrovirology 2018, 15, 76. [Google Scholar] [CrossRef]
- Ghislain, M.; Bastard, J.-P.; Meyer, L.; Capeau, J.; Fellahi, S.; Gérard, L.; May, T.; Simon, A.; Vigouroux, C.; Goujard, C.; et al. Late Antiretroviral Therapy (ART) Initiation Is Associated with Long-Term Persistence of Systemic Inflammation and Metabolic Abnormalities. PLoS ONE 2015, 10, e0144317. [Google Scholar] [CrossRef] [PubMed]
- Marcus, J.L.; Leyden, W.A.; Alexeeff, S.E.; Anderson, A.N.; Hechter, R.C.; Hu, H.; Lam, J.O.; Towner, W.J.; Yuan, Q.; Horberg, M.A.; et al. Comparison of Overall and Comorbidity-Free Life Expectancy Between Insured Adults With and Without HIV Infection, 2000–2016. JAMA Netw. Open 2020, 3, e207954. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, V.M.; Dapolito, G.; Johnson, P.R.; Elkins, W.R.; London, W.T.; Montali, R.J.; Goldstein, S.; Brown, C. Induction of AIDS by simian immunodeficiency virus from an African green monkey: Species-specific variation in pathogenicity correlates with the extent of in vivo replication. J. Virol. 1995, 69, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Telfer, P.; Reed, P.; Zhang, L.; Getti, A.; Ho, D.D.; Marx, P.A. Isolation and characterization of the first simian immunodeficiency virus from a feral sooty mangabey (Cercocebus atys) in West Africa. J. Med. Primatol. 1995, 24, 108–115. [Google Scholar] [CrossRef]
- Smith, S.M.; Makuwa, M.; Lee, F.; Gettie, A.; Russo, C.; Marx, P.A. SIVrcm infection of macaques. J. Med. Primatol. 1998, 27, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Osterhaus, A.D.; Pedersen, N.; van Amerongen, G.; Frankenhuis, M.T.; Marthas, M.; Reay, E.; Rose, T.M.; Pamungkas, J.; Bosch, M.L. Isolation and partial characterization of a lentivirus from talapoin monkeys (Myopithecus talapoin). Virology 1999, 260, 116–124. [Google Scholar] [CrossRef][Green Version]
- Takehisa, J.; Harada, Y.; Ndembi, N.; Mboudjeka, I.; Taniguchi, Y.; Ngansop, C.; Kuate, S.; Zekeng, L.; Ibuki, K.; Shimada, T.; et al. Natural infection of wild-born mandrills (Mandrillus sphinx) with two different types of simian immunodeficiency virus. AIDS Res. Hum. Retrovir. 2001, 17, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.; Fedanov, A.; Germon, S.; Kozyr, N.; Kaiser, W.J.; Garber, D.A.; McClure, H.; Feinberg, M.B.; Staprans, S.I. Divergent host responses during primary simian immunodeficiency virus SIVsm infection of natural sooty mangabey and nonnatural rhesus macaque hosts. J. Virol. 2005, 79, 4043–4054. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 integration landscape during latent and active infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef]
- Ferris, A.L.; Wells, D.W.; Guo, S.; Del Prete, G.Q.; Swanstrom, A.E.; Coffin, J.M.; Wu, X.; Lifson, J.D.; Hughes, S.H. Clonal expansion of SIV-infected cells in macaques on antiretroviral therapy is similar to that of HIV-infected cells in humans. PLoS Pathog. 2019, 15, e1007869. [Google Scholar] [CrossRef]
- Sellier, P.; Mannioui, A.; Bourry, O.; Dereuddre-Bosquet, N.; Delache, B.; Brochard, P.; Calvo, J.; Prévot, S.; Roques, P. Antiretroviral Treatment Start-Time during Primary SIVmac Infection in Macaques Exerts a Different Impact on Early Viral Replication and Dissemination. PLoS ONE 2010, 5, e10570. [Google Scholar] [CrossRef] [PubMed]
- Mannioui, A.; Bourry, O.; Sellier, P.; Delache, B.; Brochard, P.; Andrieu, T.; Vaslin, B.; Karlsson, I.; Roques, P.; Le Grand, R. Dynamics of viral replication in blood and lymphoid tissues during SIVmac251 infection of macaques. Retrovirology 2009, 6, 106. [Google Scholar] [CrossRef]
- Bourry, O.; Mannioui, A.; Sellier, P.; Roucairol, C.; Durand-Gasselin, L.; Dereuddre-Bosquet, N.; Benech, H.; Roques, P.; Le Grand, R. Effect of a short-term HAART on SIV load in macaque tissues is dependent on time of initiation and antiviral diffusion. Retrovirology 2010, 7, 78. [Google Scholar] [CrossRef]
- Canary, L.A.; Vinton, C.L.; Morcock, D.R.; Pierce, J.B.; Estes, J.D.; Brenchley, J.M.; Klatt, N.R. Rate of AIDS Progression Is Associated with Gastrointestinal Dysfunction in Simian Immunodeficiency Virus–Infected Pigtail Macaques. J. Immunol. 2013, 190, 2959–2965. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.P.; Lucero, C.M.; Turkbey, B.; Bernardo, M.L.; Morcock, D.R.; Deleage, C.; Trubey, C.M.; Smedley, J.; Klatt, N.R.; Giavedoni, L.D.; et al. Experimental colitis in SIV-uninfected rhesus macaques recapitulates important features of pathogenic SIV infection. Nat. Commun. 2015, 6, 8020. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Xu, C.; Stock, J.L.; Frank, D.N.; Ma, D.; Policicchio, B.B.; He, T.; Kristoff, J.; Cornell, E.; Haret-Richter, G.S.; et al. Antibiotic and Antiinflammatory Therapy Transiently Reduces Inflammation and Hypercoagulation in Acutely SIV-Infected Pigtailed Macaques. PLoS Pathog. 2016, 12, e1005384. [Google Scholar] [CrossRef]
- Schechter, M.E.; Andrade, B.B.; He, T.; Richter, G.H.; Tosh, K.W.; Policicchio, B.B.; Singh, A.; Raehtz, K.D.; Sheikh, V.; Ma, D.; et al. Inflammatory monocytes expressing tissue factor drive SIV and HIV coagulopathy. Sci. Transl. Med. 2017, 9, eaam5441. [Google Scholar] [CrossRef]
- Zink, M.C.; Suryanarayana, K.; Mankowski, J.L.; Shen, A.; Piatak, M., Jr.; Spelman, J.P.; Carter, D.L.; Adams, R.J.; Lifson, J.D.; Clements, J.E. High viral load in the cerebrospinal fluid and brain correlates with severity of simian immunodeficiency virus encephalitis. J. Virol. 1999, 73, 10480–10488. [Google Scholar] [CrossRef]
- Matsuda, K.; Riddick, N.E.; Lee, C.A.; Puryear, S.B.; Wu, F.; Lafont, B.A.P.; Whitted, S.; Hirsch, V.M. A SIV molecular clone that targets the CNS and induces neuroAIDS in rhesus macaques. PLoS Pathog. 2017, 13, e1006538. [Google Scholar] [CrossRef]
- Gama, L.; Abreu, C.M.; Shirk, E.N.; Price, S.L.; Li, M.; Laird, G.M.; Pate, K.A.M.; Wietgrefe, S.W.; O’Connor, S.L.; Pianowski, L.; et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS (Lond. Engl.) 2017, 31, 5–14. [Google Scholar] [CrossRef]
- Giorgi, J.V.; Hultin, L.E.; McKeating, J.A.; Johnson, T.D.; Owens, B.; Jacobson, L.P.; Shih, R.; Lewis, J.; Wiley, D.J.; Phair, J.P.; et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J. Infect. Dis. 1999, 179, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Hazenberg, M.D.; Otto, S.A.; van Benthem, B.H.; Roos, M.T.; Coutinho, R.A.; Lange, J.M.; Hamann, D.; Prins, M.; Miedema, F. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS (Lond. Engl.) 2003, 17, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, M.F.; Petitjean, G.; Dunyach-Rémy, C.; Didier, C.; Girard, P.M.; Manea, M.E.; Campa, P.; Meyer, L.; Rouzioux, C.; Lavigne, J.P.; et al. The Th17/Treg ratio, IL-1RA and sCD14 levels in primary HIV infection predict the T-cell activation set point in the absence of systemic microbial translocation. PLoS Pathog. 2013, 9, e1003453. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Paiardini, M.; Knox, K.S.; Asher, A.I.; Cervasi, B.; Asher, T.E.; Scheinberg, P.; Price, D.A.; Hage, C.A.; Kholi, L.M.; et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 2008, 112, 2826–2835. [Google Scholar] [CrossRef]
- Favre, D.; Lederer, S.; Kanwar, B.; Ma, Z.M.; Proll, S.; Kasakow, Z.; Mold, J.; Swainson, L.; Barbour, J.D.; Baskin, C.R.; et al. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 2009, 5, e1000295. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef]
- Estes, J.D.; Harris, L.D.; Klatt, N.R.; Tabb, B.; Pittaluga, S.; Paiardini, M.; Barclay, G.R.; Smedley, J.; Pung, R.; Oliveira, K.M.; et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 2010, 6, e1001052. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Douek, D.C. Microbial translocation across the GI tract. Annu. Rev. Immunol. 2012, 30, 149–173. [Google Scholar] [CrossRef]
- Kristoff, J.; Haret-Richter, G.; Ma, D.; Ribeiro, R.M.; Xu, C.; Cornell, E.; Stock, J.L.; He, T.; Mobley, A.D.; Ross, S.; et al. Early microbial translocation blockade reduces SIV-mediated inflammation and viral replication. J. Clin. Investig. 2014, 124, 2802–2806. [Google Scholar] [CrossRef]
- Kovacs, A.; Al-Harthi, L.; Christensen, S.; Mack, W.; Cohen, M.; Landay, A. CD8+ T Cell Activation in Women Coinfected with Human Immunodeficiency Virus Type 1 and Hepatitis C Virus. J. Infect. Dis. 2008, 197, 1402. [Google Scholar] [CrossRef]
- Bautista-Amorocho, H.; Castellanos-Domínguez, Y.Z.; Rodríguez-Villamizar, L.A.; Velandia-Cruz, S.A.; Becerra-Peña, J.A.; Farfán-García, A.E. Epidemiology, Risk Factors and Genotypes of HBV in HIV-Infected Patients in the Northeast Region of Colombia: High Prevalence of Occult Hepatitis B and F3 Subgenotype Dominance. PLoS ONE 2014, 9, e114272. [Google Scholar] [CrossRef]
- Sheth, P.M.; Sunderji, S.; Shin, L.Y.Y.; Rebbapragada, A.; Huibner, S.; Kimani, J.; MacDonald, K.S.; Ngugi, E.; Bwayo, J.J.; Moses, S.; et al. Coinfection with Herpes Simplex Virus Type 2 Is Associated with Reduced HIV-Specific T Cell Responses and Systemic Immune Activation. J. Infect. Dis. 2008, 197, 1394–1401. [Google Scholar] [CrossRef]
- Gianella, S.; Strain, M.C.; Rought, S.E.; Vargas, M.V.; Little, S.J.; Richman, D.D.; Spina, C.A.; Smith, D.M. Associations between virologic and immunologic dynamics in blood and in the male genital tract. J. Virol. 2012, 86, 1307–1315. [Google Scholar] [CrossRef]
- Freeman, M.L.; Mudd, J.C.; Shive, C.L.; Younes, S.A.; Panigrahi, S.; Sieg, S.F.; Lee, S.A.; Hunt, P.W.; Calabrese, L.H.; Gianella, S.; et al. CD8 T-cell expansion and inflammation linked to CMV coinfection in ART-treated HIV infection. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2016, 62, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Petrara, M.R.; Cattelan, A.M.; Zanchetta, M.; Sasset, L.; Freguja, R.; Gianesin, K.; Cecchetto, M.G.; Carmona, F.; De Rossi, A. Epstein-Barr Virus load and immune activation in Human Immunodeficiency Virus type 1-infected patients. J. Clin. Virol. 2012, 53, 195–200. [Google Scholar] [CrossRef]
- Boulougoura, A.; Sereti, I. HIV infection and immune activation: The role of coinfections. Curr Opin HIV AIDS 2016, 11, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Friis-Møller, N.; Reiss, P.; Sabin, C.A.; Weber, R.; Monforte, A.; El-Sadr, W.; Thiébaut, R.; De Wit, S.; Kirk, O.; Fontas, E.; et al. Class of antiretroviral drugs and the risk of myocardial infarction. N. Engl. J. Med. 2007, 356, 1723–1735. [Google Scholar] [CrossRef]
- Contento, R.L.; Molon, B.; Boularan, C.; Pozzan, T.; Manes, S.; Marullo, S.; Viola, A. CXCR4–CCR5: A couple modulating T cell functions. Proc. Natl. Acad. Sci.USA 2008, 105, 10101–10106. [Google Scholar] [CrossRef] [PubMed]
- Gornalusse, G.G.; Mummidi, S.; Gaitan, A.A.; Jimenez, F.; Ramsuran, V.; Picton, A.; Rogers, K.; Manoharan, M.S.; Avadhanam, N.; Murthy, K.K.; et al. Epigenetic mechanisms, T-cell activation, and CCR5 genetics interact to regulate T-cell expression of CCR5, the major HIV-1 coreceptor. Proc. Natl. Acad. Sci. USA 2015, 112, E4762–E4771. [Google Scholar] [CrossRef]
- Kawakami, K.; Scheidereit, C.; Roeder, R.G. Identification and purification of a human immunoglobulin-enhancer-binding protein (NF-kappa B) that activates transcription from a human immunodeficiency virus type 1 promoter in vitro. Proc. Natl. Acad. Sci. USA 1988, 85, 4700–4704. [Google Scholar] [CrossRef] [PubMed]
- Papagno, L.; Spina, C.A.; Marchant, A.; Salio, M.; Rufer, N.; Little, S.; Dong, T.; Chesney, G.; Waters, A.; Easterbrook, P.; et al. Immune Activation and CD8+ T-Cell Differentiation towards Senescence in HIV-1 Infection. PLOS Biol. 2004, 2, e20. [Google Scholar] [CrossRef] [PubMed]
- Cobos Jiménez, V.; Wit, F.W.; Joerink, M.; Maurer, I.; Harskamp, A.M.; Schouten, J.; Prins, M.; van Leeuwen, E.M.; Booiman, T.; Deeks, S.G.; et al. T-Cell Activation Independently Associates With Immune Senescence in HIV-Infected Recipients of Long-term Antiretroviral Treatment. J. Infect. Dis. 2016, 214, 216–225. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.-R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Zhang, Z.; Wang, X.; Fu, J.-L.; Yao, J.; Jiao, Y.; Chen, L.; Zhang, H.; Wei, J.; Jin, L.; et al. PD-1 up-regulation is correlated with HIV-specific memory CD8+ T-cell exhaustion in typical progressors but not in long-term nonprogressors. Blood 2007, 109, 4671–4678. [Google Scholar] [CrossRef] [PubMed]
- Evans, V.A.; van der Sluis, R.M.; Solomon, A.; Dantanarayana, A.; McNeil, C.; Garsia, R.; Palmer, S.; Fromentin, R.; Chomont, N.; Sékaly, R.-P.; et al. Programmed cell death-1 contributes to the establishment and maintenance of HIV-1 latency. AIDS (Lond. Engl.) 2018, 32, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- McGary, C.S.; Deleage, C.; Harper, J.; Micci, L.; Ribeiro, S.P.; Paganini, S.; Kuri-Cervantes, L.; Benne, C.; Ryan, E.S.; Balderas, R.; et al. CTLA-4+PD-1- Memory CD4+ T Cells Critically Contribute to Viral Persistence in Antiretroviral Therapy-Suppressed, SIV-Infected Rhesus Macaques. Immunity 2017, 47, 776–788.e775. [Google Scholar] [CrossRef] [PubMed]
- Schweneker, M.; Favre, D.; Martin, J.N.; Deeks, S.G.; McCune, J.M. HIV-Induced Changes in T Cell Signaling Pathways. J. Immunol. 2008, 180, 6490–6500. [Google Scholar] [CrossRef]
- Estes, J.D.; Wietgrefe, S.; Schacker, T.; Southern, P.; Beilman, G.; Reilly, C.; Milush, J.M.; Lifson, J.D.; Sodora, D.L.; Carlis, J.V.; et al. Simian immunodeficiency virus-induced lymphatic tissue fibrosis is mediated by transforming growth factor beta 1-positive regulatory T cells and begins in early infection. J. Infect. Dis. 2007, 195, 551–561. [Google Scholar] [CrossRef]
- Estes, J.D.; Haase, A.T.; Schacker, T.W. The role of collagen deposition in depleting CD4+ T cells and limiting reconstitution in HIV-1 and SIV infections through damage to the secondary lymphoid organ niche. Semin. Immunol. 2008, 20, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Smith, A.J.; Wietgrefe, S.W.; Southern, P.J.; Schacker, T.W.; Reilly, C.S.; Estes, J.D.; Burton, G.F.; Silvestri, G.; Lifson, J.D.; et al. Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J. Clin. Investig. 2011, 121, 998–1008. [Google Scholar] [CrossRef]
- Estes, J.D. Pathobiology of HIV/SIV-associated changes in secondary lymphoid tissues. Immunol Rev. 2013, 254, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Schottenfeld, D.; Beebe-Dimmer, J. Chronic inflammation: A common and important factor in the pathogenesis of neoplasia. CA: A Cancer J. Clin. 2006, 56, 69–83. [Google Scholar] [CrossRef]
- Dubrow, R.; Silverberg, M.J.; Park, L.S.; Crothers, K.; Justice, A.C. HIV infection, aging, and immune function: Implications for cancer risk and prevention. Curr. Opin. Oncol. 2012, 24, 506–516. [Google Scholar] [CrossRef]
- Triant, V.A.; Lee, H.; Hadigan, C.; Grinspoon, S.K. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J. Clin. Endocrinol. Metab. 2007, 92, 2506–2512. [Google Scholar] [CrossRef]
- Hsue, P.Y.; Deeks, S.G.; Hunt, P.W. Immunologic basis of cardiovascular disease in HIV-infected adults. J. Infect. Dis. 2012, 205 (Suppl. 3), S375–S382. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T.; Büller, H.R. Bidirectional relation between inflammation and coagulation. Circulation 2004, 109, 2698–2704. [Google Scholar] [CrossRef]
- He, T.; Falwell, E.; Brocca-Cofano, E.; Pandrea, I. Modeling aging in HIV infection in nonhuman primates to address an emerging challenge of the post-ART era. Curr. Opin. Virol 2017, 25, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, W.D.; Phillips, K.D.; Carson, J.A.; Brewer, R.B.; Durstine, J.L.; Hand, G.A. Counteracting muscle wasting in HIV-infected individuals. HIV Med. 2006, 7, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, M.; Serfaty, L.; Capeau, J. From nonalcoholic fatty liver to nonalcoholic steatohepatitis and cirrhosis in HIV-infected patients: Diagnosis and management. Curr. Opin. Infect. Dis. 2012, 25, 10–16. [Google Scholar] [CrossRef]
- Sanmarti, M.; Ibáñez, L.; Huertas, S.; Badenes, D.; Dalmau, D.; Slevin, M.; Krupinski, J.; Popa-Wagner, A.; Jaen, A. HIV-associated neurocognitive disorders. J. Mol. Psychiatry 2014, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Funderburg, N.T.; Brenchley, J.M. Microbial translocation, immune activation, and HIV disease. Trends Microbiol. 2013, 21, 6–13. [Google Scholar] [CrossRef]
- Kim, W.K.; Sun, Y.; Do, H.; Autissier, P.; Halpern, E.F.; Piatak, M., Jr.; Lifson, J.D.; Burdo, T.H.; McGrath, M.S.; Williams, K. Monocyte heterogeneity underlying phenotypic changes in monocytes according to SIV disease stage. J. Leukoc Biol 2010, 87, 557–567. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. The CD14+ CD16+ blood monocytes: Their role in infection and inflammation. J. Leukoc Biol 2007, 81, 584–592. [Google Scholar] [CrossRef]
- Dandekar, S. Pathogenesis of HIV in the gastrointestinal tract. Curr. HIV/AIDS Rep. 2007, 4, 10–15. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Price, D.A.; Douek, D.C. HIV disease: Fallout from a mucosal catastrophe? Nat. Immunol. 2006, 7, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Douek, D.C. HIV infection and the gastrointestinal immune system. Mucosal Immunol 2008, 1, 23–30. [Google Scholar] [CrossRef]
- Grossman, Z.; Meier-Schellersheim, M.; Paul, W.E.; Picker, L.J. Pathogenesis of HIV infection: What the virus spares is as important as what it destroys. Nat. Med. 2006, 12, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Douek, D.C.; Roederer, M.; Koup, R.A. Emerging Concepts in the Immunopathogenesis of AIDS. Annu. Rev. Med. 2008, 60, 471–484. [Google Scholar] [CrossRef]
- Veazey, R.S.; Mansfield, K.G.; Tham, I.C.; Carville, A.C.; Shvetz, D.E.; Forand, A.E.; Lackner, A.A. Dynamics of CCR5 expression by CD4(+) T cells in lymphoid tissues during simian immunodeficiency virus infection. J. Virol. 2000, 74, 11001–11007. [Google Scholar] [CrossRef]
- Veazey, R.S.; Marx, P.A.; Lackner, A.A. The mucosal immune system: Primary target for HIV infection and AIDS. Trends Immunol 2001, 22, 626–633. [Google Scholar] [CrossRef]
- Veazey, R.S.; Lackner, A.A. Getting to the guts of HIV pathogenesis. J. Exp. Med. 2004, 200, 697–700. [Google Scholar] [CrossRef]
- Picker, L.J.; Watkins, D.I. HIV pathogenesis: The first cut is the deepest. Nat. Immunol. 2005, 6, 430–432. [Google Scholar] [CrossRef]
- Steele, A.K.; Lee, E.J.; Manuzak, J.A.; Dillon, S.M.; Beckham, J.D.; McCarter, M.D.; Santiago, M.L.; Wilson, C.C. Microbial exposure alters HIV-1-induced mucosal CD4+ T cell death pathways Ex vivo. Retrovirology 2014, 11, 14. [Google Scholar] [CrossRef]
- Doitsh, G.; Galloway, N.L.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Paiardini, M.; Frank, I.; Pandrea, I.; Apetrei, C.; Silvestri, G. Mucosal immune dysfunction in AIDS pathogenesis. AIDS Rev. 2008, 10, 36–46. [Google Scholar] [PubMed]
- Sandler, N.G.; Douek, D.C. Microbial translocation in HIV infection: Causes, consequences and treatment opportunities. Nat. Reviews. Microbiol. 2012, 10, 655–666. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Sheikh, V.; Thompson, W.L.; Morcock, D.R.; Perez-Diez, A.; Yao, M.D.; Rupert, A.W.; Utay, N.S.; Roby, G.; Freeman, A.F.; et al. T-cell depletion in the colonic mucosa of patients with idiopathic CD4 lymphopenia. J. Infect. Dis. 2015. [Google Scholar] [CrossRef]
- Sodora, D.L.; Silvestri, G. Immune activation and AIDS pathogenesis. AIDS (Lond. Engl.) 2008, 22, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.V.; Gautam, R.; Ribeiro, R.M.; Brenchley, J.M.; Butler, I.F.; Pattison, M.; Rasmussen, T.; Marx, P.A.; Silvestri, G.; Lackner, A.A.; et al. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J. Immunol. 2007, 179, 3035–3046. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Gaufin, T.; Brenchley, J.M.; Gautam, R.; Monjure, C.; Gautam, A.; Coleman, C.; Lackner, A.A.; Ribeiro, R.M.; Douek, D.C.; et al. Cutting edge: Experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. J. Immunol. 2008, 181, 6687–6691. [Google Scholar] [CrossRef]
- Silvestri, G.; Paiardini, M.; Pandrea, I.; Lederman, M.M.; Sodora, D.L. Understanding the benign nature of SIV infection in natural hosts. J. Clin. Investig. 2007, 117, 3148–3154. [Google Scholar] [CrossRef]
- Apetrei, C.; Gormus, B.; Pandrea, I.; Metzger, M.; ten Haaft, P.; Martin, L.N.; Bohm, R.; Alvarez, X.; Koopman, G.; Murphey-Corb, M.; et al. Direct inoculation of simian immunodeficiency virus from sooty mangabeys in black mangabeys (Lophocebus aterrimus): First evidence of AIDS in a heterologous African species and different pathologic outcomes of experimental infection. J. Virol. 2004, 78, 11506–11518. [Google Scholar] [CrossRef]
- Ling, B.; Apetrei, C.; Pandrea, I.; Veazey, R.S.; Lackner, A.A.; Gormus, B.; Marx, P.A. Classic AIDS in a sooty mangabey after an 18-year natural infection. J. Virol. 2004, 78, 8902–8908. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pandrea, I.; Onanga, R.; Rouquet, P.; Bourry, O.; Ngari, P.; Wickings, E.J.; Roques, P.; Apetrei, C. Chronic SIV infection ultimately causes immunodeficiency in African non-human primates. AIDS (Lond. Engl.) 2001, 15, 2461–2462. [Google Scholar] [CrossRef]
- Pandrea, I.; Sodora, D.L.; Silvestri, G.; Apetrei, C. Into the wild: Simian immunodeficiency virus (SIV) infection in natural hosts. Trends Immunol. 2008, 29, 419–428. [Google Scholar] [CrossRef]
- Apetrei, C.; Gautam, R.; Sumpter, B.; Carter, A.C.; Gaufin, T.; Staprans, S.I.; Else, J.; Barnes, M.; Cao, R., Jr.; Garg, S.; et al. Virus subtype-specific features of natural simian immunodeficiency virus SIVsmm infection in sooty mangabeys. J. Virol. 2007, 81, 7913–7923. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Broussard, S.R.; Staprans, S.I.; White, R.; Whitehead, E.M.; Feinberg, M.B.; Allan, J.S. Simian immunodeficiency virus replicates to high levels in naturally infected African green monkeys without inducing immunologic or neurologic disease. J. Virol. 2001, 75, 2262–2275. [Google Scholar] [CrossRef]
- Gueye, A.; Diop, O.M.; Ploquin, M.J.; Kornfeld, C.; Faye, A.; Cumont, M.C.; Hurtrel, B.; Barré-Sinoussi, F.; Müller-Trutwin, M.C. Viral load in tissues during the early and chronic phase of non-pathogenic SIVagm infection. J. Med. Primatol. 2004, 33, 83–97. [Google Scholar] [CrossRef]
- Onanga, R.; Kornfeld, C.; Pandrea, I.; Estaquier, J.; Souquière, S.; Rouquet, P.; Mavoungou, V.P.; Bourry, O.; M’Boup, S.; Barré-Sinoussi, F.; et al. High levels of viral replication contrast with only transient changes in CD4(+) and CD8(+) cell numbers during the early phase of experimental infection with simian immunodeficiency virus SIVmnd-1 in Mandrillus sphinx. J. Virol. 2002, 76, 10256–10263. [Google Scholar] [CrossRef]
- Pandrea, I.; Silvestri, G.; Onanga, R.; Veazey, R.S.; Marx, P.A.; Hirsch, V.; Apetrei, C. Simian immunodeficiency viruses replication dynamics in African non-human primate hosts: Common patterns and species-specific differences. J. Med. Primatol. 2006, 35, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Souquière, S.; Onanga, R.; Makuwa, M.; Pandrea, I.; Ngari, P.; Rouquet, P.; Bourry, O.; Kazanji, M.; Apetrei, C.; Simon, F.; et al. Simian immunodeficiency virus types 1 and 2 (SIV mnd 1 and 2) have different pathogenic potentials in rhesus macaques upon experimental cross-species transmission. J. Gen. Virol. 2009, 90, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Apetrei, C.; Dufour, J.; Dillon, N.; Barbercheck, J.; Metzger, M.; Jacquelin, B.; Bohm, R.; Marx, P.A.; Barre-Sinoussi, F.; et al. Simian immunodeficiency virus SIVagm.sab infection of Caribbean African green monkeys: A new model for the study of SIV pathogenesis in natural hosts. J. Virol. 2006, 80, 4858–4867. [Google Scholar] [CrossRef][Green Version]
- Pandrea, I.; Apetrei, C.; Gordon, S.; Barbercheck, J.; Dufour, J.; Bohm, R.; Sumpter, B.; Roques, P.; Marx, P.A.; Hirsch, V.M.; et al. Paucity of CD4+CCR5+ T cells is a typical feature of natural SIV hosts. Blood 2006, 109, 1069–1076. [Google Scholar] [CrossRef]
- Pandrea, I.; Parrish, N.F.; Raehtz, K.; Gaufin, T.; Barbian, H.J.; Ma, D.; Kristoff, J.; Gautam, R.; Zhong, F.; Haret-Richter, G.S.; et al. Mucosal simian immunodeficiency virus transmission in African green monkeys: Susceptibility to infection is proportional to target cell availability at mucosal sites. J. Virol. 2012, 86, 4158–4168. [Google Scholar] [CrossRef]
- Dunham, R.; Pagliardini, P.; Gordon, S.; Sumpter, B.; Engram, J.; Moanna, A.; Paiardini, M.; Mandl, J.N.; Lawson, B.; Garg, S.; et al. The AIDS resistance of naturally SIV-infected sooty mangabeys is independent of cellular immunity to the virus. Blood 2006, 108, 209–217. [Google Scholar] [CrossRef]
- Schmitz, J.E.; Zahn, R.C.; Brown, C.R.; Rett, M.D.; Li, M.; Tang, H.; Pryputniewicz, S.; Byrum, R.A.; Kaur, A.; Montefiori, D.C.; et al. Inhibition of Adaptive Immune Responses Leads to a Fatal Clinical Outcome in SIV-Infected Pigtailed Macaques but Not Vervet African Green Monkeys. PLoS Pathog. 2009, 5, e1000691. [Google Scholar] [CrossRef]
- Zahn, R.C.; Rett, M.D.; Li, M.; Tang, H.; Korioth-Schmitz, B.; Balachandran, H.; White, R.; Pryputniewicz, S.; Letvin, N.L.; Kaur, A.; et al. Suppression of adaptive immune responses during primary SIV infection of sabaeus African green monkeys delays partial containment of viremia but does not induce disease. Blood 2010, 115, 3070–3078. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klatt, N.R.; Canary, L.A.; Vanderford, T.H.; Vinton, C.L.; Engram, J.C.; Dunham, R.M.; Cronise, H.E.; Swerczek, J.M.; Lafont, B.A.; Picker, L.J.; et al. Dynamics of simian immunodeficiency virus SIVmac239 infection in pigtail macaques. J. Virol. 2012, 86, 1203–1213. [Google Scholar] [CrossRef]
- Compton, A.A.; Emerman, M. Convergence and Divergence in the Evolution of the APOBEC3G-Vif Interaction Reveal Ancient Origins of Simian Immunodeficiency Viruses. PLoS Pathog. 2013, 9, e1003135. [Google Scholar] [CrossRef] [PubMed]
- Gifford, R.J.; Katzourakis, A.; Tristem, M.; Pybus, O.G.; Winters, M.; Shafer, R.W. A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 20362–20367. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Telfer, P.; Souquière, S.; Hunter, M.; Coleman Clint, A.; Metzger Michael, J.; Reed, P.; Makuwa, M.; Hearn, G.; Honarvar, S.; et al. Island Biogeography Reveals the Deep History of SIV. Science 2010, 329, 1487. [Google Scholar] [CrossRef] [PubMed]
- Apetrei, C.; Gaufin, T.; Gautam, R.; Vinton, C.; Hirsch, V.; Lewis, M.; Brenchley, J.; Pandrea, I. Pattern of SIVagm Infection in Patas Monkeys Suggests that Host Adaptation to Simian Immunodeficiency Virus Infection May Result in Resistance to Infection and Virus Extinction. J. Infect. Dis. 2010, 202, S371–S376. [Google Scholar] [CrossRef]
- Harris, L.D.; Tabb, B.; Sodora, D.L.; Paiardini, M.; Klatt, N.R.; Douek, D.C.; Silvestri, G.; Muller-Trutwin, M.; Vasile-Pandrea, I.; Apetrei, C.; et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J. Virol. 2010, 84, 7886–7891. [Google Scholar] [CrossRef]
- Palesch, D.; Bosinger, S.E.; Tharp, G.K.; Vanderford, T.H.; Paiardini, M.; Chahroudi, A.; Johnson, Z.P.; Kirchhoff, F.; Hahn, B.H.; Norgren, R.B.; et al. Sooty mangabey genome sequence provides insight into AIDS resistance in a natural SIV host. Nature 2018, 553, 77–81. [Google Scholar] [CrossRef]
- Riddick, N.E.; Hermann, E.A.; Loftin, L.M.; Elliott, S.T.; Wey, W.C.; Cervasi, B.; Taaffe, J.; Engram, J.C.; Li, B.; Else, J.G.; et al. A Novel CCR5 Mutation Common in Sooty Mangabeys Reveals SIVsmm Infection of CCR5-Null Natural Hosts and Efficient Alternative Coreceptor Use In Vivo. PLoS Pathog. 2010, 6, e1001064. [Google Scholar] [CrossRef] [PubMed]
- Vinton, C.; Klatt, N.R.; Harris, L.D.; Briant, J.A.; Sanders-Beer, B.E.; Herbert, R.; Woodward, R.; Silvestri, G.; Pandrea, I.; Apetrei, C.; et al. CD4-like immunological function by CD4- T cells in multiple natural hosts of simian immunodeficiency virus. J. Virol. 2011, 85, 8702–8708. [Google Scholar] [CrossRef]
- Wetzel, K.S.; Yi, Y.; Elliott, S.T.C.; Romero, D.; Jacquelin, B.; Hahn, B.H.; Muller-Trutwin, M.; Apetrei, C.; Pandrea, I.; Collman, R.G. CXCR6-Mediated Simian Immunodeficiency Virus SIVagmSab Entry into Sabaeus African Green Monkey Lymphocytes Implicates Widespread Use of Non-CCR5 Pathways in Natural Host Infections. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Paiardini, M.; Cervasi, B.; Reyes-Aviles, E.; Micci, L.; Ortiz, A.M.; Chahroudi, A.; Vinton, C.; Gordon, S.N.; Bosinger, S.E.; Francella, N.; et al. Low levels of SIV infection in sooty mangabey central memory CD4+ T cells are associated with limited CCR5 expression. Nat. Med. 2011, 17, 830–836. [Google Scholar] [CrossRef]
- Pandrea, I.; Onanga, R.; Souquiere, S.; Mouinga-Ondéme, A.; Bourry, O.; Makuwa, M.; Rouquet, P.; Silvestri, G.; Simon, F.; Roques, P.; et al. Paucity of CD4+ CCR5+ T Cells May Prevent Transmission of Simian Immunodeficiency Virus in Natural Nonhuman Primate Hosts by Breast-Feeding. J. Virol. 2008, 82, 5501–5509. [Google Scholar] [CrossRef]
- Beaumier, C.M.; Harris, L.D.; Goldstein, S.; Klatt, N.R.; Whitted, S.; McGinty, J.; Apetrei, C.; Pandrea, I.; Hirsch, V.M.; Brenchley, J.M. CD4 downregulation by memory CD4+ T cells in vivo renders African green monkeys resistant to progressive SIVagm infection. Nat. Med. 2009, 15, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.T.; Wetzel, K.S.; Francella, N.; Bryan, S.; Romero, D.C.; Riddick, N.E.; Shaheen, F.; Vanderford, T.; Derdeyn, C.A.; Silvestri, G.; et al. Dualtropic CXCR6/CCR5 Simian Immunodeficiency Virus (SIV) Infection of Sooty Mangabey Primary Lymphocytes: Distinct Coreceptor Use in Natural versus Pathogenic Hosts of SIV. J. Virol. 2015, 89, 9252–9261. [Google Scholar] [CrossRef] [PubMed]
- Huot, N.; Jacquelin, B.; Garcia-Tellez, T.; Rascle, P.; Ploquin, M.J.; Madec, Y.; Reeves, R.K.; Derreudre-Bosquet, N.; Müller-Trutwin, M. Natural killer cells migrate into and control simian immunodeficiency virus replication in lymph node follicles in African green monkeys. Nat. Med. 2017, 23, 1277–1286. [Google Scholar] [CrossRef]
- Huot, N.; Rascle, P.; Petitdemange, C.; Contreras, V.; Stürzel, C.M.; Baquero, E.; Harper, J.L.; Passaes, C.; Legendre, R.; Varet, H.; et al. SIV-induced terminally differentiated adaptive NK cells in lymph nodes associated with enhanced MHC-E restricted activity. Nat. Commun. 2021, 12, 1282. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, G.; Tincati, C.; Silvestri, G. Microbial Translocation in the Pathogenesis of HIV Infection and AIDS. Clin. Microbiol. Rev. 2013, 26, 2–18. [Google Scholar] [CrossRef]
- Lerner, A.M.; Eisinger, R.W.; Fauci, A.S. Comorbidities in Persons With HIV: The Lingering Challenge. JAMA 2020, 323, 19–20. [Google Scholar] [CrossRef]
- Morlat, P.; Roussillon, C.; Henard, S.; Salmon, D.; Bonnet, F.; Cacoub, P.; Georget, A.; Aouba, A.; Rosenthal, E.; May, T.; et al. Causes of death among HIV-infected patients in France in 2010 (national survey): Trends since 2000. AIDS (Lond. Engl.) 2014, 28, 1181–1191. [Google Scholar] [CrossRef]
- Smith, C.J.; Ryom, L.; Weber, R.; Morlat, P.; Pradier, C.; Reiss, P.; Kowalska, J.D.; de Wit, S.; Law, M.; el Sadr, W.; et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): A multicohort collaboration. Lancet (Lond. Engl. ) 2014, 384, 241–248. [Google Scholar] [CrossRef]
- Marcus, J.L.; Chao, C.R.; Leyden, W.A.; Xu, L.; Quesenberry, C.P., Jr.; Klein, D.B.; Towner, W.J.; Horberg, M.A.; Silverberg, M.J. Narrowing the Gap in Life Expectancy Between HIV-Infected and HIV-Uninfected Individuals With Access to Care. J. Acquir. Immune Defic. syndromes 2016, 73, 39–46. [Google Scholar] [CrossRef]
- Schouten, J.; Wit, F.W.; Stolte, I.G.; Kootstra, N.A.; van der Valk, M.; Geerlings, S.E.; Prins, M.; Reiss, P.; for the, A.C.S.G. Cross-sectional Comparison of the Prevalence of Age-Associated Comorbidities and Their Risk Factors Between HIV-Infected and Uninfected Individuals: The AGEhIV Cohort Study. Clin. Infect. Dis. 2014, 59, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.T.; Silvestri, G. Nonhuman primate models in AIDS research. Curr Opin HIV AIDS 2013, 8, 255–261. [Google Scholar] [CrossRef]
- Lifson, J.D.; Haigwood, N.L. Lessons in nonhuman primate models for AIDS vaccine research: From minefields to milestones. Cold Spring Harb. Perspect. Med. 2012, 2, a007310. [Google Scholar] [CrossRef] [PubMed]
- Palella, F.J., Jr.; Phair, J.P. Cardiovascular disease in HIV infection. Curr Opin HIV AIDS 2011, 6, 266–271. [Google Scholar] [CrossRef]
- Balagopal, A.; Ray, S.C.; De Oca, R.M.; Sutcliffe, C.G.; Vivekanandan, P.; Higgins, Y.; Mehta, S.H.; Moore, R.D.; Sulkowski, M.S.; Thomas, D.L.; et al. Kupffer cells are depleted with HIV immunodeficiency and partially recovered with antiretroviral immune reconstitution. AIDS (Lond. Engl.) 2009, 23, 2397–2404. [Google Scholar] [CrossRef]
- Tuyama, A.C.; Hong, F.; Saiman, Y.; Wang, C.; Ozkok, D.; Mosoian, A.; Chen, P.; Chen, B.K.; Klotman, M.E.; Bansal, M.B. Human immunodeficiency virus (HIV)-1 infects human hepatic stellate cells and promotes collagen I and monocyte chemoattractant protein-1 expression: Implications for the pathogenesis of HIV/hepatitis C virus-induced liver fibrosis. Hepatol. (Baltim. Md.) 2010, 52, 612–622. [Google Scholar] [CrossRef]
- Ahsan, M.H.; Gill, A.F.; Alvarez, X.; Lackner, A.A.; Veazey, R.S. Kinetics of liver macrophages (Kupffer cells) in SIV-infected macaques. Virology 2013, 446, 77–85. [Google Scholar] [CrossRef]
- Justice, A.C.; Freiberg, M.S.; Tracy, R.; Kuller, L.; Tate, J.P.; Goetz, M.B.; Fiellin, D.A.; Vanasse, G.J.; Butt, A.A.; Rodriguez-Barradas, M.C.; et al. Does an index composed of clinical data reflect effects of inflammation, coagulation, and monocyte activation on mortality among those aging with HIV? Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2012, 54, 984–994. [Google Scholar] [CrossRef]
- Violi, F.; Ferro, D.; Basili, S.; Saliola, M.; Quintarelli, C.; Alessandri, C.; Cordova, C. Association between low-grade disseminated intravascular coagulation and endotoxemia in patients with liver cirrhosis. Gastroenterology 1995, 109, 531–539. [Google Scholar] [CrossRef]
- Shmagel, K.V.; Saidakova, E.V.; Shmagel, N.G.; Korolevskaya, L.B.; Chereshnev, V.A.; Robinson, J.; Grivel, J.C.; Douek, D.C.; Margolis, L.; Anthony, D.D.; et al. Systemic inflammation and liver damage in HIV/hepatitis C virus coinfection. HIV Med. 2016, 17, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Happel, K.I.; Amedee, A.M.; Bagby, G.J.; Nelson, S. Alcohol’s role in HIV transmission and disease progression. Alcohol Res. Health 2010, 33, 203–218. [Google Scholar] [PubMed]
- Zhang, L.; Dailey Peter, J.; Gettie, A.; Blanchard, J.; Ho David, D. The Liver Is a Major Organ for Clearing Simian Immunodeficiency Virus in Rhesus Monkeys. J. Virol. 2002, 76, 5271–5273. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.H.; Gill, A.F.; Lackner, A.A.; Veazey, R.S. Acute and chronic T cell dynamics in the livers of simian immunodeficiency virus-infected macaques. J. Virol. 2012, 86, 5244–5252. [Google Scholar] [CrossRef]
- Schmitz, J.E.; Kuroda, M.J.; Veazey, R.S.; Seth, A.; Taylor, W.M.; Nickerson, C.E.; Lifton, M.A.; Dailey, P.J.; Forman, M.A.; Racz, P.; et al. Simian Immunodeficiency Virus (SIV)-Specific CTL Are Present in Large Numbers in Livers of SIV-Infected Rhesus Monkeys. J. Immunol. 2000, 164, 6015–6019. [Google Scholar] [CrossRef]
- Kunisaki, K.M. Will expanded ART use reduce the burden of HIV-associated chronic lung disease? Curr Opin HIV AIDS 2014, 9, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Sugimoto, C.; Arainga, M.; Midkiff, C.C.; Liu, D.X.; Alvarez, X.; Lackner, A.A.; Kim, W.-K.; Didier, E.S.; Kuroda, M.J. Preferential Destruction of Interstitial Macrophages over Alveolar Macrophages as a Cause of Pulmonary Disease in Simian Immunodeficiency Virus–Infected Rhesus Macaques. J. Immunol. 2015, 195, 4884–4891. [Google Scholar] [CrossRef]
- Campbell, L.J.; Ibrahim, F.; Fisher, M.; Holt, S.G.; Hendry, B.M.; Post, F.A. Spectrum of chronic kidney disease in HIV-infected patients. HIV Med. 2009, 10, 329–336. [Google Scholar] [CrossRef]
- Kalyesubula, R.; Wearne, N.; Semitala, F.C.; Bowa, K. HIV-Associated Renal and Genitourinary Comorbidities in Africa. JAIDS J. Acquir. Immune Defic. Syndr. 2014, 67, S68–S78. [Google Scholar] [CrossRef]
- Ryom, L.; Mocroft, A.; Lundgren, J.D. Antiretroviral therapy, immune suppression and renal impairment in HIV-positive persons. Curr Opin HIV AIDS 2014, 9, 41–47. [Google Scholar] [CrossRef]
- Stephens, E.B.; Tian, C.; Dalton, S.B.; Gattone, V.H., 2nd. Simian-human immunodeficiency virus-associated nephropathy in macaques. AIDS Res. Hum. Retrovir. 2000, 16, 1295–1306. [Google Scholar] [CrossRef]
- Clarke, C.L.; Eckhaus, M.A.; Zerfas, P.M.; Elkins, W.R. Peripheral edema with hypoalbuminemia in a nonhuman primate infected with simian-human immunodeficiency virus: A case report. J. Am. Assoc. Lab. Anim. Sci. JAALAS 2008, 47, 42–48. [Google Scholar] [PubMed]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Holt, J.L.; Kraft-Terry, S.D.; Chang, L. Neuroimaging studies of the aging HIV-1-infected brain. J. Neurovirology 2012, 18, 291–302. [Google Scholar] [CrossRef][Green Version]
- Williams, R.; Bokhari, S.; Silverstein, P.; Pinson, D.; Kumar, A.; Buch, S. Nonhuman primate models of NeuroAIDS. J. Neurovirol 2008, 14, 292–300. [Google Scholar] [CrossRef]
- Beck, S.E.; Queen, S.E.; Metcalf Pate, K.A.; Mangus, L.M.; Abreu, C.M.; Gama, L.; Witwer, K.W.; Adams, R.J.; Zink, M.C.; Clements, J.E.; et al. An SIV/macaque model targeted to study HIV-associated neurocognitive disorders. J. Neurovirology 2018, 24, 204–212. [Google Scholar] [CrossRef]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Pan, X.; Baldauf, H.-M.; Keppler, O.T.; Fackler, O.T. Restrictions to HIV-1 replication in resting CD4+ T lymphocytes. Cell Res. 2013, 23, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Davey, R.T.; Engel, D.; Lane, H.C.; Fauci, A.S. Re-emergence of HIV after stopping therapy. Nature 1999, 401, 874–875. [Google Scholar] [CrossRef]
- Chun, T.-W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.M.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [PubMed]
- Perelson, A.S.; Essunger, P.; Cao, Y.; Vesanen, M.; Hurley, A.; Saksela, K.; Markowitz, M.; Ho, D.D. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 1997, 387, 188–191. [Google Scholar] [CrossRef]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Engel, D.; Berrey, M.M.; Shea, T.; Corey, L.; Fauci, A.S. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef]
- Archin, N.M.; Kirchherr, J.L.; Sung, J.A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba, A.D.; Kuruc, J.D.; et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Investig. 2017, 127, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog. 2014, 10, e1004071. [Google Scholar] [CrossRef]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. mBio 2015, 6. [Google Scholar] [CrossRef]
- Donahue, D.A.; Kuhl, B.D.; Sloan, R.D.; Wainberg, M.A. The viral protein Tat can inhibit the establishment of HIV-1 latency. J. Virol. 2012, 86, 3253–3263. [Google Scholar] [CrossRef]
- Borducchi, E.N.; Cabral, C.; Stephenson, K.E.; Liu, J.; Abbink, P.; Ng’ang’a, D.; Nkolola, J.P.; Brinkman, A.L.; Peter, L.; Lee, B.C.; et al. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV-infected rhesus monkeys. Nature 2016, 540, 284. [Google Scholar] [CrossRef]
- Tsai, A.; Irrinki, A.; Kaur, J.; Cihlar, T.; Kukolj, G.; Sloan, D.D.; Murry, J.P. Toll-Like Receptor 7 Agonist GS-9620 Induces HIV Expression and HIV-Specific Immunity in Cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy. J. Virol. 2017, 91, e02166-16. [Google Scholar] [CrossRef]
- Murray, S.M.; Down, C.M.; Boulware, D.R.; Stauffer, W.M.; Cavert, W.P.; Schacker, T.W.; Brenchley, J.M.; Douek, D.C. Reduction of Immune Activation with Chloroquine Therapy during Chronic HIV Infection. J. Virol. 2010, 84, 12082–12086. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, L.; von Stockenstrom, S.; Faria, N.R.; Sinclair, E.; Bacchetti, P.; Killian, M.; Epling, L.; Tan, A.; Ho, T.; Lemey, P.; et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc. Natl. Acad. Sci. 2013, 110, E4987–E4996. [Google Scholar] [CrossRef]
- Porichis, F.; Hart, M.G.; Zupkosky, J.; Barblu, L.; Kwon, D.S.; McMullen, A.; Brennan, T.; Ahmed, R.; Freeman, G.J.; Kavanagh, D.G.; et al. Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J. Virol. 2014, 88, 2508–2518. [Google Scholar] [CrossRef]
- Palmer, B.E.; Neff, C.P.; LeCureux, J.; Ehler, A.; Dsouza, M.; Remling-Mulder, L.; Korman, A.J.; Fontenot, A.P.; Akkina, R. In vivo blockade of the PD-1 receptor suppresses HIV-1 viral loads and improves CD4+ T cell levels in humanized mice. J. Immunol. 2013, 190, 211–219. [Google Scholar] [CrossRef]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M.; et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef]
- Zhang, Z.-N.; Zhu, M.-L.; Chen, Y.-H.; Fu, Y.-J.; Zhang, T.-W.; Jiang, Y.-J.; Chu, Z.-X.; Shang, H. Elevation of Tim-3 and PD-1 Expression on T Cells Appears Early in HIV Infection, and Differential Tim-3 and PD-1 Expression Patterns Can Be Induced by Common γ-Chain Cytokines. BioMed Res. Int. 2015, 2015, 916936. [Google Scholar] [CrossRef] [PubMed]
- Peretz, Y.; He, Z.; Shi, Y.; Yassine-Diab, B.; Goulet, J.-P.; Bordi, R.; Filali-Mouhim, A.; Loubert, J.-B.; El-Far, M.; Dupuy, F.P.; et al. CD160 and PD-1 Co-Expression on HIV-Specific CD8 T Cells Defines a Subset with Advanced Dysfunction. PLoS Pathog. 2012, 8, e1002840. [Google Scholar] [CrossRef] [PubMed]
- Vandergeeten, C.; Fromentin, R.; DaFonseca, S.; Lawani, M.B.; Sereti, I.; Lederman, M.M.; Ramgopal, M.; Routy, J.P.; Sekaly, R.P.; Chomont, N. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood 2013, 121, 4321–4329. [Google Scholar] [CrossRef]
- Seay, K.; Church, C.; Zheng, J.H.; Deneroff, K.; Ochsenbauer, C.; Kappes, J.C.; Liu, B.; Jeng, E.K.; Wong, H.C.; Goldstein, H. In Vivo Activation of Human NK Cells by Treatment with an Interleukin-15 Superagonist Potently Inhibits Acute In Vivo HIV-1 Infection in Humanized Mice. J. Virol. 2015, 89, 6264–6274. [Google Scholar] [CrossRef]
- Younes, S.A.; Freeman, M.L.; Mudd, J.C.; Shive, C.L.; Reynaldi, A.; Panigrahi, S.; Estes, J.D.; Deleage, C.; Lucero, C.; Anderson, J.; et al. IL-15 promotes activation and expansion of CD8+ T cells in HIV-1 infection. J. Clin. Investig. 2016, 126, 2745–2756. [Google Scholar] [CrossRef]
- Mendez-Lagares, G.; Lu, D.; Merriam, D.; Baker, C.A.; Villinger, F.; Van Rompay, K.K.A.; McCune, J.M.; Hartigan-O’Connor, D.J. IL-21 Therapy Controls Immune Activation and Maintains Antiviral CD8(+) T Cell Responses in Acute Simian Immunodeficiency Virus Infection. AIDS Res. Hum. Retrovir. 2017, 33, S81–S92. [Google Scholar] [CrossRef] [PubMed]
- Hale, M.; Mesojednik, T.; Romano Ibarra, G.S.; Sahni, J.; Bernard, A.; Sommer, K.; Scharenberg, A.M.; Rawlings, D.J.; Wagner, T.A. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol. Ther. : J. Am. Soc. Gene Ther. 2017, 25, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Hütter, G.; Bodor, J.; Ledger, S.; Boyd, M.; Millington, M.; Tsie, M.; Symonds, G. CCR5 Targeted Cell Therapy for HIV and Prevention of Viral Escape. Viruses 2015, 7, 4186–4203. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, J.M.; Angin, M.; Passaes, C.; Salgado, M.; Monceaux, V.; Knops, E.; Kobbe, G.; Jensen, B.; Christopeit, M.; Kroger, N.; et al. Vulnerability to reservoir reseeding due to high immune activation after allogeneic hematopoietic stem cell transplantation in individuals with HIV-1. Sci. Transl Med. 2020, 12. [Google Scholar] [CrossRef]
- Saez-Cirion, A.; Muller-Trutwin, M. The Yellow Brick Road towards HIV Eradication. Trends Immunol 2019, 40, 465–467. [Google Scholar] [CrossRef]
- Dufour, C.; Gantner, P.; Fromentin, R.; Chomont, N. The multifaceted nature of HIV latency. J. Clin. Investig. 2020, 130, 3381–3390. [Google Scholar] [CrossRef]
- Margolis, D.M.; Archin, N.M.; Cohen, M.S.; Eron, J.J.; Ferrari, G.; Garcia, J.V.; Gay, C.L.; Goonetilleke, N.; Joseph, S.B.; Swanstrom, R.; et al. Curing HIV: Seeking to Target and Clear Persistent Infection. Cell 2020, 181, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Massanella, M.; Fromentin, R.; Chomont, N. Residual inflammation and viral reservoirs: Alliance against an HIV cure. Curr Opin HIV AIDS 2016, 11, 234–241. [Google Scholar] [CrossRef]
- Passaes, C.P.; Saez-Cirion, A. HIV cure research: Advances and prospects. Virology 2014, 454–455, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Fromentin, R.; Chomont, N. HIV persistence in subsets of CD4+ T cells: 50 shades of reservoirs. Semin. Immunol. 2020, 101438. [Google Scholar] [CrossRef] [PubMed]
- Rabezanahary, H.; Moukambi, F.; Palesch, D.; Clain, J.; Racine, G.; Andreani, G.; Benmadid-Laktout, G.; Zghidi-Abouzid, O.; Soundaramourty, C.; Tremblay, C.; et al. Despite early antiretroviral therapy effector memory and follicular helper CD4 T cells are major reservoirs in visceral lymphoid tissues of SIV-infected macaques. Mucosal Immunol. 2020, 13, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Delobel, P.; Sandres-Sauné, K.; Cazabat, M.; L’Faqihi, F.-E.; Aquilina, C.; Obadia, M.; Pasquier, C.; Marchou, B.; Massip, P.; Izopet, J. Persistence of distinct HIV-1 populations in blood monocytes and naive and memory CD4 T cells during prolonged suppressive HAART. AIDS (Lond. Engl.) 2005, 19, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Buzon, M.J.; Sun, H.; Li, C.; Shaw, A.; Seiss, K.; Ouyang, Z.; Martin-Gayo, E.; Leng, J.; Henrich, T.J.; Li, J.Z.; et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat. Med. 2014, 20, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, A.J.; Sivanandham, R.; Pandrea, I.; Chougnet, C.A.; Apetrei, C. Regulatory T Cells As Potential Targets for HIV Cure Research. Front. Immunol 2018, 9, 734. [Google Scholar] [CrossRef]
- Pallikkuth, S.; Sharkey, M.; Babic, D.Z.; Gupta, S.; Stone, G.W.; Fischl, M.A.; Stevenson, M.; Pahwa, S. Peripheral T Follicular Helper Cells Are the Major HIV Reservoir within Central Memory CD4 T Cells in Peripheral Blood from Chronically HIV-Infected Individuals on Combination Antiretroviral Therapy. J. Virol. 2016, 90, 2718–2728. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.E.; Jaworowski, A.; Hearps, A.C. The HIV Reservoir in Monocytes and Macrophages. Front. Immunol. 2019, 10, 1435. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, H.; Herbst, H.; Niedobitek, G.; Foss, H.D.; Stein, H. Follicular dendritic cells are a major reservoir for human immunodeficiency virus type 1 in lymphoid tissues facilitating infection of CD4+ T-helper cells. Am. J. Pathol 1992, 140, 15–22. [Google Scholar]
- Badia, R.; Ballana, E.; Castellví, M.; García-Vidal, E.; Pujantell, M.; Clotet, B.; Prado, J.G.; Puig, J.; Martínez, M.A.; Riveira-Muñoz, E.; et al. CD32 expression is associated to T-cell activation and is not a marker of the HIV-1 reservoir. Nat. Commun. 2018, 9, 2739. [Google Scholar] [CrossRef]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müßig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef]
- Henrich, T.J.; Hanhauser, E.; Marty, F.M.; Sirignano, M.N.; Keating, S.; Lee, T.H.; Robles, Y.P.; Davis, B.T.; Li, J.Z.; Heisey, A.; et al. Antiretroviral-free hiv-1 remission and viral rebound after allogeneic stem cell transplantation: Report of 2 cases. Ann. Intern. Med. 2014, 161, 319–327. [Google Scholar] [CrossRef]
- Persaud, D.; Gay, H.; Ziemniak, C.; Chen, Y.H.; Piatak, M.J.; Chun, T.-W.; Strain, M.; Richman, D.; Luzuriaga, K. Absence of Detectable HIV-1 Viremia after Treatment Cessation in an Infant. N. Engl. J. Med. 2013, 369, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, K.; Gay, H.; Ziemniak, C.; Sanborn, K.B.; Somasundaran, M.; Rainwater-Lovett, K.; Mellors, J.W.; Rosenbloom, D.; Persaud, D. Viremic Relapse after HIV-1 Remission in a Perinatally Infected Child. N. Engl. J. Med. 2015, 372, 786–788. [Google Scholar] [CrossRef]
- Sáez-Cirión, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-Treatment HIV-1 Controllers with a Long-Term Virological Remission after the Interruption of Early Initiated Antiretroviral Therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef]
- Violari, A.; Cotton, M.; Kuhn, L.; Schramm, D.; Paximadis, M.; Shalekoff, S.; Dias, B.d.C.; Otwombe, K.; Liberty, A.; McIntyre, J.; et al. Viral and host characterists of a child with perinatal HIV-1 following a prolonged period after ART cessation in the CHER trial. In Proceedings of the International AIDS Society 2017, Paris, France, 23–26 July 2017. [Google Scholar]
- Cummins, N.W.; Rizza, S.; Litzow, M.R.; Hua, S.; Lee, G.Q.; Einkauf, K.; Chun, T.-W.; Rhame, F.; Baker, J.V.; Busch, M.P.; et al. Extensive virologic and immunologic characterization in an HIV-infected individual following allogeneic stem cell transplant and analytic cessation of antiretroviral therapy: A case study. PLoS Med. 2017, 14, e1002461. [Google Scholar] [CrossRef]
- Folks, T.; Powell, D.M.; Lightfoote, M.M.; Benn, S.; Martin, M.A.; Fauci, A.S. Induction of HTLV-III/LAV from a nonvirus-producing T-cell line: Implications for latency. Science 1986, 231, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Siekevitz, M.; Josephs, S.F.; Dukovich, M.; Peffer, N.; Wong-Staal, F.; Greene, W.C. Activation of the HIV-1 LTR by T cell mitogens and the trans-activator protein of HTLV-I. Science 1987, 238, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Duh, E.J.; Maury, W.J.; Folks, T.M.; Fauci, A.S.; Rabson, A.B. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc. Natl. Acad. Sci. USA 1989, 86, 5974–5978. [Google Scholar] [CrossRef]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S.Y. HIV-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell 1990, 61, 213–222. [Google Scholar] [CrossRef]
- Bukrinsky, M.I.; Stanwick, T.L.; Dempsey, M.P.; Stevenson, M. Quiescent T Lymphocytes as an Inducible Virus Reservoir in HIV-1 Infection. Science 1991, 254, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Sahu, G.K.; Lee, K.; Ji, J.; Braciale, V.; Baron, S.; Cloyd, M.W. A novel in vitro system to generate and study latently HIV-infected long-lived normal CD4+ T-lymphocytes. Virology 2006, 355, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Bosque, A.; Planelles, V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65. [Google Scholar] [CrossRef]
- Burke, B.; Brown, H.J.; Marsden, M.D.; Bristol, G.; Vatakis, D.N.; Zack, J.A. Primary cell model for activation-inducible human immunodeficiency virus. J. Virol. 2007, 81, 7424–7434. [Google Scholar] [CrossRef][Green Version]
- Marini, A.; Harper, J.M.; Romerio, F. An in vitro system to model the establishment and reactivation of HIV-1 latency. J. Immunol. 2008, 181, 7713–7720. [Google Scholar] [CrossRef]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV Latency in Primary CD4+ Cells Is due to Epigenetic Transcriptional Silencing and P-TEFb Restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef]
- Yang, H.C.; Xing, S.; Shan, L.; O’Connell, K.; Dinoso, J.; Shen, A.; Zhou, Y.; Shrum, C.K.; Han, Y.; Liu, J.O.; et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J. Clin. Investig. 2009, 119, 3473–3486. [Google Scholar] [CrossRef]
- Yang, H.-C. Primary cell models of HIV latency. Curr. Opin. HIV AIDS 2011, 6, 62–67. [Google Scholar] [CrossRef]
- Schröder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S.; et al. Retroviral DNA integration: Viral and cellular determinants of target-site selection. PLoS Pathog. 2006, 2, e60. [Google Scholar] [CrossRef]
- Han, Y.; Lassen, K.; Monie, D.; Sedaghat, A.R.; Shimoji, S.; Liu, X.; Pierson, T.C.; Margolick, J.B.; Siliciano, R.F.; Siliciano, J.D. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. J. Virol. 2004, 78, 6122–6133. [Google Scholar] [CrossRef]
- Verdin, E.; Paras Jr, P.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef] [PubMed]
- Keedy, K.S.; Archin, N.M.; Gates, A.T.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J. Virol. 2009, 83, 4749–4756. [Google Scholar] [CrossRef]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Lusic, M.; Marcello, A.; Cereseto, A.; Giacca, M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003, 22, 6550–6561. [Google Scholar] [CrossRef]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Jeng, M.; Camus, G.; Gramatica, A.; Schwarzer, R.; Johnson, J.R.; Hull, P.A.; Montano, M.; Sakane, N.; Pagans, S.; et al. SMYD2-Mediated Histone Methylation Contributes to HIV-1 Latency. Cell Host Microbe 2017, 21, 569–579. [Google Scholar] [CrossRef]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of Histone H3 Lysine 9 (H3K9) Methyltransferase G9a in the Maintenance of HIV-1 Latency and Its Reactivation by BIX01294 *. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [PubMed]
- Chéné, I.d.; Basyuk, E.; Lin, Y.-L.; Triboulet, R.; Knezevich, A.; Chable-Bessia, C.; Mettling, C.; Baillat, V.; Reynes, J.; Corbeau, P.; et al. Suv39H1 and HP1γ are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26, 424–435. [Google Scholar] [CrossRef]
- He, G.; Margolis, D.M. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef]
- Williams, S.A.; Chen, L.-F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef] [PubMed]
- Greger, I.H.; Demarchi, F.; Giacca, M.; Proudfoot, N.J. Transcriptional interference perturbs the binding of Sp1 to the HIV-1 promoter. Nucleic Acids Res. 1998, 26, 1294–1301. [Google Scholar] [CrossRef]
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional Interference Antagonizes Proviral Gene Expression to Promote HIV Latency. Cell Host Microbe 2008, 4, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Lin, Y.B.; An, W.; Xu, J.; Yang, H.-C.; O’Connell, K.; Dordai, D.; Boeke, J.D.; Siliciano, J.D.; Siliciano, R.F. Orientation-Dependent Regulation of Integrated HIV-1 Expression by Host Gene Transcriptional Readthrough. Cell Host Microbe 2008, 4, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Sedore, S.C.; Byers, S.A.; Biglione, S.; Price, J.P.; Maury, W.J.; Price, D.H. Manipulation of P-TEFb control machinery by HIV: Recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007, 35, 4347–4358. [Google Scholar] [CrossRef]
- Muniz, L.; Egloff, S.; Ughy, B.; Jády, B.E.; Kiss, T. Controlling Cellular P-TEFb Activity by the HIV-1 Transcriptional Transactivator Tat. PLoS Pathog. 2010, 6, e1001152. [Google Scholar] [CrossRef]
- Parada, C.A.; Roeder, R.G. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature 1996, 384, 375–378. [Google Scholar] [CrossRef]
- Kim, Y.K.; Bourgeois, C.F.; Isel, C.; Churcher, M.J.; Karn, J. Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol. Cell Biol. 2002, 22, 4622–4637. [Google Scholar] [CrossRef]
- Bourgeois, C.F.; Kim, Y.K.; Churcher, M.J.; West, M.J.; Karn, J. Spt5 cooperates with human immunodeficiency virus type 1 Tat by preventing premature RNA release at terminator sequences. Mol. Cell Biol. 2002, 22, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar] [CrossRef]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.-C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Justement, J.S.; Moir, S.; Hallahan, C.W.; Maenza, J.; Mullins, J.I.; Collier, A.C.; Corey, L.; Fauci, A.S. Decay of the HIV Reservoir in Patients Receiving Antiretroviral Therapy for Extended Periods: Implications for Eradication of Virus. J. Infect. Dis. 2007, 195, 1762–1764. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.T.; Cyktor, J.C.; Bosch, R.J.; Mar, H.; Laird, G.M.; Martin, A.; Collier, A.C.; Riddler, S.A.; Macatangay, B.J.; Rinaldo, C.R.; et al. Selective Decay of Intact HIV-1 Proviral DNA on Antiretroviral Therapy. J. Infect. Dis. 2021, 223, 225–233. [Google Scholar] [CrossRef]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef]
- Kuzmichev, Y.V.; Veenhuis, R.T.; Pohlmeyer, C.W.; Garliss, C.C.; Walker-Sperling, V.E.; Blankson, J.N. A CD3/CD28 microbead-based HIV-1 viral outgrowth assay. J. Virus Erad. 2017, 3, 85–89. [Google Scholar] [CrossRef]
- Patel, S.S.; Duby, A.D.; Thiele, D.L.; Lipsky, P.E. Phenotypic and functional characterization of human T cell clones. J. Immunol. 1988, 141, 3726–3736. [Google Scholar]
- Beliakova-Bethell, N.; Hezareh, M.; Wong, J.K.; Strain, M.C.; Lewinski, M.K.; Richman, D.D.; Spina, C.A. Relative efficacy of T cell stimuli as inducers of productive HIV-1 replication in latently infected CD4 lymphocytes from patients on suppressive cART. Virology 2017, 508, 127–133. [Google Scholar] [CrossRef]
- Laird, G.M.; Eisele, E.E.; Rabi, S.A.; Lai, J.; Chioma, S.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Rapid quantification of the latent reservoir for HIV-1 using a viral outgrowth assay. PLoS Pathog. 2013, 9, e1003398. [Google Scholar] [CrossRef] [PubMed]
- Pollack, R.A.; Jones, R.B.; Pertea, M.; Bruner, K.M.; Martin, A.R.; Thomas, A.S.; Capoferri, A.A.; Beg, S.A.; Huang, S.-H.; Karandish, S.; et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 2017, 21, 494–506. [Google Scholar] [CrossRef]
- Ho, Y.-C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.S.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef]
- Hosmane, N.N.; Kwon, K.J.; Bruner, K.M.; Capoferri, A.A.; Beg, S.; Rosenbloom, D.I.; Keele, B.F.; Ho, Y.C.; Siliciano, J.D.; Siliciano, R.F. Proliferation of latently infected CD4(+) T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 2017, 214, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Bui, J.K.; Sobolewski, M.D.; Keele, B.F.; Spindler, J.; Musick, A.; Wiegand, A.; Luke, B.T.; Shao, W.; Hughes, S.H.; Coffin, J.M.; et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Pathog. 2017, 13, e1006283. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gladden, A.D.; Altfeld, M.; Kaldor, J.M.; Cooper, D.A.; Kelleher, A.D.; Allen, T.M. Rapid reversion of sequence polymorphisms dominates early human immunodeficiency virus type 1 evolution. J. Virol. 2007, 81, 193–201. [Google Scholar] [CrossRef]
- Wang, Z.; Simonetti, F.R.; Siliciano, R.F.; Laird, G.M. Measuring replication competent HIV-1: Advances and challenges in defining the latent reservoir. Retrovirology 2018, 15, 21. [Google Scholar] [CrossRef]
- Hiener, B.; Horsburgh, B.A.; Eden, J.S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4(+) T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Q.; Orlova-Fink, N.; Einkauf, K.; Chowdhury, F.Z.; Sun, X.; Harrington, S.; Kuo, H.H.; Hua, S.; Chen, H.R.; Ouyang, Z.; et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J. Clin. Investig. 2017, 127, 2689–2696. [Google Scholar] [CrossRef]
- Kijak, G.H.; Sanders-Buell, E.; Pham, P.; Harbolick, E.A.; Oropeza, C.; O’Sullivan, A.M.; Bose, M.; Beckett, C.G.; Milazzo, M.; Robb, M.L.; et al. Next-generation sequencing of HIV-1 single genome amplicons. Biomol Detect. Quantif. 2019, 17, 100080. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.; López, P.; Sánchez, R.; Yamamura, Y.; Rivera-Amill, V. Sanger and Next Generation Sequencing Approaches to Evaluate HIV-1 Virus in Blood Compartments. Int. J. Env. Res. Public Health 2018, 15, 1697. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.R.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125. [Google Scholar] [CrossRef]
- Kinloch, N.N.; Ren, Y.; Conce Alberto, W.D.; Dong, W.; Khadka, P.; Huang, S.H.; Mota, T.M.; Wilson, A.; Shahid, A.; Kirkby, D.; et al. HIV-1 diversity considerations in the application of the Intact Proviral DNA Assay (IPDA). Nat. Commun. 2021, 12, 165. [Google Scholar] [CrossRef]
- Gaebler, C.; Lorenzi, J.C.C.; Oliveira, T.Y.; Nogueira, L.; Ramos, V.; Lu, C.-L.; Pai, J.A.; Mendoza, P.; Jankovic, M.; Caskey, M.; et al. Combination of quadruplex qPCR and next-generation sequencing for qualitative and quantitative analysis of the HIV-1 latent reservoir. J. Exp. Med. 2019, 216, 2253–2264. [Google Scholar] [CrossRef]
- Eriksson, S.; Graf, E.H.; Dahl, V.; Strain, M.C.; Yukl, S.A.; Lysenko, E.S.; Bosch, R.J.; Lai, J.; Chioma, S.; Emad, F.; et al. Comparative Analysis of Measures of Viral Reservoirs in HIV-1 Eradication Studies. PLoS Pathog. 2013, 9, e1003174. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8788. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, H.; Smith, M.; Adelsberger, J.W.; Izumi, T.; Scrimieri, F.; Sherman, B.T.; Rehm, C.A.; Imamichi, T.; Pau, A.; Catalfamo, M.; et al. Defective HIV-1 proviruses produce viral proteins. Proc. Natl. Acad. Sci. USA 2020, 117, 3704–3710. [Google Scholar] [CrossRef]
- Apetrei, C.; Pandrea, I.; Mellors, J.W. Nonhuman primate models for HIV cure research. PLoS Pathog. 2012, 8, e1002892. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.K.; Archin, N.M.; Cheema, M.; Dahl, N.P.; Garcia, J.V.; Margolis, D.M. Latent HIV-1 infection of resting CD4(+) T cells in the humanized Rag2(-)/(-) gammac(-)/(-) mouse. J. Virol. 2012, 86, 114–120. [Google Scholar] [CrossRef]
- Denton, P.W.; Olesen, R.; Choudhary, S.K.; Archin, N.M.; Wahl, A.; Swanson, M.D.; Chateau, M.; Nochi, T.; Krisko, J.F.; Spagnuolo, R.A.; et al. Generation of HIV latency in humanized BLT mice. J. Virol. 2012, 86, 630–634. [Google Scholar] [CrossRef]
- Honeycutt, J.B.; Wahl, A.; Baker, C.; Spagnuolo, R.A.; Foster, J.; Zakharova, O.; Wietgrefe, S.; Caro-Vegas, C.; Madden, V.; Sharpe, G.; et al. Macrophages sustain HIV replication in vivo independently of T cells. J. Clin. Investig. 2016, 126, 1353–1366. [Google Scholar] [CrossRef]
- Olesen, R.; Swanson, M.D.; Kovarova, M.; Nochi, T.; Chateau, M.; Honeycutt, J.B.; Long, J.M.; Denton, P.W.; Hudgens, M.G.; Richardson, A.; et al. ART influences HIV persistence in the female reproductive tract and cervicovaginal secretions. J. Clin. Investig. 2016, 126, 892–904. [Google Scholar] [CrossRef]
- Garcia, J.V. In vivo platforms for analysis of HIV persistence and eradication. J. Clin. Investig. 2016, 126, 424–431. [Google Scholar] [CrossRef]
- Nishimura, Y.; Sadjadpour, R.; Mattapallil, J.J.; Igarashi, T.; Lee, W.; Buckler-White, A.; Roederer, M.; Chun, T.W.; Martin, M.A. High frequencies of resting CD4+ T cells containing integrated viral DNA are found in rhesus macaques during acute lentivirus infections. Proc. Natl. Acad. Sci. USA 2009, 106, 8015–8020. [Google Scholar] [CrossRef] [PubMed]
- Shen, A.; Zink, M.C.; Mankowski, J.L.; Chadwick, K.; Margolick, J.B.; Carruth, L.M.; Li, M.; Clements, J.E.; Siliciano, R.F. Resting CD4+ T lymphocytes but not thymocytes provide a latent viral reservoir in a simian immunodeficiency virus-Macaca nemestrina model of human immunodeficiency virus type 1-infected patients on highly active antiretroviral therapy. J. Virol. 2003, 77, 4938–4949. [Google Scholar] [CrossRef]
- Crise, B.; Li, Y.; Yuan, C.; Morcock, D.R.; Whitby, D.; Munroe, D.J.; Arthur, L.O.; Wu, X. Simian immunodeficiency virus integration preference is similar to that of human immunodeficiency virus type 1. J. Virol. 2005, 79, 12199–12204. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.A.; Gama, L.; Dudaronek, J.M.; Voelker, T.; Tarwater, P.M.; Clements, J.E. Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus-macaque model. J. Infect. Dis. 2006, 193, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Shen, A.; Yang, H.C.; Zhou, Y.; Chase, A.J.; Boyer, J.D.; Zhang, H.; Margolick, J.B.; Zink, M.C.; Clements, J.E.; Siliciano, R.F. Novel pathway for induction of latent virus from resting CD4(+) T cells in the simian immunodeficiency virus/macaque model of human immunodeficiency virus type 1 latency. J. Virol. 2007, 81, 1660–1670. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Norelli, S.; Chirullo, B.; Della Corte, A.; Collins, M.; Yalley-Ogunro, J.; Greenhouse, J.; Iraci, N.; Acosta, E.P.; Barreca, M.L.; et al. A highly intensified ART regimen induces long-term viral suppression and restriction of the viral reservoir in a simian AIDS model. PLoS Pathog. 2012, 8, e1002774. [Google Scholar] [CrossRef]
- Del Prete, G.Q.; Smedley, J.; Macallister, R.; Jones, G.S.; Li, B.; Hattersley, J.; Zheng, J.; Piatak, M., Jr.; Keele, B.F.; Hesselgesser, J.; et al. Short Communication: Comparative Evaluation of Coformulated Injectable Combination Antiretroviral Therapy Regimens in Simian Immunodeficiency Virus-Infected Rhesus Macaques. AIDS Res. Hum. Retrovir. 2016, 32, 163–168. [Google Scholar] [CrossRef]
- Dinoso, J.B.; Kim, S.Y.; Wiegand, A.M.; Palmer, S.E.; Gange, S.J.; Cranmer, L.; O’Shea, A.; Callender, M.; Spivak, A.; Brennan, T.; et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 9403–9408. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Bosch, R.J.; Aga, E.; Albrecht, M.; Demeter, L.M.; Dykes, C.; Bastow, B.; Para, M.; Lai, J.; Siliciano, R.F.; et al. No Evidence for Decay of the Latent Reservoir in HIV-1–Infected Patients Receiving Intensive Enfuvirtide-Containing Antiretroviral Therapy. J. Infect. Dis. 2010, 201, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.T.; Zheng, L.; Bosch, R.J.; Chan, E.S.; Margolis, D.M.; Read, S.; Kallungal, B.; Palmer, S.; Medvik, K.; Lederman, M.M.; et al. The Effect of Raltegravir Intensification on Low-level Residual Viremia in HIV-Infected Patients on Antiretroviral Therapy: A Randomized Controlled Trial. PLOS Med. 2010, 7, e1000321. [Google Scholar] [CrossRef]
- Hammer, S.M.; Ribaudo, H.; Bassett, R.; Mellors, J.W.; Demeter, L.M.; Coombs, R.W.; Currier, J.; Morse, G.D.; Gerber, J.G.; Martinez, A.I.; et al. A randomized, placebo-controlled trial of abacavir intensification in HIV-1-infected adults with virologic suppression on a protease inhibitor-containing regimen. HIV Clin. Trials 2010, 11, 312–324. [Google Scholar] [CrossRef]
- Mousseau, G.; Mediouni, S.; Valente, S.T. Targeting HIV Transcription: The Quest for a Functional Cure. Curr. Top. Microbiol. Immunol. 2015, 389, 121–145. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Wagner, B.; Anton, P.A.; Mitsuyasu, R.T.; Scadden, D.T.; Huang, C.; Macken, C.; Richman, D.D.; Christopherson, C.; June, C.H.; et al. A Phase II Randomized Study of HIV-Specific T-Cell Gene Therapy in Subjects with Undetectable Plasma Viremia on Combination Antiretroviral Therapy. Mol. Ther. 2002, 5, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Zhen, A.; Peterson, C.W.; Carrillo, M.A.; Reddy, S.S.; Youn, C.S.; Lam, B.B.; Chang, N.Y.; Martin, H.A.; Rick, J.W.; Kim, J.; et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog. 2017, 13, e1006753. [Google Scholar] [CrossRef]
- Cummins, N.W.; Sainski, A.M.; Dai, H.; Natesampillai, S.; Pang, Y.P.; Bren, G.D.; de Araujo Correia, M.C.M.; Sampath, R.; Rizza, S.A.; O’Brien, D.; et al. Prime, Shock, and Kill: Priming CD4 T Cells from HIV Patients with a BCL-2 Antagonist before HIV Reactivation Reduces HIV Reservoir Size. J. Virol. 2016, 90, 4032–4048. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Sainski-Nguyen, A.M.; Natesampillai, S.; Aboulnasr, F.; Kaufmann, S.; Badley, A.D. Maintenance of the HIV Reservoir Is Antagonized by Selective BCL2 Inhibition. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ghneim, K.; Sok, D.; Bosche, W.J.; Li, Y.; Chipriano, E.; Berkemeier, B.; Oswald, K.; Borducchi, E.; Cabral, C.; et al. Antibody-mediated protection against SHIV challenge includes systemic clearance of distal virus. Science 2016, 353, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pegu, A.; Rao, E.; Doria-Rose, N.; Beninga, J.; McKee, K.; Lord, D.M.; Wei, R.R.; Deng, G.; Louder, M.; et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science 2017, 358, 85–90. [Google Scholar] [CrossRef]
- Ng’uni, T.; Chasara, C.; Ndhlovu, Z.M. Major Scientific Hurdles in HIV Vaccine Development: Historical Perspective and Future Directions. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- He, T.; Brocca-Cofano, E.; Policicchio, B.B.; Sivanandham, R.; Gautam, R.; Raehtz, K.D.; Xu, C.; Pandrea, I.; Apetrei, C. Cutting Edge: T Regulatory Cell Depletion Reactivates Latent Simian Immunodeficiency Virus (SIV) in Controller Macaques While Boosting SIV-Specific T Lymphocytes. J. Immunol. 2016. [Google Scholar] [CrossRef]
- Sivanandham, R.; Kleinman, A.J.; Sette, P.; Brocca-Cofano, E.; Kilapandal Venkatraman, S.M.; Policicchio, B.B.; He, T.; Xu, C.; Swarthout, J.; Wang, Z.; et al. Nonhuman Primate Testing of the Impact of Different Regulatory T Cell Depletion Strategies on Reactivation and Clearance of Latent Simian Immunodeficiency Virus. J. Virol. 2020, 94, e00533-00520. [Google Scholar] [CrossRef] [PubMed]
- Wightman, F.; Solomon, A.; Kumar, S.S.; Urriola, N.; Gallagher, K.; Hiener, B.; Palmer, S.; McNeil, C.; Garsia, R.; Lewin, S.R. Effect of ipilimumab on the HIV reservoir in an HIV-infected individual with metastatic melanoma. AIDS (Lond. Engl.) 2015, 29, 504–506. [Google Scholar] [CrossRef]
- Hryniewicz, A.; Boasso, A.; Edghill-Smith, Y.; Vaccari, M.; Fuchs, D.; Venzon, D.; Nacsa, J.; Betts, M.R.; Tsai, W.-P.; Heraud, J.-M.; et al. CTLA-4 blockade decreases TGF-β, IDO, and viral RNA expression in tissues of SIVmac251-infected macaques. Blood 2006, 108, 3834–3842. [Google Scholar] [CrossRef] [PubMed]
- Petrovas, C.; Casazza, J.P.; Brenchley, J.M.; Price, D.A.; Gostick, E.; Adams, W.C.; Precopio, M.L.; Schacker, T.; Roederer, M.; Douek, D.C.; et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006, 203, 2281–2292. [Google Scholar] [CrossRef] [PubMed]
- Velu, V.; Kannanganat, S.; Ibegbu, C.; Chennareddi, L.; Villinger, F.; Freeman, G.J.; Ahmed, R.; Amara, R.R. Elevated expression levels of inhibitory receptor programmed death 1 on simian immunodeficiency virus-specific CD8 T cells during chronic infection but not after vaccination. J. Virol. 2007, 81, 5819–5828. [Google Scholar] [CrossRef]
- Blazkova, J.; Chun, T.-W.; Belay, B.W.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.; Hallahan, C.W.; Moir, S.; Wender, P.A.; et al. Effect of Histone Deacetylase Inhibitors on HIV Production in Latently Infected, Resting CD4+ T Cells From Infected Individuals Receiving Effective Antiretroviral Therapy. J. Infect. Dis. 2012, 206, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Cillo, A.R.; Sobolewski, M.D.; Bosch, R.J.; Fyne, E.; Piatak, M.; Coffin, J.M.; Mellors, J.W. Quantification of HIV-1 latency reversal in resting CD4+ T cells from patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2014, 111, 7078–7083. [Google Scholar] [CrossRef]
- Gutierrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Perez-Elias, M.J.; Martin, M.E.; Barbas, C.; Ruiperez, J.; Munoz, E.; Munoz-Fernandez, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS (Lond. Engl.) 2016, 30, 1385–1392. [Google Scholar] [CrossRef]
- Prins, J.M.; Jurriaans, S.; van Praag, R.M.; Blaak, H.; van Rij, R.; Schellekens, P.T.; ten Berge, I.J.; Yong, S.L.; Fox, C.H.; Roos, M.T.; et al. Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS (Lond. Engl.) 1999, 13, 2405–2410. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone Deacetylase Inhibitors Impair the Elimination of HIV-Infected Cells by Cytotoxic T-Lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.-C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-Specific Cytolytic T Lymphocytes Facilitates Elimination of Latent Viral Reservoir after Virus Reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef]
- Policicchio, B.B.; Xu, C.; Brocca-Cofano, E.; Raehtz, K.D.; He, T.; Ma, D.; Li, H.; Sivanandham, R.; Haret-Richter, G.S.; Dunsmore, T.; et al. Multi-dose Romidepsin Reactivates Replication Competent SIV in Post-antiretroviral Rhesus Macaque Controllers. PLoS Pathog. 2016, 12, e1005879. [Google Scholar] [CrossRef]
- Migueles, S.A.; Weeks, K.A.; Nou, E.; Berkley, A.M.; Rood, J.E.; Osborne, C.M.; Hallahan, C.W.; Cogliano-Shutta, N.A.; Metcalf, J.A.; McLaughlin, M.; et al. Defective human immunodeficiency virus-specific CD8+ T-cell polyfunctionality, proliferation, and cytotoxicity are not restored by antiretroviral therapy. J. Virol. 2009, 83, 11876–11889. [Google Scholar] [CrossRef]
- Ananworanich, J.; Chomont, N.; Fletcher, J.L.; Pinyakorn, S.; Schuetz, A.; Sereti, I.; Rerknimitr, R.; Dewar, R.; Kroon, E.; Vandergeeten, C.; et al. Markers of HIV reservoir size and immune activation after treatment in acute HIV infection with and without raltegravir and maraviroc intensification. J. Virus Erad. 2015, 1, 116–122. [Google Scholar] [CrossRef]
- Cohen, Y.Z.; Caskey, M. Broadly neutralizing antibodies for treatment and prevention of HIV-1 infection. Curr Opin HIV AIDS 2018, 13, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.X.; Cannon, P.M. The clinical applications of genome editing in HIV. Blood 2016, 127, 2546–2552. [Google Scholar] [CrossRef]
- Duarte, R.; Labopin, M.; Badoglio, M.; Cahn, J.-Y.; Leblond, V.; Milpied, N.; Peniket, A.; Passweg, J.; Richard, C.; Russell, N.; et al. Allogeneic Transplantation in Patients with HIV-Infection: A Pair Matched Cohort Study by the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant. 2020, 55, 22–23. [Google Scholar]
- Dornadula, G.; Zhang, H.; VanUitert, B.; Stern, J.; Livornese, J.L.; Ingerman, M.J.; Witek, J.; Kedanis, R.J.; Natkin, J.; DeSimone, J.; et al. Residual HIV-1 RNA in Blood Plasma of Patients Taking Suppressive Highly Active Antiretroviral Therapy. JAMA 1999, 282, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Maldarelli, F.; Wiegand, A.; Bernstein, B.; Hanna, G.J.; Brun, S.C.; Kempf, D.J.; Mellors, J.W.; Coffin, J.M.; King, M.S. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 3879–3884. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Coombs, R.W.; Chan, E.S.; Bosch, R.J.; Zheng, L.; Margolis, D.M.; Read, S.; Kallungal, B.; Chang, M.; Goecker, E.A.; et al. No effect of raltegravir intensification on viral replication markers in the blood of HIV-1-infected patients receiving antiretroviral therapy. J. Acquir. Immune Defic. Syndr. (1999) 2012, 59, 229–235. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; McMahon, J.H.; Chang, J.J.; Audsley, J.; Rhodes, A.; Tennakoon, S.; Dantanarayana, A.; Spelman, T.; Schmidt, T.; Kent, S.J.; et al. The effect of antiretroviral intensification with dolutegravir on residual virus replication in HIV-infected individuals: A randomised, placebo-controlled, double-blind trial. Lancet. HIV 2018, 5, e221–e230. [Google Scholar] [CrossRef]
- Chaillon, A.; Gianella, S.; Lada, S.M.; Perez-Santiago, J.; Jordan, P.; Ignacio, C.; Karris, M.; Richman, D.D.; Mehta, S.R.; Little, S.J.; et al. Size, Composition, and Evolution of HIV DNA Populations during Early Antiretroviral Therapy and Intensification with Maraviroc. J. Virol. 2018, 92, e01589-01517. [Google Scholar] [CrossRef]
- Kim, C.J.; Rousseau, R.; Huibner, S.; Kovacs, C.; Benko, E.; Shahabi, K.; Kandel, G.; Ostrowski, M.; Kaul, R. Impact of intensified antiretroviral therapy during early HIV infection on gut immunology and inflammatory blood biomarkers. AIDS (Lond. Engl.) 2017, 31. [Google Scholar] [CrossRef]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Holland, E.C. HIV-1 tat trans-activation requires the loop sequence within tar. Nature 1988, 334, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, C.L.; Symonds, G.; Kent, S.J.; Kelleher, A.D. Block and Lock HIV Cure Strategies to Control the Latent Reservoir. Front. Cell Infect. Microbiol. 2020, 10, 424. [Google Scholar] [CrossRef]
- Meredith, L.W.; Sivakumaran, H.; Major, L.; Suhrbier, A.; Harrich, D. Potent inhibition of HIV-1 replication by a Tat mutant. PLoS ONE 2009, 4, e7769. [Google Scholar] [CrossRef]
- Jin, H.; Li, D.; Sivakumaran, H.; Lor, M.; Rustanti, L.; Cloonan, N.; Wani, S.; Harrich, D. Shutdown of HIV-1 Transcription in T Cells by Nullbasic, a Mutant Tat Protein. mBio 2016, 7, e00518-00516. [Google Scholar] [CrossRef]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef]
- Vargas, B.; Giacobbi, N.S.; Sanyal, A.; Venkatachari, N.J.; Han, F.; Gupta, P.; Sluis-Cremer, N. Inhibitors of Signaling Pathways That Block Reversal of HIV-1 Latency. Antimicrob Agents Chemother 2019, 63, e01744-18. [Google Scholar] [CrossRef]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, 2510. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lei, R.; Le Duff, Y.; Li, J.; Guo, F.; Wainberg, M.A.; Liang, C. The CRISPR/Cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology 2015, 12, 22. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef] [PubMed]
- Mout, R.; Ray, M.; Lee, Y.-W.; Scaletti, F.; Rotello, V.M. In Vivo Delivery of CRISPR/Cas9 for Therapeutic Gene Editing: Progress and Challenges. Bioconjugate Chem. 2017, 28, 880–884. [Google Scholar] [CrossRef]
- Leslie, G.J.; Wang, J.; Richardson, M.W.; Haggarty, B.S.; Hua, K.L.; Duong, J.; Secreto, A.J.; Jordon, A.P.; Romano, J.; Kumar, K.E.; et al. Potent and Broad Inhibition of HIV-1 by a Peptide from the gp41 Heptad Repeat-2 Domain Conjugated to the CXCR4 Amino Terminus. PLoS Pathog. 2016, 12, e1005983. [Google Scholar] [CrossRef]
- Maldini, C.R.; Claiborne, D.T.; Okawa, K.; Chen, T.; Dopkin, D.L.; Shan, X.; Power, K.A.; Trifonova, R.T.; Krupp, K.; Phelps, M.; et al. Dual CD4-based CAR T cells with distinct costimulatory domains mitigate HIV pathogenesis in vivo. Nat. Med. 2020, 26, 1776–1787. [Google Scholar] [CrossRef]
- Ventoso, I.; Navarro, J.; Munoz, M.A.; Carrasco, L. Involvement of HIV-1 protease in virus-induced cell killing. Antivir. Res. 2005, 66, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Algeciras-Schimnich, A.; Belzacq-Casagrande, A.-S.; Bren, G.D.; Nie, Z.; Taylor, J.A.; Rizza, S.A.; Brenner, C.; Badley, A.D. Analysis of HIV Protease Killing Through Caspase 8 Reveals a Novel Interaction Between Caspase 8 and Mitochondria. Open Virol. J. 2007, 1, 39–46. [Google Scholar] [CrossRef]
- Sainski, A.M.; Natesampillai, S.; Cummins, N.W.; Bren, G.D.; Taylor, J.; Saenz, D.T.; Poeschla, E.M.; Badley, A.D. The HIV-1-Specific Protein Casp8p41 Induces Death of Infected Cells through Bax/Bak. J. Virol. 2011, 85, 7965–7975. [Google Scholar] [CrossRef]
- Sainski, A.M.; Dai, H.; Natesampillai, S.; Pang, Y.-P.; Bren, G.D.; Cummins, N.W.; Correia, C.; Meng, X.W.; Tarara, J.E.; Ramirez-Alvarado, M.; et al. Casp8p41 generated by HIV protease kills CD4 T cells through direct Bak activation. J. Cell Biol. 2014, 206, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Natesampillai, S.; Cummins, N.W.; Nie, Z.; Sampath, R.; Baker, J.V.; Henry, K.; Pinzone, M.; O’Doherty, U.; Polley, E.C.; Bren, G.D.; et al. HIV Protease-Generated Casp8p41, When Bound and Inactivated by Bcl2, Is Degraded by the Proteasome. J. Virol. 2018, 92, e00037-18. [Google Scholar] [CrossRef] [PubMed]
- Alto, A.; Natesampillai, S.; Chandrasekar, A.P.; Krogman, A.; Misra, A.; Shweta, F.; VanLith, C.; Yao, J.D.; Cummins, N.W.; Badley, A.D. The Combination of Venetoclax and Ixazomib Selectively and Efficiently Kills HIV-Infected Cell Lines but Has Unacceptable Toxicity in Primary Cell Models. J. Virol. 2021, 95, e00138-00121. [Google Scholar] [CrossRef]
- Ren, Y.; Huang, S.H.; Patel, S.; Alberto, W.D.C.; Magat, D.; Ahimovic, D.; Macedo, A.B.; Durga, R.; Chan, D.; Zale, E.; et al. BCL-2 antagonism sensitizes cytotoxic T cell–resistant HIV reservoirs to elimination ex vivo. J. Clin. Investig. 2020, 130, 2542–2559. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Alegre, M.L.; Vandenabeele, P.; Depierreux, M.; Florquin, S.; Deschodt-Lanckman, M.; Flamand, V.; Moser, M.; Leo, O.; Urbain, J.; Fiers, W.; et al. Cytokine release syndrome induced by the 145-2C11 anti-CD3 monoclonal antibody in mice: Prevention by high doses of methylprednisolone. J. Immunol. 1991, 146, 1184–1191. [Google Scholar] [PubMed]
- Hütter, G. Stem cell transplantation in strategies for curing HIV/AIDS. AIDS Res. Ther. 2016, 13, 31. [Google Scholar] [CrossRef]
- Kordelas, L.; Verheyen, J.; Beelen, D.W.; Horn, P.A.; Heinold, A.; Kaiser, R.; Trenschel, R.; Schadendorf, D.; Dittmer, U.; Esser, S. Shift of HIV tropism in stem-cell transplantation with CCR5 Delta32 mutation. N. Engl. J. Med. 2014, 371, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Koelsch, K.K.; Rasmussen, T.A.; Hey-Nguyen, W.J.; Pearson, C.; Xu, Y.; Bailey, M.; Marks, K.H.; Sasson, S.C.; Taylor, M.S.; Tantau, R.; et al. Impact of Allogeneic Hematopoietic Stem Cell Transplantation on the HIV Reservoir and Immune Response in 3 HIV-Infected Individuals. J. Acquir. Immune Defic. Syndr. 2017, 75, 328–337. [Google Scholar] [CrossRef]
- Burton, D.R.; Barbas, C.F.; Persson, M.A.; Koenig, S.; Chanock, R.M.; Lerner, R.A. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc. Natl. Acad. Sci. USA 1991, 88, 10134–10137. [Google Scholar] [CrossRef]
- Klasse, P.J. Neutralization of Virus Infectivity by Antibodies: Old Problems in New Perspectives. Adv. Biol. 2014, 2014, 157895. [Google Scholar] [CrossRef]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar] [CrossRef] [PubMed]
- Forthal, D.; Hope, T.J.; Alter, G. New paradigms for functional HIV-specific nonneutralizing antibodies. Curr. Opin. HIV AIDS 2013, 8, 393–401. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Z.-Y.; Li, Y.; Hogerkorp, C.-M.; Schief, W.R.; Seaman, M.S.; Zhou, T.; Schmidt, S.D.; Wu, L.; Xu, L.; et al. Rational Design of Envelope Identifies Broadly Neutralizing Human Monoclonal Antibodies to HIV-1. Science 2010, 329, 856–861. [Google Scholar] [CrossRef]
- McCoy, L.E. The expanding array of HIV broadly neutralizing antibodies. Retrovirology 2018, 15, 70. [Google Scholar] [CrossRef]
- Moldt, B.; Rakasz, E.G.; Schultz, N.; Chan-Hui, P.-Y.; Swiderek, K.; Weisgrau, K.L.; Piaskowski, S.M.; Bergman, Z.; Watkins, D.I.; Poignard, P.; et al. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 18921–18925. [Google Scholar] [CrossRef]
- Shingai, M.; Donau, O.K.; Plishka, R.J.; Buckler-White, A.; Mascola, J.R.; Nabel, G.J.; Nason, M.C.; Montefiori, D.; Moldt, B.; Poignard, P.; et al. Passive transfer of modest titers of potent and broadly neutralizing anti-HIV monoclonal antibodies block SHIV infection in macaques. J. Exp. Med. 2014, 211, 2061–2074. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O.; Wang, L.; Joyce, M.G.; Yang, Z.-Y.; Balazs, A.B.; Cheng, C.; Ko, S.-Y.; Kong, W.-P.; Rudicell, R.S.; Georgiev, I.S.; et al. Broadly Neutralizing Human Immunodeficiency Virus Type 1 Antibody Gene Transfer Protects Nonhuman Primates from Mucosal Simian-Human Immunodeficiency Virus Infection. J. Virol. 2015, 89, 8334–8345. [Google Scholar] [CrossRef] [PubMed]
- Gautam, R.; Nishimura, Y.; Pegu, A.; Nason, M.C.; Klein, F.; Gazumyan, A.; Golijanin, J.; Buckler-White, A.; Sadjadpour, R.; Wang, K.; et al. A single injection of anti-HIV-1 antibodies protects against repeated SHIV challenges. Nature 2016, 533, 105–109. [Google Scholar] [CrossRef]
- Julg, B.; Sok, D.; Schmidt, S.D.; Abbink, P.; Newman, R.M.; Broge, T.; Linde, C.; Nkolola, J.; Le, K.; Su, D.; et al. Protective Efficacy of Broadly Neutralizing Antibodies with Incomplete Neutralization Activity against Simian-Human Immunodeficiency Virus in Rhesus Monkeys. J. Virol. 2017, 91, e01187-01117. [Google Scholar] [CrossRef]
- Julg, B.; Tartaglia, L.J.; Keele, B.F.; Wagh, K.; Pegu, A.; Sok, D.; Abbink, P.; Schmidt, S.D.; Wang, K.; Chen, X.; et al. Broadly neutralizing antibodies targeting the HIV-1 envelope V2 apex confer protection against a clade C SHIV challenge. Sci. Transl. Med. 2017, 9, eaal1321. [Google Scholar] [CrossRef]
- Ledgerwood, J.E.; Coates, E.E.; Yamshchikov, G.; Saunders, J.G.; Holman, L.; Enama, M.E.; DeZure, A.; Lynch, R.M.; Gordon, I.; Plummer, S.; et al. Safety, pharmacokinetics and neutralization of the broadly neutralizing HIV-1 human monoclonal antibody VRC01 in healthy adults. Clin. Exp. Immunol. 2015, 182, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Gaudinski, M.R.; Coates, E.E.; Houser, K.V.; Chen, G.L.; Yamshchikov, G.; Saunders, J.G.; Holman, L.A.; Gordon, I.; Plummer, S.; Hendel, C.S.; et al. Safety and pharmacokinetics of the Fc-modified HIV-1 human monoclonal antibody VRC01LS: A Phase 1 open-label clinical trial in healthy adults. PLOS Med. 2018, 15, e1002493. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, L.; Ledgerwood, J.; Grunenberg, N.; Bailer, R.; Isaacs, A.; Seaton, K.; Mayer, K.H.; Capparelli, E.; Corey, L.; et al. Population pharmacokinetics analysis of VRC01, an HIV-1 broadly neutralizing monoclonal antibody, in healthy adults. mAbs 2017, 9, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Niessl, J.; Baxter, A.E.; Mendoza, P.; Jankovic, M.; Cohen, Y.Z.; Butler, A.L.; Lu, C.-L.; Dubé, M.; Shimeliovich, I.; Gruell, H.; et al. Combination anti-HIV-1 antibody therapy is associated with increased virus-specific T cell immunity. Nat. Med. 2020, 26, 222–227. [Google Scholar] [CrossRef]
- Caskey, M.; Klein, F.; Lorenzi, J.C.C.; Seaman, M.S.; West, A.P.; Buckley, N.; Kremer, G.; Nogueira, L.; Braunschweig, M.; Scheid, J.F.; et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 2015, 522, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Caskey, M.; Schoofs, T.; Gruell, H.; Settler, A.; Karagounis, T.; Kreider, E.F.; Murrell, B.; Pfeifer, N.; Nogueira, L.; Oliveira, T.Y.; et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 2017, 23, 185–191. [Google Scholar] [CrossRef]
- Crowell, T.A.; Colby, D.J.; Pinyakorn, S.; Sacdalan, C.; Pagliuzza, A.; Intasan, J.; Benjapornpong, K.; Tangnaree, K.; Chomchey, N.; Kroon, E.; et al. Safety and efficacy of VRC01 broadly neutralising antibodies in adults with acutely treated HIV (RV397): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet. HIV 2019, 6, e297–e306. [Google Scholar] [CrossRef]
- Reh, L.; Magnus, C.; Schanz, M.; Weber, J.; Uhr, T.; Rusert, P.; Trkola, A. Capacity of Broadly Neutralizing Antibodies to Inhibit HIV-1 Cell-Cell Transmission Is Strain- and Epitope-Dependent. PLoS Pathog. 2015, 11, e1004966. [Google Scholar] [CrossRef]
- Li, H.; Zony, C.; Chen, P.; Chen, B.K. Reduced Potency and Incomplete Neutralization of Broadly Neutralizing Antibodies against Cell-to-Cell Transmission of HIV-1 with Transmitted Founder Envs. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Parsons, M.S.; Lloyd, S.B.; Lee, W.S.; Kristensen, A.B.; Amarasena, T.; Center, R.J.; Keele, B.F.; Lifson, J.D.; LaBranche, C.C.; Montefiori, D.; et al. Partial efficacy of a broadly neutralizing antibody against cell-associated SHIV infection. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Walker, L.M.; Huber, M.; Doores, K.J.; Falkowska, E.; Pejchal, R.; Julien, J.-P.; Wang, S.-K.; Ramos, A.; Chan-Hui, P.-Y.; Moyle, M.; et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 2011, 477, 466–470. [Google Scholar] [CrossRef]
- Kong, R.; Louder, M.K.; Wagh, K.; Bailer, R.T.; deCamp, A.; Greene, K.; Gao, H.; Taft, J.D.; Gazumyan, A.; Liu, C.; et al. Improving Neutralization Potency and Breadth by Combining Broadly Reactive HIV-1 Antibodies Targeting Major Neutralization Epitopes. J. Virol. 2015, 89, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Wagh, K.; Seaman, M.S.; Zingg, M.; Fitzsimons, T.; Barouch, D.H.; Burton, D.R.; Connors, M.; Ho, D.D.; Mascola, J.R.; Nussenzweig, M.C.; et al. Potential of conventional & bispecific broadly neutralizing antibodies for prevention of HIV-1 subtype A, C & D infections. PLoS Pathog. 2018, 14, e1006860. [Google Scholar] [CrossRef]
- Nishimura, Y.; Gautam, R.; Chun, T.-W.; Sadjadpour, R.; Foulds, K.E.; Shingai, M.; Klein, F.; Gazumyan, A.; Golijanin, J.; Donaldson, M.; et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature 2017, 543, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Donau, O.K.; Dias, J.; Ferrando-Martinez, S.; Jesteadt, E.; Sadjadpour, R.; Gautam, R.; Buckler-White, A.; Geleziunas, R.; Koup, R.A.; et al. Immunotherapy during the acute SHIV infection of macaques confers long-term suppression of viremia. J. Exp. Med. 2020, 218. [Google Scholar] [CrossRef] [PubMed]
- Borducchi, E.N.; Liu, J.; Nkolola, J.P.; Cadena, A.M.; Yu, W.-H.; Fischinger, S.; Broge, T.; Abbink, P.; Mercado, N.B.; Chandrashekar, A.; et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 2018, 563, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.C.; Schuetz, A.; Imerbsin, R.; Silsorn, D.; Pegu, A.; Inthawong, D.; Sopanaporn, J.; Visudhiphan, P.; Chuenarom, W.; Keawboon, B.; et al. TLR7 agonist, N6-LS and PGT121 delayed viral rebound in SHIV-infected macaques after antiretroviral therapy interruption. PLoS Pathog. 2021, 17, e1009339. [Google Scholar] [CrossRef]
- Lim, S.-Y.; Osuna, C.E.; Lee, J.; Silva-Ayala, D.; Vikhe, P.; Chen, E.; Lundgren, S.; Eliot, M.; Schalk, D.; Schultz-Darken, N.; et al. Combination IL-15 Therapy in a SHIV NHP Model. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 10 December 2021. [Google Scholar]
- Gebara, N.Y.; El Kamari, V.; Rizk, N. HIV-1 elite controllers: An immunovirological review and clinical perspectives. J. Virus Erad. 2019, 5, 163–166. [Google Scholar] [CrossRef]
- Gray, G.; Buchbinder, S.; Duerr, A. Overview of STEP and Phambili trial results: Two phase IIb test-of-concept studies investigating the efficacy of MRK adenovirus type 5 gag/pol/nef subtype B HIV vaccine. Curr. Opin. HIV AIDS 2010, 5, 357–361. [Google Scholar] [CrossRef]
- Bekker, L.-G.; Moodie, Z.; Grunenberg, N.; Laher, F.; Tomaras, G.D.; Cohen, K.W.; Allen, M.; Malahleha, M.; Mngadi, K.; Daniels, B.; et al. Subtype C ALVAC-HIV and bivalent subtype C gp120/MF59 HIV-1 vaccine in low-risk, HIV-uninfected, South African adults: A phase 1/2 trial. Lancet. HIV 2018, 5, e366–e378. [Google Scholar] [CrossRef]
- Laher, F.; Moodie, Z.; Cohen, K.W.; Grunenberg, N.; Bekker, L.-G.; Allen, M.; Frahm, N.; Yates, N.L.; Morris, L.; Malahleha, M.; et al. Safety and immune responses after a 12-month booster in healthy HIV-uninfected adults in HVTN 100 in South Africa: A randomized double-blind placebo-controlled trial of ALVAC-HIV (vCP2438) and bivalent subtype C gp120/MF59 vaccines. PLoS Med. 2020, 17, e1003038. [Google Scholar] [CrossRef]
- Gray, G.E.; Bekker, L.-G.; Laher, F.; Malahleha, M.; Allen, M.; Moodie, Z.; Grunenberg, N.; Huang, Y.; Grove, D.; Prigmore, B.; et al. Vaccine Efficacy of ALVAC-HIV and Bivalent Subtype C gp120–MF59 in Adults. N. Engl. J. Med. 2021, 384, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Jawad, S.; Ondondo, B.; van Hateren, A.; Gardner, A.; Elliott, T.; Korber, B.; Hanke, T. Increased Valency of Conserved-mosaic Vaccines Enhances the Breadth and Depth of Epitope Recognition. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 375–384. [Google Scholar] [CrossRef]
- Barouch, D.H.; Tomaka, F.L.; Wegmann, F.; Stieh, D.J.; Alter, G.; Robb, M.L.; Michael, N.L.; Peter, L.; Nkolola, J.P.; Borducchi, E.N.; et al. Evaluation of a mosaic HIV-1 vaccine in a multicentre, randomised, double-blind, placebo-controlled, phase 1/2a clinical trial (APPROACH) and in rhesus monkeys (NHP 13-19). Lancet (Lond. Engl. ) 2018, 392, 232–243. [Google Scholar] [CrossRef]
- Fischer, W.; Perkins, S.; Theiler, J.; Bhattacharya, T.; Yusim, K.; Funkhouser, R.; Kuiken, C.; Haynes, B.; Letvin, N.L.; Walker, B.D.; et al. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat. Med. 2007, 13, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Johnson & Johnson and Global Partners Announce Results from Phase 2b Imbokodo HIV Vaccine Clinical Trial in Young Women in Sub-Saharan Africa. Available online: https://www.jnj.com/johnson-johnson-and-global-partners-announce-results-from-phase-2b-imbokodo-hiv-vaccine-clinical-trial-in-young-women-in-sub-saharan-africa (accessed on 13 September 2021).
- Mothe, B.; Climent, N.; Plana, M.; Rosàs, M.; Jiménez, J.L.; Muñoz-Fernández, M.; Puertas, M.C.; Carrillo, J.; Gonzalez, N.; León, A.; et al. Safety and immunogenicity of a modified vaccinia Ankara-based HIV-1 vaccine (MVA-B) in HIV-1-infected patients alone or in combination with a drug to reactivate latent HIV-1. J. Antimicrob. Chemother. 2015, 70, 1833–1842. [Google Scholar] [CrossRef]
- Hansen, S.G.; Vieville, C.; Whizin, N.; Coyne-Johnson, L.; Siess, D.C.; Drummond, D.D.; Legasse, A.W.; Axthelm, M.K.; Oswald, K.; Trubey, C.M.; et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med. 2009, 15, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Piatak, M., Jr.; Ventura, A.B.; Hughes, C.M.; Gilbride, R.M.; Ford, J.C.; Oswald, K.; Shoemaker, R.; Li, Y.; Lewis, M.S.; et al. Immune clearance of highly pathogenic SIV infection. Nature 2013, 502, 100–104. [Google Scholar] [CrossRef]
- Murray, S.E.; Nesterenko, P.A.; Vanarsdall, A.L.; Munks, M.W.; Smart, S.M.; Veziroglu, E.M.; Sagario, L.C.; Lee, R.; Claas, F.H.J.; Doxiadis, I.I.N.; et al. Fibroblast-adapted human CMV vaccines elicit predominantly conventional CD8 T cell responses in humans. J. Exp. Med. 2017, 214, 1889–1899. [Google Scholar] [CrossRef]
- Combadière, B.; Beaujean, M.; Chaudesaigues, C.; Vieillard, V. Peptide-Based Vaccination for Antibody Responses Against HIV. Vaccines 2019, 7, 105. [Google Scholar] [CrossRef]
- Belyakov, I.M.; Derby, M.A.; Ahlers, J.D.; Kelsall, B.L.; Earl, P.; Moss, B.; Strober, W.; Berzofsky, J.A. Mucosal immunization with HIV-1 peptide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc. Natl. Acad. Sci. USA 1998, 95, 1709–1714. [Google Scholar] [CrossRef]
- Kelleher, A.D.; Emery, S.; Cunningham, P.; Duncombe, C.; Carr, A.; Golding, H.; Forde, S.; Hudson, J.; Roggensack, M.; Forrest, B.D.; et al. Safety and immunogenicity of UBI HIV-1MN octameric V3 peptide vaccine administered by subcutaneous injection. AIDS Res. Hum. Retrovir. 1997, 13, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Gorse, G.J.; Keefer, M.C.; Belshe, R.B.; Matthews, T.J.; Forrest, B.D.; Hsieh, R.H.; Koff, W.C.; Hanson, C.V.; Dolin, R.; Weinhold, K.J.; et al. A dose-ranging study of a prototype synthetic HIV-1MN V3 branched peptide vaccine. The National Institute of Allergy and Infectious Diseases AIDS Vaccine Evaluation Group. J. Infect. Dis. 1996, 173, 330–339. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Flynn, N.M.; Forthal, D.N.; Harro, C.D.; Judson, F.N.; Mayer, K.H.; Para, M.F. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J. Infect. Dis. 2005, 191, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Vieillard, V.; Combadière, B.; Tubiana, R.; Launay, O.; Pialoux, G.; Cotte, L.; Girard, P.-M.; Simon, A.; Dudoit, Y.; Reynes, J.; et al. HIV therapeutic vaccine enhances non-exhausted CD4+ T cells in a randomised phase 2 trial. NPJ Vaccines 2019, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Pollard, R.B.; Rockstroh, J.K.; Pantaleo, G.; Asmuth, D.M.; Peters, B.; Lazzarin, A.; Garcia, F.; Ellefsen, K.; Podzamczer, D.; van Lunzen, J.; et al. Safety and efficacy of the peptide-based therapeutic vaccine for HIV-1, Vacc-4x: A phase 2 randomised, double-blind, placebo-controlled trial. Lancet. Infect. Dis. 2014, 14, 291–300. [Google Scholar] [CrossRef]
- Rockstroh, J.K.; Asmuth, D.; Pantaleo, G.; Clotet, B.; Podzamczer, D.; van Lunzen, J.; Arastéh, K.; Mitsuyasu, R.; Peters, B.; Silvia, N.; et al. Re-boost immunizations with the peptide-based therapeutic HIV vaccine, Vacc-4x, restores geometric mean viral load set-point during treatment interruption. PLoS ONE 2019, 14, e0210965. [Google Scholar] [CrossRef]
- Chupradit, K.; Moonmuang, S.; Nangola, S.; Kitidee, K.; Yasamut, U.; Mougel, M.; Tayapiwatana, C. Current Peptide and Protein Candidates Challenging HIV Therapy beyond the Vaccine Era. Viruses 2017, 9, 281. [Google Scholar] [CrossRef]
- Dando, T.M.; Perry, C.M. Enfuvirtide. Drugs 2003, 63, 2755–2766. [Google Scholar] [CrossRef]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef]
- Bi, W.; Xu, W.; Cheng, L.; Xue, J.; Wang, Q.; Yu, F.; Xia, S.; Wang, Q.; Li, G.; Qin, C.; et al. IgG Fc-binding motif-conjugated HIV-1 fusion inhibitor exhibits improved potency and in vivo half-life: Potential application in combination with broad neutralizing antibodies. PLoS Pathog. 2019, 15, e1008082. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.A.; Bischof, G.F.; Shin, Y.C.; Lauer, W.A.; Gonzalez-Nieto, L.; Watkins, D.I.; Rakasz, E.G.; Lifson, J.D.; Desrosiers, R.C. Vaccine protection against SIVmac239 acquisition. Proc. Natl. Acad. Sci. USA 2019, 116, 1739–1744. [Google Scholar] [CrossRef]
- Martins, M.A.; Gonzalez-Nieto, L.; Ricciardi, M.J.; Bailey, V.K.; Dang, C.M.; Bischof, G.F.; Pedreño-Lopez, N.; Pauthner, M.G.; Burton, D.R.; Parks, C.L.; et al. Rectal Acquisition of Simian Immunodeficiency Virus (SIV) SIVmac239 Infection despite Vaccine-Induced Immune Responses against the Entire SIV Proteome. J. Virol. 2020, 94, e00979-00920. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fernandez, M.E.; Presicce, P.; Chougnet, C.A. Homeostasis and function of regulatory T cells in HIV/SIV infection. J. Virol. 2012, 86, 10262–10269. [Google Scholar] [CrossRef]
- Tran, T.A.; de Goër de Herve, M.G.; Hendel-Chavez, H.; Dembele, B.; Le Névot, E.; Abbed, K.; Pallier, C.; Goujard, C.; Gasnault, J.; Delfraissy, J.F.; et al. Resting regulatory CD4 T cells: A site of HIV persistence in patients on long-term effective antiretroviral therapy. PLoS ONE 2008, 3, e3305. [Google Scholar] [CrossRef] [PubMed]
- Allers, K.; Loddenkemper, C.; Hofmann, J.; Unbehaun, A.; Kunkel, D.; Moos, V.; Kaup, F.J.; Stahl-Hennig, C.; Sauermann, U.; Epple, H.J.; et al. Gut mucosal FOXP3+ regulatory CD4+ T cells and Nonregulatory CD4+ T cells are differentially affected by simian immunodeficiency virus infection in rhesus macaques. J. Virol. 2010, 84, 3259–3269. [Google Scholar] [CrossRef]
- Bi, X.; Suzuki, Y.; Gatanaga, H.; Oka, S. High frequency and proliferation of CD4+ FOXP3+ Treg in HIV-1-infected patients with low CD4 counts. Eur. J. Immunol. 2009, 39, 301–309. [Google Scholar] [CrossRef]
- Weiss, L.; Donkova-Petrini, V.; Caccavelli, L.; Balbo, M.; Carbonneil, C.; Levy, Y. Human immunodeficiency virus-driven expansion of CD4+CD25+ regulatory T cells, which suppress HIV-specific CD4 T-cell responses in HIV-infected patients. Blood 2004, 104, 3249–3256. [Google Scholar] [CrossRef]
- Nikolova, M.; Lelievre, J.D.; Carriere, M.; Bensussan, A.; Levy, Y. Regulatory T cells differentially modulate the maturation and apoptosis of human CD8+ T-cell subsets. Blood 2009, 113, 4556–4565. [Google Scholar] [CrossRef]
- Nilsson, J.; Boasso, A.; Velilla, P.A.; Zhang, R.; Vaccari, M.; Franchini, G.; Shearer, G.M.; Andersson, J.; Chougnet, C. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood 2006, 108, 3808–3817. [Google Scholar] [CrossRef]
- Elahi, S.; Dinges, W.L.; Lejarcegui, N.; Laing, K.J.; Collier, A.C.; Koelle, D.M.; McElrath, M.J.; Horton, H. Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat. Med. 2011, 17, 989–995. [Google Scholar] [CrossRef]
- Queen, C.; Schneider, W.P.; Selick, H.E.; Payne, P.W.; Landolfi, N.F.; Duncan, J.F.; Avdalovic, N.M.; Levitt, M.; Junghans, R.P.; Waldmann, T.A. A humanized antibody that binds to the interleukin 2 receptor. Proc. Natl. Acad. Sci. USA 1989, 86, 10029–10033. [Google Scholar] [CrossRef]
- Williams, D.P.; Parker, K.; Bacha, P.; Bishai, W.; Borowski, M.; Genbauffe, F.; Strom, T.B.; Murphy, J.R. Diphtheria toxin receptor binding domain substitution with interleukin-2: Genetic construction and properties of a diphtheria toxin-related interleukin-2 fusion protein. Protein Eng. 1987, 1, 493–498. [Google Scholar] [CrossRef]
- Collier, R.J. Diphtheria toxin: Mode of action and structure. Bacteriol. Rev. 1975, 39, 54–85. [Google Scholar] [CrossRef]
- Atchison, E.; Eklund, J.; Martone, B.; Wang, L.; Gidron, A.; Macvicar, G.; Rademaker, A.; Goolsby, C.; Marszalek, L.; Kozlowski, J.; et al. A pilot study of denileukin diftitox (DD) in combination with high-dose interleukin-2 (IL-2) for patients with metastatic renal cell carcinoma (RCC). J. Immuno. (Hagerstown Md. 1997) 2010, 33, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Kaminetzky, D.; Hymes, K.B. Denileukin diftitox for the treatment of cutaneous T-cell lymphoma. Biol. Targets Ther. 2008, 2, 717–724. [Google Scholar]
- Baur, A.S.; Lutz, M.B.; Schierer, S.; Beltrame, L.; Theiner, G.; Zinser, E.; Ostalecki, C.; Heidkamp, G.; Haendle, I.; Erdmann, M.; et al. Denileukin diftitox (ONTAK) induces a tolerogenic phenotype in dendritic cells and stimulates survival of resting Treg. Blood 2013, 122, 2185–2194. [Google Scholar] [CrossRef]
- Foss, F.M.; Sjak-Shie, N.; Goy, A.; Jacobsen, E.; Advani, R.; Smith, M.R.; Komrokji, R.; Pendergrass, K.; Bolejack, V. A multicenter phase II trial to determine the safety and efficacy of combination therapy with denileukin diftitox and cyclophosphamide, doxorubicin, vincristine and prednisone in untreated peripheral T-cell lymphoma: The CONCEPT study. Leuk. Lymphoma 2013, 54, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Telang, S.; Rasku, M.A.; Clem, A.L.; Carter, K.; Klarer, A.C.; Badger, W.R.; Milam, R.A.; Rai, S.N.; Pan, J.; Gragg, H.; et al. Phase II trial of the regulatory T cell-depleting agent, denileukin diftitox, in patients with unresectable stage IV melanoma. BMC Cancer 2011, 11, 515. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, Q.; Zhang, H.; Bronson, R.T.; Madsen, J.C.; Sachs, D.H.; Huang, C.A.; Wang, Z. Ontak-like human IL-2 fusion toxin. J. Immunol. Methods 2017, 448, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Peraino, J.S.; Zhang, H.; Rajasekera, P.V.; Wei, M.; Madsen, J.C.; Sachs, D.H.; Huang, C.A.; Wang, Z. Diphtheria toxin-based bivalent human IL-2 fusion toxin with improved efficacy for targeting human CD25(+) cells. J. Immunol. Methods 2014, 405, 57–66. [Google Scholar] [CrossRef][Green Version]
- Ishida, T.; Ueda, R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer Sci. 2006, 97, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in tumor immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef]
- Iellem, A.; Mariani, M.; Lang, R.; Recalde, H.; Panina-Bordignon, P.; Sinigaglia, F.; D’Ambrosio, D. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J. Exp. Med. 2001, 194, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, L.; Vanhorn-Ali, Z.; Alkhatib, G. Multiple determinants are involved in HIV coreceptor use as demonstrated by CCR4/CCL22 interaction in peripheral blood mononuclear cells (PBMCs). J. Leukoc. Biol 2002, 72, 1063–1074. [Google Scholar] [PubMed]
- Wang, Z.; Pratts, S.G.; Zhang, H.; Spencer, P.J.; Yu, R.; Tonsho, M.; Shah, J.A.; Tanabe, T.; Powell, H.R.; Huang, C.A.; et al. Treg depletion in non-human primates using a novel diphtheria toxin-based anti-human CCR4 immunotoxin. Mol. Oncol. 2016, 10, 553–565. [Google Scholar] [CrossRef]
- Ni, X.; Jorgensen, J.L.; Goswami, M.; Challagundla, P.; Decker, W.K.; Kim, Y.H.; Duvic, M.A. Reduction of regulatory T cells by Mogamulizumab, a defucosylated anti-CC chemokine receptor 4 antibody, in patients with aggressive/refractory mycosis fungoides and Sezary syndrome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Ishida, T.; Hatake, K.; Taniwaki, M.; Ando, K.; Tobinai, K.; Fujimoto, K.; Yamamoto, K.; Miyamoto, T.; Uike, N.; et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 1157–1163. [Google Scholar] [CrossRef]
- Coe, D.; Begom, S.; Addey, C.; White, M.; Dyson, J.; Chai, J.-G. Depletion of regulatory T cells by anti-GITR mAb as a novel mechanism for cancer immunotherapy. Cancer Immunol. Immunother. 2010, 59, 1367–1377. [Google Scholar] [CrossRef]
- Nocentini, G.; Giunchi, L.; Ronchetti, S.; Krausz, L.T.; Bartoli, A.; Moraca, R.; Migliorati, G.; Riccardi, C. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 6216–6221. [Google Scholar] [CrossRef]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, F.; Youngnak, P.; Hashiguchi, M.; Nishioka, T.; Takahashi, T.; Sakaguchi, S.; Ishikawa, I.; Azuma, M. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J. Immunol. (Baltimore Md. 1950) 2004, 172, 7306–7314. [Google Scholar] [CrossRef]
- Stephens, G.L.; McHugh, R.S.; Whitters, M.J.; Young, D.A.; Luxenberg, D.; Carreno, B.M.; Collins, M.; Shevach, E.M. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J. Immunol. (Baltimore Md. 1950) 2004, 173, 5008–5020. [Google Scholar] [CrossRef] [PubMed]
- Mahne, A.E.; Mauze, S.; Joyce-Shaikh, B.; Xia, J.; Bowman, E.P.; Beebe, A.M.; Cua, D.J.; Jain, R. Dual Roles for Regulatory T-cell Depletion and Costimulatory Signaling in Agonistic GITR Targeting for Tumor Immunotherapy. Cancer Res. 2017, 77, 1108–1118. [Google Scholar] [CrossRef]
- Lahey, T.P.; Loisel, S.D.; Wieland-Alter, W. Glucocorticoid-induced tumor necrosis factor receptor family-related protein triggering enhances HIV-specific CD4+ T cell cytokine secretion and protects HIV-specific CD4+ T cells from apoptosis. J. Infect. Dis. 2007, 196, 43–49. [Google Scholar] [CrossRef]
- Schoenhals, J.E.; Cushman, T.R.; Barsoumian, H.B.; Li, A.; Cadena, A.P.; Niknam, S.; Younes, A.I.; Caetano, M.D.S.; Cortez, M.A.; Welsh, J.W. Anti-glucocorticoid-induced Tumor Necrosis Factor-Related Protein (GITR) Therapy Overcomes Radiation-Induced Treg Immunosuppression and Drives Abscopal Effects. Front. Immunol 2018, 9, 2170. [Google Scholar] [CrossRef]
- Shrimali, R.; Ahmad, S.; Berrong, Z.; Okoev, G.; Matevosyan, A.; Razavi, G.S.E.; Petit, R.; Gupta, S.; Mkrtichyan, M.; Khleif, S.N. Agonist anti-GITR antibody significantly enhances the therapeutic efficacy of Listeria monocytogenes-based immunotherapy. J. Immunol. Therapy Cancer 2017, 5, 64. [Google Scholar] [CrossRef]
- Appelbaum, F.R.; Sullivan, K.M.; Buckner, C.D.; Clift, R.A.; Deeg, H.J.; Fefer, A.; Hill, R.; Mortimer, J.; Neiman, P.E.; Sanders, J.E. Treatment of malignant lymphoma in 100 patients with chemotherapy, total body irradiation, and marrow transplantation. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1987, 5, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- McCune, W.J.; Golbus, J.; Zeldes, W.; Bohlke, P.; Dunne, R.; Fox, D.A. Clinical and Immunologic Effects of Monthly Administration of Intravenous Cyclophosphamide in Severe Systemic Lupus Erythematosus. N. Engl. J. Med. 1988, 318, 1423–1431. [Google Scholar] [CrossRef]
- Gladstone, D.E.; Prestrud, A.A.; Pradhan, A.; Styler, M.J.; Topolsky, D.L.; Crilley, P.A.; Hoch, S.; Huppert, A.; Brodsky, I. High-dose cyclophosphamide for severe systemic lupus erythematosus. Lupus 2002, 11, 405–410. [Google Scholar] [CrossRef]
- Petri, M.; Brodsky, R.A.; Jones, R.J.; Gladstone, D.; Fillius, M.; Magder, L.S. High Dose Cyclophosphamide versus Monthly Intravenous Cyclophosphamide for Systemic Lupus Erythematosus. Arthritis Rheum. 2010, 62, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Glode, L.M.; Barqawi, A.; Crighton, F.; Crawford, E.D.; Kerbel, R. Metronomic therapy with cyclophosphamide and dexamethasone for prostate carcinoma. Cancer 2003, 98, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Lutsiak, M.E.C.; Semnani, R.T.; De Pascalis, R.; Kashmiri, S.V.S.; Schlom, J.; Sabzevari, H. Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005, 105, 2862–2868. [Google Scholar] [CrossRef] [PubMed]
- Ikezawa, Y.; Nakazawa, M.; Tamura, C.; Takahashi, K.; Minami, M.; Ikezawa, Z. Cyclophosphamide decreases the number, percentage and the function of CD25+ CD4+ regulatory T cells, which suppress induction of contact hypersensitivity. J. Dermatol. Sci. 2005, 39, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Heylmann, D.; Bauer, M.; Becker, H.; van Gool, S.; Bacher, N.; Steinbrink, K.; Kaina, B. Human CD4+CD25+ Regulatory T Cells Are Sensitive to Low Dose Cyclophosphamide: Implications for the Immune Response. PLoS ONE 2013, 8, e83384. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Cao, Y.; Lei, Z.; Yang, Z.; Zhang, B.; Huang, B. Selective Depletion of CD4+CD25+Foxp3+ Regulatory T Cells by Low-Dose Cyclophosphamide Is Explained by Reduced Intracellular ATP Levels. Cancer Res. 2010, 70, 4850–4858. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef]
- Kleinman, A.J.; Sivanandham, R.; Sette, P.; Brocca-Cofano, E.; McAndrews, C.; Keele, B.F.; Pandrea, I.; Apetrei, C. Lack of Specific Regulatory T Cell Depletion and Cytoreduction Associated with Extensive Toxicity After Administration of Low and High Doses of Cyclophosphamide. AIDS Res. Hum. Retrovir. 2021. [Google Scholar] [CrossRef]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.D.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral Immune Evasion Due to Persistence of Activated T Cells Without Effector Function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Chew, G.M.; Fujita, T.; Webb, G.M.; Burwitz, B.J.; Wu, H.L.; Reed, J.S.; Hammond, K.B.; Clayton, K.L.; Ishii, N.; Abdel-Mohsen, M.; et al. TIGIT Marks Exhausted T Cells, Correlates with Disease Progression, and Serves as a Target for Immune Restoration in HIV and SIV Infection. PLoS Pathog. 2016, 12, e1005349. [Google Scholar] [CrossRef]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E.; et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog. 2016, 12, e1005761. [Google Scholar] [CrossRef] [PubMed]
- Vali, B.; Jones, R.B.; Sakhdari, A.; Sheth, P.M.; Clayton, K.; Yue, F.-Y.; Gyenes, G.; Wong, D.; Klein, M.B.; Saeed, S.; et al. HCV-specific T cells in HCV/HIV co-infection show elevated frequencies of dual Tim-3/PD-1 expression that correlate with liver disease progression. Eur. J. Immunol. 2010, 40, 2493–2505. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.; Hoffmann, M.; Pace, M.; Williams, J.P.; Thornhill, J.; Hamlyn, E.; Meyerowitz, J.; Willberg, C.; Koelsch, K.K.; Robinson, N.; et al. Immunological biomarkers predict HIV-1 viral rebound after treatment interruption. Nat. Commun. 2015, 6, 8495. [Google Scholar] [CrossRef]
- Kaufmann, D.E.; Kavanagh, D.G.; Pereyra, F.; Zaunders, J.J.; Mackey, E.W.; Miura, T.; Palmer, S.; Brockman, M.; Rathod, A.; Piechocka-Trocha, A.; et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat. Immunol. 2007, 8, 1246–1254. [Google Scholar] [CrossRef]
- Elrefaei, M.; Burke, C.M.; Baker, C.A.R.; Jones, N.G.; Bousheri, S.; Bangsberg, D.R.; Cao, H. HIV-Specific TGF-β-Positive CD4(+) T Cells Do Not Express Regulatory Surface Markers and Are Regulated by CTLA-4. AIDS Res. Hum. Retrovir. 2010, 26, 329–337. [Google Scholar] [CrossRef]
- Cecchinato, V.; Tryniszewska, E.; Ma, Z.M.; Vaccari, M.; Boasso, A.; Tsai, W.-P.; Petrovas, C.; Fuchs, D.; Heraud, J.-M.; Venzon, D.; et al. Immune Activation Driven by CTLA-4 Blockade Augments Viral Replication at Mucosal Sites in Simian Immunodeficiency Virus Infection. J. Immunol. 2008, 180, 5439–5447. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.E.; Poteet, E.C.; Liu, D.; Chen, C.; LaBranche, C.C.; Stanfield-Oakley, S.A.; Montefiori, D.C.; Ferrari, G.; Yao, Q. CTLA-4 Blockade, during HIV Virus-Like Particles Immunization, Alters HIV-Specific B-Cell Responses. Vaccines 2020, 8, 284. [Google Scholar] [CrossRef]
- Le Garff, G.; Samri, A.; Lambert-Niclot, S.; Even, S.; Lavolé, A.; Cadranel, J.; Spano, J.-P.; Autran, B.; Marcelin, A.-G.; Guihot, A. Transient HIV-specific T cells increase and inflammation in an HIV-infected patient treated with nivolumab. AIDS (Lond. Engl.) 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Guihot, A.; Marcelin, A.G.; Massiani, M.A.; Samri, A.; Soulié, C.; Autran, B.; Spano, J.P. Drastic decrease of the HIV reservoir in a patient treated with nivolumab for lung cancer. Ann. Oncol. 2018, 29, 517–518. [Google Scholar] [CrossRef]
- Scully, E.P.; Rutishauser, R.L.; Simoneau, C.R.; Delagrèverie, H.; Euler, Z.; Thanh, C.; Li, J.Z.; Hartig, H.; Bakkour, S.; Busch, M.; et al. Inconsistent HIV reservoir dynamics and immune responses following anti-PD-1 therapy in cancer patients with HIV infection. Ann. Oncol. 2018, 29, 2141–2142. [Google Scholar] [CrossRef]
- Fromentin, R.; DaFonseca, S.; Costiniuk, C.T.; El-Far, M.; Procopio, F.A.; Hecht, F.M.; Hoh, R.; Deeks, S.G.; Hazuda, D.J.; Lewin, S.R.; et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4+ T cells from ART-suppressed individuals. Nat. Commun. 2019, 10, 814. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef]
- Gupta, P.K.; Godec, J.; Wolski, D.; Adland, E.; Yates, K.; Pauken, K.E.; Cosgrove, C.; Ledderose, C.; Junger, W.G.; Robson, S.C.; et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog. 2015, 11, e1005177. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, H.H.; Tu, B.; Zhou, M.J.; Hu, W.; Fu, Y.L.; Li, X.Y.; Yang, T.; Song, J.W.; Fan, X.; et al. Reversal of the CD8(+) T-Cell Exhaustion Induced by Chronic HIV-1 Infection Through Combined Blockade of the Adenosine and PD-1 Pathways. Front. Immunol 2021, 12, 687296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, S.; Hwang, I.; Tough, D.F.; Sprent, J. Potent and Selective Stimulation of Memory-Phenotype CD8+ T Cells In Vivo by IL-15. Immunity 1998, 8, 591–599. [Google Scholar] [CrossRef]
- Lodolce, J.P.; Boone, D.L.; Chai, S.; Swain, R.E.; Dassopoulos, T.; Trettin, S.; Ma, A. IL-15 Receptor Maintains Lymphoid Homeostasis by Supporting Lymphocyte Homing and Proliferation. Immunity 1998, 9, 669–676. [Google Scholar] [CrossRef]
- Mueller, Y.M.; Bojczuk, P.M.; Halstead, E.S.; Kim, A.H.J.; Witek, J.; Altman, J.D.; Katsikis, P.D. IL-15 enhances survival and function of HIV-specific CD8+ T cells. Blood 2003, 101, 1024–1029. [Google Scholar] [CrossRef]
- Garrido, C.; Abad-Fernandez, M.; Tuyishime, M.; Pollara, J.J.; Ferrari, G.; Soriano-Sarabia, N.; Margolis, D.M. Interleukin-15-Stimulated Natural Killer Cells Clear HIV-1-Infected Cells following Latency Reversal Ex Vivo. J. Virol. 2018, 92, e00235-00218. [Google Scholar] [CrossRef] [PubMed]
- Lugli, E.; Mueller, Y.M.; Lewis, M.G.; Villinger, F.; Katsikis, P.D.; Roederer, M. IL-15 delays suppression and fails to promote immune reconstitution in virally suppressed chronically SIV-infected macaques. Blood 2011, 118, 2520–2529. [Google Scholar] [CrossRef]
- Mueller, Y.M.; Petrovas, C.; Bojczuk, P.M.; Dimitriou, I.D.; Beer, B.; Silvera, P.; Villinger, F.; Cairns, J.S.; Gracely, E.J.; Lewis, M.G.; et al. Interleukin-15 increases effector memory CD8+ t cells and NK Cells in simian immunodeficiency virus-infected macaques. J. Virol. 2005, 79, 4877–4885. [Google Scholar] [CrossRef]
- Harwood, O.; O’Connor, S. Therapeutic Potential of IL-15 and N-803 in HIV/SIV Infection. Viruses 2021, 13, 1750. [Google Scholar] [CrossRef]
- Watson, D.C.; Moysi, E.; Valentin, A.; Bergamaschi, C.; Devasundaram, S.; Fortis, S.P.; Bear, J.; Chertova, E.; Bess, J., Jr.; Sowder, R.; et al. Treatment with native heterodimeric IL-15 increases cytotoxic lymphocytes and reduces SHIV RNA in lymph nodes. PLoS Pathog. 2018, 14, e1006902. [Google Scholar] [CrossRef]
- Zhu, X.; Marcus, W.D.; Xu, W.; Lee, H.-i.; Han, K.; Egan, J.O.; Yovandich, J.L.; Rhode, P.R.; Wong, H.C. Novel Human Interleukin-15 Agonists. J. Immunol. 2009, 183, 3598–3607. [Google Scholar] [CrossRef]
- Jones, R.B.; Mueller, S.; O’Connor, R.; Rimpel, K.; Sloan, D.D.; Karel, D.; Wong, H.C.; Jeng, E.K.; Thomas, A.S.; Whitney, J.B.; et al. A Subset of Latency-Reversing Agents Expose HIV-Infected Resting CD4+ T-Cells to Recognition by Cytotoxic T-Lymphocytes. PLoS Pathog. 2016, 12, e1005545. [Google Scholar] [CrossRef]
- Ellis-Connell, A.L.; Balgeman, A.J.; Zarbock, K.R.; Barry, G.; Weiler, A.; Egan, J.O.; Jeng, E.K.; Friedrich, T.; Miller, J.S.; Haase, A.T.; et al. ALT-803 Transiently Reduces Simian Immunodeficiency Virus Replication in the Absence of Antiretroviral Treatment. J. Virol. 2018, 92, e01748-01717. [Google Scholar] [CrossRef]
- McBrien, J.B.; Mavigner, M.; Franchitti, L.; Smith, S.A.; White, E.; Tharp, G.K.; Walum, H.; Busman-Sahay, K.; Aguilera-Sandoval, C.R.; Thayer, W.O.; et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature 2020, 578, 154–159. [Google Scholar] [CrossRef] [PubMed]
- McBrien, J.B.; Wong, A.K.H.; White, E.; Carnathan, D.G.; Lee, J.H.; Safrit, J.T.; Vanderford, T.H.; Paiardini, M.; Chahroudi, A.; Silvestri, G. Combination of CD8beta Depletion and Interleukin-15 Superagonist N-803 Induces Virus Reactivation in Simian-Human Immunodeficiency Virus-Infected, Long-Term ART-Treated Rhesus Macaques. J. Virol. 2020, 94, e00755-00720. [Google Scholar] [CrossRef] [PubMed]
- Webb, G.M.; Molden, J.; Busman-Sahay, K.; Abdulhaqq, S.; Wu, H.L.; Weber, W.C.; Bateman, K.B.; Reed, J.S.; Northrup, M.; Maier, N.; et al. The human IL-15 superagonist N-803 promotes migration of virus-specific CD8+ T and NK cells to B cell follicles but does not reverse latency in ART-suppressed, SHIV-infected macaques. PLoS Pathog. 2020, 16, e1008339. [Google Scholar] [CrossRef] [PubMed]
- Pallikkuth, S.; Parmigiani, A.; Pahwa, S. The role of interleukin-21 in HIV infection. Cytokine Growth Factor Rev. 2012, 23, 173–180. [Google Scholar] [CrossRef][Green Version]
- White, L.; Krishnan, S.; Strbo, N.; Liu, H.; Kolber, M.A.; Lichtenheld, M.G.; Pahwa, R.N.; Pahwa, S. Differential effects of IL-21 and IL-15 on perforin expression, lysosomal degranulation, and proliferation in CD8 T cells of patients with human immunodeficiency virus-1 (HIV). Blood 2007, 109, 3873–3880. [Google Scholar] [CrossRef] [PubMed]
- Strbo, N.; de Armas, L.; Liu, H.; Kolber, M.A.; Lichtenheld, M.; Pahwa, S. IL-21 augments natural killer effector functions in chronically HIV-infected individuals. AIDS (Lond. Engl.) 2008, 22, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Pallikkuth, S.; Rogers, K.; Villinger, F.; Dosterii, M.; Vaccari, M.; Franchini, G.; Pahwa, R.; Pahwa, S. Interleukin-21 administration to rhesus macaques chronically infected with simian immunodeficiency virus increases cytotoxic effector molecules in T cells and NK cells and enhances B cell function without increasing immune activation or viral replication. Vaccine 2011, 29, 9229–9238. [Google Scholar] [CrossRef]
- Micci, L.; Ryan, E.S.; Fromentin, R.; Bosinger, S.E.; Harper, J.L.; He, T.; Paganini, S.; Easley, K.A.; Chahroudi, A.; Benne, C.; et al. Interleukin-21 combined with ART reduces inflammation and viral reservoir in SIV-infected macaques. J. Clin. Investig. 2015, 125, 4497–4513. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Huot, N.; Micci, L.; Tharp, G.; King, C.; Rascle, P.; Shenvi, N.; Wang, H.; Galardi, C.; Upadhyay, A.A.; et al. IL-21 and IFNα therapy rescues terminally differentiated NK cells and limits SIV reservoir in ART-treated macaques. Nat. Commun. 2021, 12, 2866. [Google Scholar] [CrossRef] [PubMed]
- Ylisastigui, L.; Archin, N.M.; Lehrman, G.; Bosch, R.J.; Margolis, D.M. Coaxing HIV-1 from resting CD4 T cells: Histone deacetylase inhibition allows latent viral expression. AIDS (Lond. Engl.) 2004, 18, 1101–1108. [Google Scholar] [CrossRef]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of Latent HIV Induced by the Potent HDAC Inhibitor Suberoylanilide Hydroxamic Acid. AIDS Res. Hum. Retrovir. 2009, 25, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef]
- Kulkosky, J.; Culnan, D.M.; Roman, J.; Dornadula, G.; Schnell, M.; Boyd, M.R.; Pomerantz, R.J. Prostratin: Activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 2001, 98, 3006–3015. [Google Scholar] [CrossRef]
- Korin, Y.D.; Brooks, D.G.; Brown, S.; Korotzer, A.; Zack, J.A. Effects of Prostratin on T-Cell Activation and Human Immunodeficiency Virus Latency. J. Virol. 2002, 76, 8118–8123. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Bosque, A.; Balch, A.H.; Smyth, D.; Martins, L.; Planelles, V. Ex Vivo Bioactivity and HIV-1 Latency Reversal by Ingenol Dibenzoate and Panobinostat in Resting CD4(+) T Cells from Aviremic Patients. Antimicrob. Agents Chemother. 2015, 59, 5984–5991. [Google Scholar] [CrossRef]
- Jiang, G.; Mendes, E.A.; Kaiser, P.; Wong, D.P.; Tang, Y.; Cai, I.; Fenton, A.; Melcher, G.P.; Hildreth, J.E.K.; Thompson, G.R.; et al. Synergistic Reactivation of Latent HIV Expression by Ingenol-3-Angelate, PEP005, Targeted NF-kB Signaling in Combination with JQ1 Induced p-TEFb Activation. PLoS Pathog. 2015, 11, e1005066. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lu, P.; Qu, X.; Shen, Y.; Zeng, H.; Zhu, X.; Zhu, Y.; Li, X.; Wu, H.; Xu, J.; et al. Reactivation of HIV-1 from Latency by an Ingenol Derivative from Euphorbia Kansui. Sci. Rep. 2017, 7, 9451. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, X.; Yang, X.; Lu, P.; Wang, Y.; Jiang, Z.; Pan, H.; Zhao, L.; Zhu, Y.; Khan, I.U.; et al. Dual effects of the novel ingenol derivatives on the acute and latent HIV-1 infections. Antivir. Res. 2019, 169, 104555. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Sampey, G.C.; Irlbeck, D.M.; Browne, E.P.; Kanke, M.; McAllister, A.B.; Ferris, R.G.; Brehm, J.H.; Favre, D.; Routy, J.-P.; Jones, C.D.; et al. The SMAC Mimetic AZD5582 is a Potent HIV Latency Reversing Agent. bioRxiv 2018, 312447. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kanuma, T.; Takahama, S.; Okamura, T.; Moriishi, E.; Ishii, K.J.; Terahara, K.; Yasutomi, Y. STING agonists activate latently infected cells and enhance SIV-specific responses ex vivo in naturally SIV controlled cynomolgus macaques. Sci. Rep. 2019, 9, 5917. [Google Scholar] [CrossRef]
- Thibault, S.; Imbeault, M.; Tardif, M.R.; Tremblay, M.J. TLR5 stimulation is sufficient to trigger reactivation of latent HIV-1 provirus in T lymphoid cells and activate virus gene expression in central memory CD4+ T cells. Virology 2009, 389, 20–25. [Google Scholar] [CrossRef]
- Novis, C.L.; Archin, N.M.; Buzon, M.J.; Verdin, E.; Round, J.L.; Lichterfeld, M.; Margolis, D.M.; Planelles, V.; Bosque, A. Reactivation of latent HIV-1 in central memory CD4+T cells through TLR-1/2 stimulation. Retrovirology 2013, 10, 119. [Google Scholar] [CrossRef]
- Li, P.; Kaiser, P.; Lampiris, H.W.; Kim, P.; Yukl, S.A.; Havlir, D.V.; Greene, W.C.; Wong, J.K. Stimulating the RIG-I pathway to kill cells in the latent HIV reservoir following viral reactivation. Nat. Med. 2016, 22, 807–811. [Google Scholar] [CrossRef]
- Bam, R.A.; Hansen, D.; Irrinki, A.; Mulato, A.; Jones, G.S.; Hesselgesser, J.; Frey, C.R.; Cihlar, T.; Yant, S.R. TLR7 Agonist GS-9620 Is a Potent Inhibitor of Acute HIV-1 Infection in Human Peripheral Blood Mononuclear Cells. Antimicrob. Agents Chemother. 2017, 61, e01369-01316. [Google Scholar] [CrossRef]
- Macedo, A.B.; Novis, C.L.; De Assis, C.M.; Sorensen, E.S.; Moszczynski, P.; Huang, S.-H.; Ren, Y.; Spivak, A.M.; Jones, R.B.; Planelles, V.; et al. Dual TLR2 and TLR7 agonists as HIV latency-reversing agents. JCI Insight 2018, 3, e122673. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- McGhee, J.D.; Felsenfeld, G. Nucleosome structure. Annu. Rev. Biochem. 1980, 49, 1115–1156. [Google Scholar] [CrossRef]
- Norton, V.G.; Marvin, K.W.; Yau, P.; Bradbury, E.M. Nucleosome linking number change controlled by acetylation of histones H3 and H4. J. Biol. Chem. 1990, 265, 19848–19852. [Google Scholar] [CrossRef]
- Lee, D.Y.; Hayes, J.J.; Pruss, D.; Wolffe, A.P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 1993, 72, 73–84. [Google Scholar] [CrossRef]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef]
- Lu, H.K.; Gray, L.R.; Wightman, F.; Ellenberg, P.; Khoury, G.; Cheng, W.J.; Mota, T.M.; Wesselingh, S.; Gorry, P.R.; Cameron, P.U.; et al. Ex vivo response to histone deacetylase (HDAC) inhibitors of the HIV long terminal repeat (LTR) derived from HIV-infected patients on antiretroviral therapy. PLoS ONE 2014, 9, e113341. [Google Scholar] [CrossRef]
- Kim, H.-J.; Bae, S.-C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Kroon, E.D.M.B.; Ananworanich, J.; Pagliuzza, A.; Rhodes, A.; Phanuphak, N.; Trautmann, L.; Mitchell, J.L.; Chintanaphol, M.; Intasan, J.; Pinyakorn, S.; et al. A randomized trial of vorinostat with treatment interruption after initiating antiretroviral therapy during acute HIV-1 infection. J. Virus Erad. 2020, 6, 100004. [Google Scholar] [CrossRef]
- Tsai, P.; Wu, G.; Baker, C.E.; Thayer, W.O.; Spagnuolo, R.A.; Sanchez, R.; Barrett, S.; Howell, B.; Margolis, D.; Hazuda, D.J.; et al. In vivo analysis of the effect of panobinostat on cell-associated HIV RNA and DNA levels and latent HIV infection. Retrovirology 2016, 13, 36. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Søgaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef]
- Nakajima, H.; Kim, Y.B.; Terano, H.; Yoshida, M.; Horinouchi, S. FR901228, a Potent Antitumor Antibiotic, Is a Novel Histone Deacetylase Inhibitor. Exp. Cell Res. 1998, 241, 126–133. [Google Scholar] [CrossRef]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.-H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (Depsipeptide) as a Natural Prodrug That Inhibits Class I Histone Deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Exp. Rev. Anticancer. 2010, 10, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, G.Q.; Oswald, K.; Lara, A.; Shoemaker, R.; Smedley, J.; Macallister, R.; Coalter, V.; Wiles, A.; Wiles, R.; Li, Y.; et al. Elevated plasma viral loads in romidepsin treated SIV-infected rhesus macaques on suppressive combination antiretroviral therapy. Antimicrob. Agents Chemother. 2015. [Google Scholar] [CrossRef]
- Jonsson, K.L.; Tolstrup, M.; Vad-Nielsen, J.; Kjaer, K.; Laustsen, A.; Andersen, M.N.; Rasmussen, T.A.; Sogaard, O.S.; Ostergaard, L.; Denton, P.W.; et al. Histone deacetylase inhibitor romidepsin inhibits de novo HIV-1 infections. Antimicrob. Agents Chemother. 2015, 59, 3984–3994. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Umbert, M.; Ruiz-Riol, M.; Fernandez, M.A.; Marszalek, M.; Coll, P.; Manzardo, C.; Cedeno, S.; Miro, J.M.; Clotet, B.; Hanke, T.; et al. In vivo Effects of Romidepsin on T-Cell Activation, Apoptosis and Function in the BCN02 HIV-1 Kick&Kill Clinical Trial. Front Immunol. 2020, 11, 418. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Fenard, D.; Bisgrove, D.; Verdin, E.; Greene, W.C. Prostratin antagonizes HIV latency by activating NF-kappaB. J. Biol. Chem. 2004, 279, 42008–42017. [Google Scholar] [CrossRef]
- Chowdhury, I.H.; Koyanagi, Y.; Kobayashi, S.; Hamamoto, Y.; Yoshiyama, H.; Yoshida, T.; Yamamoto, N. The phorbol ester TPA strongly inhibits HIV-1-induced syncytia formation but enhances virus production: Possible involvement of protein kinase C pathway. Virology 1990, 176, 126–132. [Google Scholar] [CrossRef]
- Bögi, K.; Lorenzo, P.S.; Szállási, Z.; Acs, P.; Wagner, G.S.; Blumberg, P.M. Differential selectivity of ligands for the C1a and C1b phorbol ester binding domains of protein kinase Cdelta: Possible correlation with tumor-promoting activity. Cancer Res. 1998, 58, 1423–1428. [Google Scholar] [PubMed]
- DeChristopher, B.A.; Loy, B.A.; Marsden, M.D.; Schrier, A.J.; Zack, J.A.; Wender, P.A. Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat. Chem. 2012, 4, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Marsden, M.D.; Wu, X.; Navab, S.M.; Loy, B.A.; Schrier, A.J.; DeChristopher, B.A.; Shimizu, A.J.; Hardman, C.T.; Ho, S.; Ramirez, C.M.; et al. Characterization of designed, synthetically accessible bryostatin analog HIV latency reversing agents. Virology 2018, 520, 83–93. [Google Scholar] [CrossRef]
- Brogdon, J.; Ziani, W.; Wang, X.; Veazey, R.S.; Xu, H. In vitro effects of the small-molecule protein kinase C agonists on HIV latency reactivation. Sci. Rep. 2016, 6, 39032. [Google Scholar] [CrossRef] [PubMed]
- McKernan, L.N.; Momjian, D.; Kulkosky, J. Protein Kinase C: One Pathway towards the Eradication of Latent HIV-1 Reservoirs. Adv. Virol. 2012, 2012, 805347. [Google Scholar] [CrossRef] [PubMed]
- Albert, B.J.; Niu, A.; Ramani, R.; Marshall, G.R.; Wender, P.A.; Williams, R.M.; Ratner, L.; Barnes, A.B.; Kyei, G.B. Combinations of isoform-targeted histone deacetylase inhibitors and bryostatin analogues display remarkable potency to activate latent HIV without global T-cell activation. Sci. Rep. 2017, 7, 7456. [Google Scholar] [CrossRef]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.S.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency–reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef]
- Sloane, J.L.; Benner, N.L.; Keenan, K.N.; Zang, X.; Soliman, M.S.A.; Wu, X.; Dimapasoc, M.; Chun, T.-W.; Marsden, M.D.; Zack, J.A.; et al. Prodrugs of PKC modulators show enhanced HIV latency reversal and an expanded therapeutic window. Proc. Natl. Acad. Sci. USA 2020, 117, 10688–10698. [Google Scholar] [CrossRef]
- Adolf, W.; Chanai, S.; Hecker, E. 3-0-angeloylingenol, the toxic and skin irritant factor from latex of euphorbia anitiquorum l.(euphorbiaceae) and from a derived thai purgative and anthelimintic (vermifuge) drug. Sci. Asia 1983, 9, 81–88. [Google Scholar] [CrossRef]
- Kedei, N.; Lundberg, D.J.; Toth, A.; Welburn, P.; Garfield, S.H.; Blumberg, P.M. Characterization of the Interaction of Ingenol 3-Angelate with Protein Kinase C. Cancer Res. 2004, 64, 3243–3255. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Mendes, E.A.; Kaiser, P.; Sankaran-Walters, S.; Tang, Y.; Weber, M.G.; Melcher, G.P.; Thompson, G.R., III.; Tanuri, A.; Pianowski, L.F.; et al. Reactivation of HIV latency by a newly modified Ingenol derivative via protein kinase Cδ–NF-κB signaling. AIDS (Lond. Engl.) 2014, 28. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.M.; Price, S.L.; Shirk, E.N.; Cunha, R.D.; Pianowski, L.F.; Clements, J.E.; Tanuri, A.; Gama, L. Dual role of novel ingenol derivatives from Euphorbia tirucalli in HIV replication: Inhibition of de novo infection and activation of viral LTR. PLoS ONE 2014, 9, e97257. [Google Scholar] [CrossRef]
- Fujiwara, M.; Ijichi, K.; Tokuhisa, K.; Katsuura, K.; Wang, G.Y.S.; Uemura, D.; Shigeta, S.; Konno, K.; Yokota, T.; Baba, M. Ingenol Derivatives are Highly Potent and Selective Inhibitors of HIV Replication in Vitro. Antivir. Chem. Chemother. 1996, 7, 230–236. [Google Scholar] [CrossRef]
- Ersvaer, E.; Kittang, A.O.; Hampson, P.; Sand, K.; Gjertsen, B.T.; Lord, J.M.; Bruserud, Ø. The Protein Kinase C Agonist PEP005 (Ingenol 3-Angelate) in the Treatment of Human Cancer: A Balance between Efficacy and Toxicity. Toxins 2010, 2, 174–194. [Google Scholar] [CrossRef]
- Pandeló José, D.; Bartholomeeusen, K.; da Cunha, R.D.; Abreu, C.M.; Glinski, J.; da Costa, T.B.; Bacchi Rabay, A.F.; Pianowski Filho, L.F.; Dudycz, L.W.; Ranga, U.; et al. Reactivation of latent HIV-1 by new semi-synthetic ingenol esters. Virology 2014, 462–463, 328–339. [Google Scholar] [CrossRef]
- Jiang, G.; Maverakis, E.; Cheng, M.Y.; Elsheikh, M.M.; Deleage, C.; Méndez-Lagares, G.; Shimoda, M.; Yukl, S.A.; Hartigan-O’Connor, D.J.; Thompson, G.R., III; et al. Disruption of latent HIV in vivo during the clearance of actinic keratosis by ingenol mebutate. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef]
- Lu, P.; Qu, X.; Shen, Y.; Jiang, Z.; Wang, P.; Zeng, H.; Ji, H.; Deng, J.; Yang, X.; Li, X.; et al. The BET inhibitor OTX015 reactivates latent HIV-1 through P-TEFb. Sci. Rep. 2016, 6, 24100. [Google Scholar] [CrossRef]
- Huang, H.; Liu, S.; Jean, M.; Simpson, S.; Huang, H.; Merkley, M.; Hayashi, T.; Kong, W.; Rodriguez-Sanchez, I.; Zhang, X.; et al. A Novel Bromodomain Inhibitor Reverses HIV-1 Latency through Specific Binding with BRD4 to Promote Tat and P-TEFb Association. Front. Microbiol 2017, 8, 1035. [Google Scholar] [CrossRef]
- Zhang, X.-x.; Lin, J.; Liang, T.-z.; Duan, H.; Tan, X.-h.; Xi, B.-m.; Li, L.; Liu, S.-w. The BET bromodomain inhibitor apabetalone induces apoptosis of latent HIV-1 reservoir cells following viral reactivation. Acta Pharmacol. Sin. 2019, 40, 98–110. [Google Scholar] [CrossRef]
- Liang, T.; Zhang, X.; Lai, F.; Lin, J.; Zhou, C.; Xu, X.; Tan, X.; Liu, S.; Li, L. A novel bromodomain inhibitor, CPI-203, serves as an HIV-1 latency-reversing agent by activating positive transcription elongation factor b. Biochem. Pharmacol. 2019, 164, 237–251. [Google Scholar] [CrossRef]
- Gallastegui, E.; Marshall, B.; Vidal, D.; Sanchez-Duffhues, G.; Collado, J.A.; Alvarez-Fernandez, C.; Luque, N.; Terme, J.M.; Gatell, J.M.; Sanchez-Palomino, S.; et al. Combination of biological screening in a cellular model of viral latency and virtual screening identifies novel compounds that reactivate HIV-1. J. Virol. 2012, 86, 3795–3808. [Google Scholar] [CrossRef]
- Abner, E.; Stoszko, M.; Zeng, L.; Chen, H.C.; Izquierdo-Bouldstridge, A.; Konuma, T.; Zorita, E.; Fanunza, E.; Zhang, Q.; Mahmoudi, T.; et al. A New Quinoline BRD4 Inhibitor Targets a Distinct Latent HIV-1 Reservoir for Reactivation from Other “Shock” Drugs. J. Virol. 2018, 92, e02056-17. [Google Scholar] [CrossRef] [PubMed]
- Vamos, M.; Welsh, K.; Finlay, D.; Lee, P.S.; Mace, P.D.; Snipas, S.J.; Gonzalez, M.L.; Ganji, S.R.; Ardecky, R.J.; Riedl, S.J.; et al. Expedient Synthesis of Highly Potent Antagonists of Inhibitor of Apoptosis Proteins (IAPs) with Unique Selectivity for ML-IAP. ACS Chem. Biol. 2013, 8, 725–732. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Finlay, D.; Vamos, M.; González-López, M.; Ardecky, R.J.; Ganji, S.R.; Yuan, H.; Su, Y.; Cooley, T.R.; Hauser, C.T.; Welsh, K.; et al. Small-Molecule IAP Antagonists Sensitize Cancer Cells to TRAIL-Induced Apoptosis: Roles of XIAP and cIAPs. Mol. Cancer Ther. 2014, 13, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The noncanonical NF-κB pathway. Immunol Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef]
- Pache, L.; Dutra, S.; Spivak, M.; John, M.; Murry, P.; Hwang, Y.; Maestre, M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S.I.; Matsuda, K.; Tsuchiya, K.; Gatanaga, H.; Oka, S.; Yoshimura, K.; Mitsuya, H.; Maeda, K. Combination of a Latency-Reversing Agent With a Smac Mimetic Minimizes Secondary HIV-1 Infection in vitro. Front. Microbiol. 2018, 9, 2022. [Google Scholar] [CrossRef]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef]
- Mavigner, M.; Liao, L.E.; Brooks, A.D.; Ke, R.; Mattingly, C.; Schoof, N.; McBrien, J.; Carnathan, D.; Liang, S.; Vanderford, T.H.; et al. CD8 lymphocyte depletion enhances the latency reversal activity of the SMAC mimetic AZD5582 in ART-suppressed SIV-infected rhesus macaques. J. Virol. 2021, 95, e01429-01420. [Google Scholar] [CrossRef] [PubMed]
- Pache, L.; Marsden, M.D.; Teriete, P.; Portillo, A.J.; Heimann, D.; Kim, J.T.; Soliman, M.S.A.; Dimapasoc, M.; Carmona, C.; Celeridad, M.; et al. Pharmacological Activation of Non-canonical NF-κB Signaling Activates Latent HIV-1 Reservoirs In Vivo. Cell Rep. Med. 2020, 1, 100037. [Google Scholar] [CrossRef] [PubMed]
- Takahama, S.; Yamamoto, T. Pattern Recognition Receptor Ligands as an Emerging Therapeutic Agent for Latent HIV-1 Infection. Front. Cell. Infect. Microbiol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Kuse, N.; Sun, X.; Akahoshi, T.; Lissina, A.; Yamamoto, T.; Appay, V.; Takiguchi, M. Priming of HIV-1-specific CD8(+) T cells with strong functional properties from naive T cells. EBioMedicine 2019, 42, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Aroh, C.; Wang, Z.; Dobbs, N.; Luo, M.; Chen, Z.; Gao, J.; Yan, N. Innate Immune Activation by cGMP-AMP Nanoparticles Leads to Potent and Long-Acting Antiretroviral Response against HIV-1. J. Immunol. 2017, 199, 3840–3848. [Google Scholar] [CrossRef] [PubMed]
- Palermo, E.; Acchioni, C.; Di Carlo, D.; Zevini, A.; Muscolini, M.; Ferrari, M.; Castiello, L.; Virtuoso, S.; Borsetti, A.; Antonelli, G.; et al. Activation of Latent HIV-1 T Cell Reservoirs with a Combination of Innate Immune and Epigenetic Regulators. J. Virol. 2019, 93, e01194-01119. [Google Scholar] [CrossRef] [PubMed]
- Mavigner, M.; Brooks, A.D.; Koblansky, A.; Galardi, C.; Madsen, H.; Margolis, D.M.; Silvestri, G.; Chahroudi, A. 26 - Sting agonist as a kick and kill agent to target the HIV reservoir. J. Virus Erad. 2019, 5, 14–15. [Google Scholar] [CrossRef]
- Lisziewicz, J.; Sun, D.; Weichold, F.F.; Thierry, A.R.; Lusso, P.; Tang, J.; Gallo, R.C.; Agrawal, S. Antisense oligodeoxynucleotide phosphorothioate complementary to Gag mRNA blocks replication of human immunodeficiency virus type 1 in human peripheral blood cells. Proc. Natl. Acad. Sci. USA 1994, 91, 7942–7946. [Google Scholar] [CrossRef] [PubMed]
- Martinsen, J.T.; Gunst, J.D.; Højen, J.F.; Tolstrup, M.; Søgaard, O.S. The Use of Toll-Like Receptor Agonists in HIV-1 Cure Strategies. Front. Immunol. 2020, 11, 1112. [Google Scholar] [CrossRef]
- Equils, O.; Schito, M.L.; Karahashi, H.; Madak, Z.; Yarali, A.; Michelsen, K.S.; Sher, A.; Arditi, M. Toll-like receptor 2 (TLR2) and TLR9 signaling results in HIV-long terminal repeat trans-activation and HIV replication in HIV-1 transgenic mouse spleen cells: Implications of simultaneous activation of TLRs on HIV replication. J. Immunol. (Baltimore, Md. 1950) 2003, 170, 5159–5164. [Google Scholar] [CrossRef]
- Séréni, D.; Tubiana, R.; Lascoux, C.; Katlama, C.; Taulera, O.; Bourque, A.; Cohen, A.; Dvorchik, B.; Martin, R.R.; Tournerie, C.; et al. Pharmacokinetics and Tolerability of Intravenous Trecovirsen (GEM®91), an Antisense Phosphorothioate Oligonucleotide, in HIV-Positive Subjects. J. Clin. Pharmacol. 1999, 39, 47–54. [Google Scholar] [CrossRef]
- Agrawal, S.; Martin, R.R. Was Induction of HIV-1 Through TLR9? J. Immunol. 2003, 171, 1621–1622. [Google Scholar] [CrossRef]
- Winckelmann, A.A.; Munk-Petersen, L.V.; Rasmussen, T.A.; Melchjorsen, J.; Hjelholt, T.J.; Montefiori, D.; Østergaard, L.; Søgaard, O.S.; Tolstrup, M. Administration of a Toll-Like Receptor 9 Agonist Decreases the Proviral Reservoir in Virologically Suppressed HIV-Infected Patients. PLoS ONE 2013, 8, e62074. [Google Scholar] [CrossRef]
- Schmidt, M.; Hagner, N.; Marco, A.; König-Merediz, S.A.; Schroff, M.; Wittig, B. Design and Structural Requirements of the Potent and Safe TLR-9 Agonistic Immunomodulator MGN1703. Nucleic Acid Ther. 2015, 25, 130–140. [Google Scholar] [CrossRef]
- Offersen, R.; Nissen, S.K.; Rasmussen, T.A.; Ostergaard, L.; Denton, P.W.; Sogaard, O.S.; Tolstrup, M. A Novel Toll-Like Receptor 9 Agonist, MGN1703, Enhances HIV-1 Transcription and NK Cell-Mediated Inhibition of HIV-1-Infected Autologous CD4+ T Cells. J. Virol. 2016, 90, 4441–4453. [Google Scholar] [CrossRef]
- Vibholm, L.; Schleimann, M.H.; Højen, J.F.; Benfield, T.; Offersen, R.; Rasmussen, K.; Olesen, R.; Dige, A.; Agnholt, J.; Grau, J.; et al. Short-Course Toll-Like Receptor 9 Agonist Treatment Impacts Innate Immunity and Plasma Viremia in Individuals With Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2017, 64, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Vibholm, L.K.; Konrad, C.V.; Schleimann, M.H.; Frattari, G.; Winckelmann, A.; Klastrup, V.; Jensen, N.M.; Jensen, S.S.; Schmidt, M.; Wittig, B.; et al. Effects of 24-week Toll-like receptor 9 agonist treatment in HIV type 1+ individuals. AIDS (Lond. Engl.) 2019, 33. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, G.Q.; Alvord, W.G.; Li, Y.; Deleage, C.; Nag, M.; Oswald, K.; Thomas, J.A.; Pyle, C.; Bosche, W.J.; Coalter, V.; et al. TLR7 agonist administration to SIV-infected macaques receiving early initiated cART does not induce plasma viremia. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Riddler, S.; Para, M.; Benson, C.; Mills, A.; Ramgopal, M.; DeJesus, E.; Brinson, C.; Cyktor, J.; Mellors, J.; Guo, S. Vesatolimod (GS-9620) is safe and pharmacodynamically active in HIV-infected individuals. J. Int. AIDS Soc. 2019, 22, 43. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol 2016, 16, 553–565. [Google Scholar] [CrossRef]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, D.-t.; Liu, X.-g. mTOR Signaling in T Cell Immunity and Autoimmunity. Int. Rev. Immunol. 2015, 34, 50–66. [Google Scholar] [CrossRef]
- Trautmann, L.; Mbitikon-Kobo, F.-M.; Goulet, J.-P.; Peretz, Y.; Shi, Y.; Van Grevenynghe, J.; Procopio, F.A.; Boulassel, M.R.; Routy, J.-P.; Chomont, N.; et al. Profound metabolic, functional, and cytolytic differences characterize HIV-specific CD8 T cells in primary and chronic HIV infection. Blood 2012, 120, 3466–3477. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef]
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity 2014, 41, 802–814. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef]
- Angin, M.; Volant, S.; Passaes, C.; Lecuroux, C.; Monceaux, V.; Dillies, M.-A.; Valle-Casuso, J.C.; Pancino, G.; Vaslin, B.; Le Grand, R.; et al. Metabolic plasticity of HIV-specific CD8+ T cells is associated with enhanced antiviral potential and natural control of HIV-1 infection. Nat. Metab. 2019, 1, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29, 611–626.e615. [Google Scholar] [CrossRef]
- van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef]
- Igarashi, T.; Endo, Y.; Englund, G.; Sadjadpour, R.; Matano, T.; Buckler, C.; Buckler-White, A.; Plishka, R.; Theodore, T.; Shibata, R.; et al. Emergence of a highly pathogenic simian/human immunodeficiency virus in a rhesus macaque treated with anti-CD8 mAb during a primary infection with a nonpathogenic virus. Proc. Natl. Acad. Sci. USA 1999, 96, 14049–14054. [Google Scholar] [CrossRef]
- Igarashi, T.; Brown, C.R.; Endo, Y.; Buckler-White, A.; Plishka, R.; Bischofberger, N.; Hirsch, V.; Martin, M.A. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4+ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): Implications for HIV-1 infections of humans. Proc. Natl. Acad. Sci. USA 2001, 98, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, J.M.; Fox, C.; Wahl, S.M. Macrophages as a Source of HIV During Opportunistic Infections. Science (N. Y.) 1997, 276, 1857–1861. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.E.; Babas, T.; Mankowski, J.L.; Suryanarayana, K.; Piatak, M., Jr.; Tarwater, P.M.; Lifson, J.D.; Zink, M.C. The central nervous system as a reservoir for simian immunodeficiency virus (SIV): Steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J. Infect. Dis. 2002, 186, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Zalar, A.; Figueroa, M.I.; Ruibal-Ares, B.; Baré, P.; Cahn, P.; de Bracco, M.M.d.E.; Belmonte, L. Macrophage HIV-1 infection in duodenal tissue of patients on long term HAART. Antivir. Res. 2010, 87, 269–271. [Google Scholar] [CrossRef]
- Yukl, S.A.; Sinclair, E.; Somsouk, M.; Hunt, P.W.; Epling, L.; Killian, M.; Girling, V.; Li, P.; Havlir, D.V.; Deeks, S.G.; et al. A comparison of methods for measuring rectal HIV levels suggests that HIV DNA resides in cells other than CD4+ T cells, including myeloid cells. AIDS (Lond. Engl.) 2014, 28, 439–442. [Google Scholar] [CrossRef]
- Honeycutt, J.B.; Thayer, W.O.; Baker, C.E.; Ribeiro, R.M.; Lada, S.M.; Cao, Y.; Cleary, R.A.; Hudgens, M.G.; Richman, D.D.; Garcia, J.V. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2017, 23, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Busca, A.; Saxena, M.; Kumar, A. Critical role for antiapoptotic Bcl-xL and Mcl-1 in human macrophage survival and cellular IAP1/2 (cIAP1/2) in resistance to HIV-Vpr-induced apoptosis. J. Biol. Chem. 2012, 287, 15118–15133. [Google Scholar] [CrossRef]
- Rappaport, J.; Volsky, D.J. Role of the macrophage in HIV-associated neurocognitive disorders and other comorbidities in patients on effective antiretroviral treatment. J. Neurovirol. 2015, 21, 235–241. [Google Scholar] [CrossRef]
- Vojnov, L.; Martins, M.A.; Bean, A.T.; Veloso de Santana, M.G.; Sacha, J.B.; Wilson, N.A.; Bonaldo, M.C.; Galler, R.; Stevenson, M.; Watkins, D.I. The majority of freshly sorted simian immunodeficiency virus (SIV)-specific CD8(+) T cells cannot suppress viral replication in SIV-infected macrophages. J. Virol. 2012, 86, 4682–4687. [Google Scholar] [CrossRef]
- Clayton, K.L.; Collins, D.R.; Lengieza, J.; Ghebremichael, M.; Dotiwala, F.; Lieberman, J.; Walker, B.D. Resistance of HIV-infected macrophages to CD8(+) T lymphocyte-mediated killing drives activation of the immune system. Nat. Immunol. 2018, 19, 475–486. [Google Scholar] [CrossRef]
- Mailliard, R.B.; Smith, K.N.; Fecek, R.J.; Rappocciolo, G.; Nascimento, E.J.M.; Marques, E.T.; Watkins, S.C.; Mullins, J.I.; Rinaldo, C.R. Selective Induction of CTL Helper Rather Than Killer Activity by Natural Epitope Variants Promotes Dendritic Cell–Mediated HIV-1 Dissemination. J. Immunol. 2013, 191, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.N.; Mailliard, R.B.; Piazza, P.A.; Fischer, W.; Korber, B.T.; Fecek, R.J.; Ratner, D.; Gupta, P.; Mullins, J.I.; Rinaldo, C.R. Effective Cytotoxic T Lymphocyte Targeting of Persistent HIV-1 during Antiretroviral Therapy Requires Priming of Naive CD8+ T Cells. mBio 2016, 7, e00473-16. [Google Scholar] [CrossRef]
- Smith, K.N.; Mailliard, R.B.; Rinaldo, C.R. Programming T cell Killers for an HIV Cure: Teach the New Dogs New Tricks and Let the Sleeping Dogs Lie. Forum Immunopathol. Dis. Ther. 2015, 6, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; To, R.K.; Zhang, G.; Spector, S.A. SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected macrophages. Cell Death Dis. 2020, 11, 590. [Google Scholar] [CrossRef]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746. [Google Scholar] [CrossRef]
- Sanyal, A.; Mailliard, R.B.; Rinaldo, C.R.; Ratner, D.; Ding, M.; Chen, Y.; Zerbato, J.M.; Giacobbi, N.S.; Venkatachari, N.J.; Patterson, B.K.; et al. Novel assay reveals a large, inducible, replication-competent HIV-1 reservoir in resting CD4(+) T cells. Nat. Med. 2017, 23, 885–889. [Google Scholar] [CrossRef]
- Battivelli, E.; Dahabieh, M.S.; Abdel-Mohsen, M.; Svensson, J.P.; Tojal Da Silva, I.; Cohn, L.B.; Gramatica, A.; Deeks, S.; Greene, W.C.; Pillai, S.K.; et al. Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4(+) T cells. Elife 2018, 7, e34655. [Google Scholar] [CrossRef]
- Paiardini, M.; Dhodapkar, K.; Harper, J.; Deeks, S.G.; Ahmed, R. Editorial: HIV and Cancer Immunotherapy: Similar Challenges and Converging Approaches. Front. Immunol 2020, 11, 519. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Carballo, D.; Saka, S.; Acikelli, A.H.; Homp, E.; Erwes, J.; Demmig, R.; Klein, J.; Schröer, K.; Malak, S.; D’Souza, F.; et al. Enhanced antitumoral activity of TLR7 agonists via activation of human endogenous retroviruses by HDAC inhibitors. Commun. Biol. 2021, 4, 276. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Spolski, R.; Finkelstein, S.E.; Oh, S.; Kovanen, P.E.; Hinrichs, C.S.; Pise-Masison, C.A.; Radonovich, M.F.; Brady, J.N.; Restifo, N.P.; et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J. Exp. Med. 2005, 201, 139–148. [Google Scholar] [CrossRef]
- Strengell, M.; Matikainen, S.; Sirén, J.; Lehtonen, A.; Foster, D.; Julkunen, I.; Sareneva, T. IL-21 in Synergy with IL-15 or IL-18 Enhances IFN-γ Production in Human NK and T Cells. J. Immunol. 2003, 170, 5464–5469. [Google Scholar] [CrossRef] [PubMed]
- Pouw, N.; Treffers-Westerlaken, E.; Kraan, J.; Wittink, F.; ten Hagen, T.; Verweij, J.; Debets, R. Combination of IL-21 and IL-15 enhances tumour-specific cytotoxicity and cytokine production of TCR-transduced primary T cells. Cancer Immunol. Immunother. CII 2010, 59, 921–931. [Google Scholar] [CrossRef]
- Kinter, A.L.; Godbout, E.J.; McNally, J.P.; Sereti, I.; Roby, G.A.; O’Shea, M.A.; Fauci, A.S. The Common γ-Chain Cytokines IL-2, IL-7, IL-15, and IL-21 Induce the Expression of Programmed Death-1 and Its Ligands. J. Immunol. 2008, 181, 6738–6746. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: A non-randomised, open-label, phase 1b trial. Lancet. Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef]
- Drusbosky, L.; Nangia, C.; Nguyen, A.; Szeto, C.; Newton, Y.; Spilman, P.; Reddy, S.B. Complete response to avelumab and IL-15 superagonist N-803 with Abraxane in Merkel cell carcinoma: A case study. J. ImmunoTherapy Cancer 2020, 8, e001098. [Google Scholar] [CrossRef]
- Knudson, K.M.; Hicks, K.C.; Alter, S.; Schlom, J.; Gameiro, S.R. Mechanisms involved in IL-15 superagonist enhancement of anti-PD-L1 therapy. J. Immunol. Therapy Cancer 2019, 7, 82. [Google Scholar] [CrossRef]
- Jochems, C.; Tritsch, S.R.; Knudson, K.M.; Gameiro, S.R.; Rumfield, C.S.; Pellom, S.T.; Morillon, Y.M.; Newman, R.; Marcus, W.; Szeto, C.; et al. The multi-functionality of N-809, a novel fusion protein encompassing anti-PD-L1 and the IL-15 superagonist fusion complex. OncoImmunology 2019, 8, e1532764. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cong, Y.; Jia, M.; He, Q.; Zhong, H.; Zhao, Y.; Li, H.; Yan, M.; You, J.; Liu, J.; et al. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat. Commun. 2021, 12, 951. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleinman, A.J.; Pandrea, I.; Apetrei, C. So Pathogenic or So What?—A Brief Overview of SIV Pathogenesis with an Emphasis on Cure Research. Viruses 2022, 14, 135. https://doi.org/10.3390/v14010135
Kleinman AJ, Pandrea I, Apetrei C. So Pathogenic or So What?—A Brief Overview of SIV Pathogenesis with an Emphasis on Cure Research. Viruses. 2022; 14(1):135. https://doi.org/10.3390/v14010135
Chicago/Turabian StyleKleinman, Adam J., Ivona Pandrea, and Cristian Apetrei. 2022. "So Pathogenic or So What?—A Brief Overview of SIV Pathogenesis with an Emphasis on Cure Research" Viruses 14, no. 1: 135. https://doi.org/10.3390/v14010135
APA StyleKleinman, A. J., Pandrea, I., & Apetrei, C. (2022). So Pathogenic or So What?—A Brief Overview of SIV Pathogenesis with an Emphasis on Cure Research. Viruses, 14(1), 135. https://doi.org/10.3390/v14010135