Abstract
CRISPR/Cas technology has revolutionized the fields of the genome- and epigenome-editing by supplying unparalleled control over genomic sequences and expression. Lentiviral vector (LV) systems are one of the main delivery vehicles for the CRISPR/Cas systems due to (i) its ability to carry bulky and complex transgenes and (ii) sustain robust and long-term expression in a broad range of dividing and non-dividing cells in vitro and in vivo. It is thus reasonable that substantial effort has been allocated towards the development of the improved and optimized LV systems for effective and accurate gene-to-cell transfer of CRISPR/Cas tools. The main effort on that end has been put towards the improvement and optimization of the vector’s expression, development of integrase-deficient lentiviral vector (IDLV), aiming to minimize the risk of oncogenicity, toxicity, and pathogenicity, and enhancing manufacturing protocols for clinical applications required large-scale production. In this review, we will devote attention to (i) the basic biology of lentiviruses, and (ii) recent advances in the development of safer and more efficient CRISPR/Cas vector systems towards their use in preclinical and clinical applications. In addition, we will discuss in detail the recent progress in the repurposing of CRISPR/Cas systems related to base-editing and prime-editing applications.
1. HIV-1 Based Vectors: Basic Biology
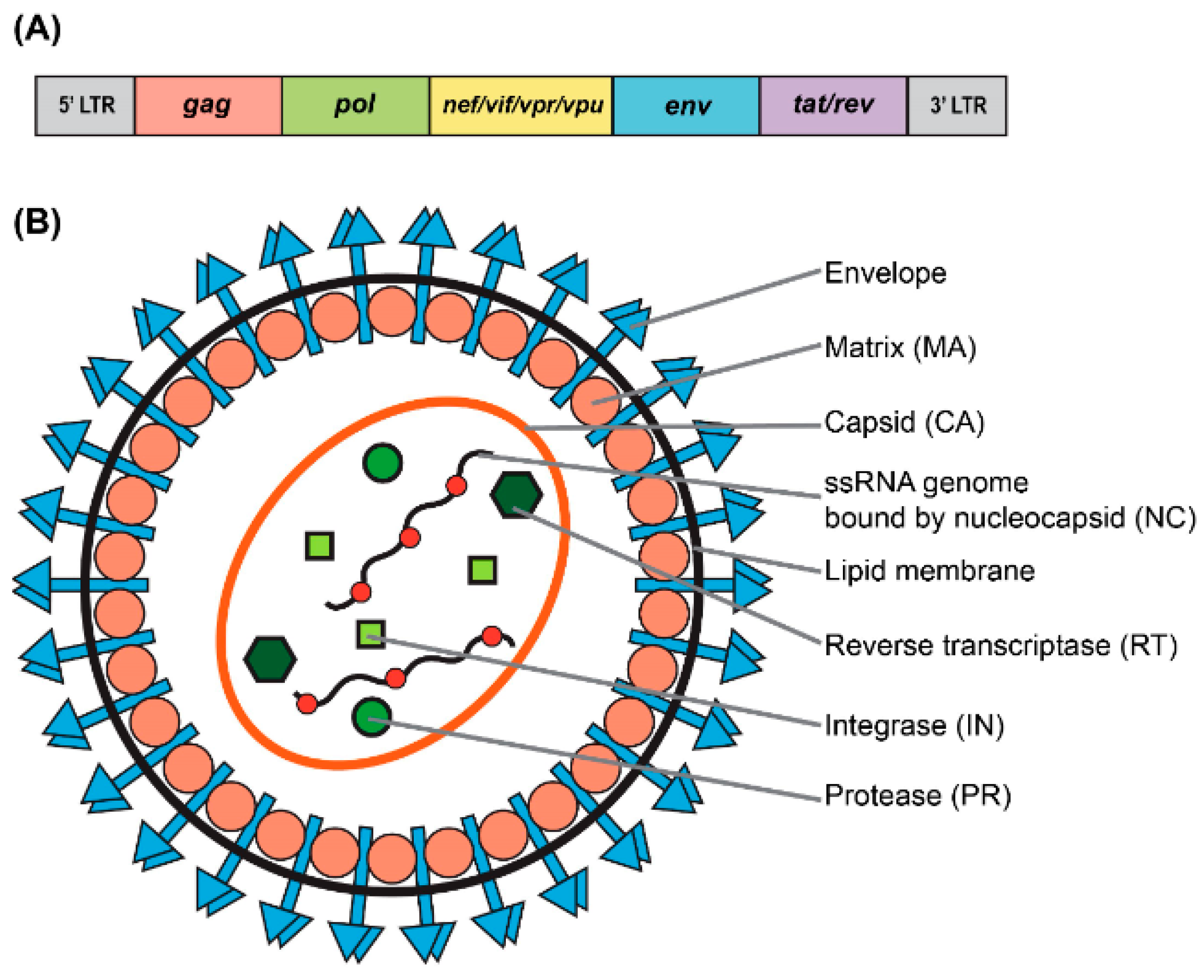
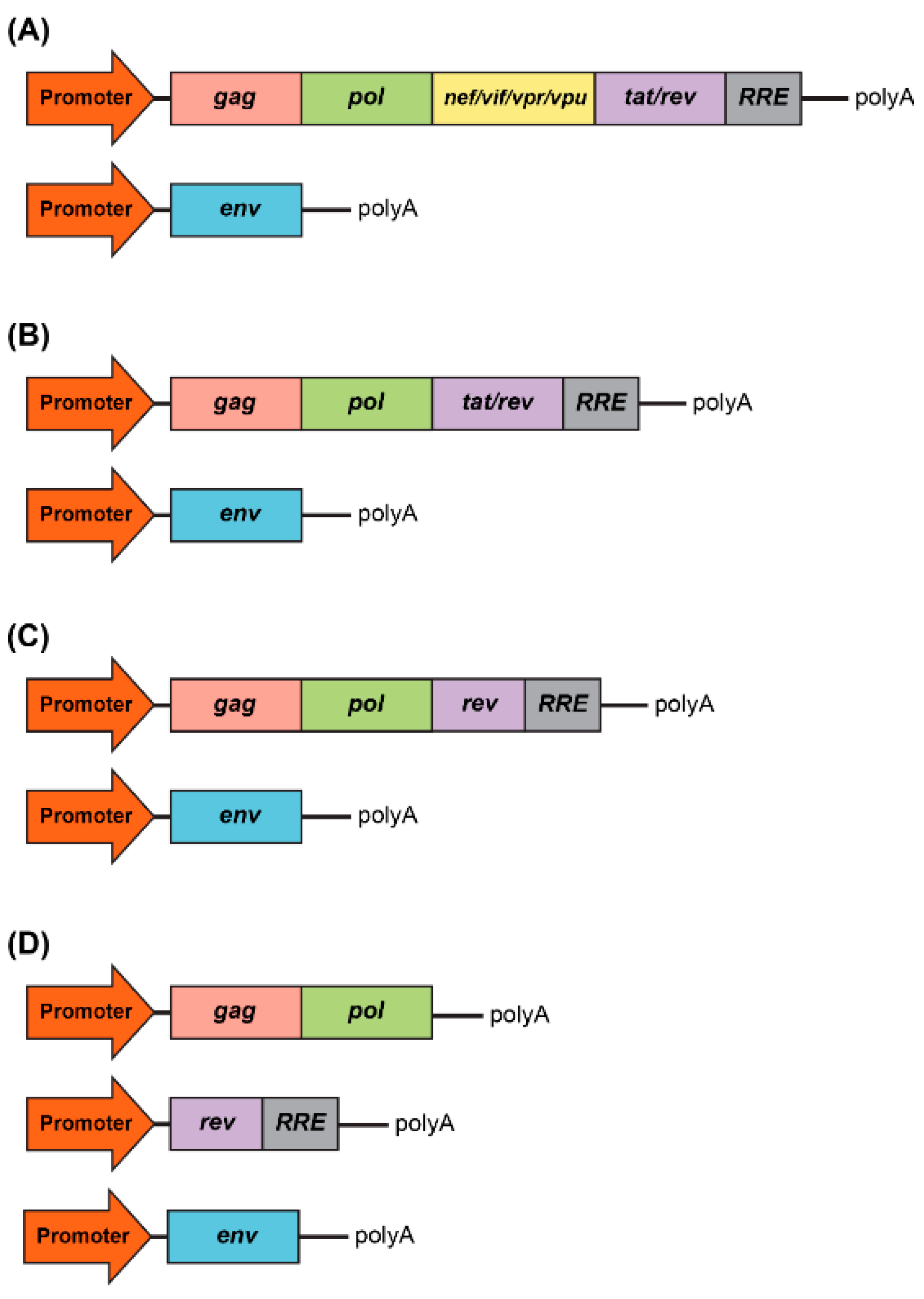
As a type of simple retrovirus, HIV-1 derived lentiviruses are capable to hijack host-mediated machinery to sustain an efficient nuclear import across the intact nuclear membrane [1]. This feature has allowed them to efficiently transduce nondividing and terminally differentiated cells (e.g., postmitotic neurons, hepatocytes, or macrophages) with superb efficiency (reviewed in [2]). The long-lasting effect of the viral transduction supports long-term production of the therapeutic gene-of-interest (thus providing permanent steady-state “dosing” after a single administration of the virus) which is essential for gene therapy applications. LV genome occupies ~10.7 kb of single-stranded RNA (ssRNA) placed inside a lipid-enriched spheric capsid of ~100 nm in diameter (Figure 1). The viral genome encodes both structural and enzymatic genes including gag and pol. The polycistronic gag gene encodes three products, namely matrix (MA), capsid (CA), and nucleoproteins (NC). The polycistronic pol gene supplies three viral enzymes, namely reverse transcriptase (RT), protease (PR), and integrase (IN) (Figure 1A). LV is an enveloped virus that uses a glycoprotein envelope to attach and enter the host cell. The creation of heterologous envelopes used for viral particle pseudotyping was one of the main progresses in the field that allowed to dramatically diversify and extend the tropism of transduction. Additionally, the supplementation of viral particles with heterologous envelopes has positively impacted vector safety (reviewed in [2]). As mentioned above, LVs are capable of being efficiently pseudotyped with a broad range of heterologous envelopes, enabling broad viral tropism. For example, LV supplemented with Mokola virus (MV), Ross River virus (RRV), and Rabies virus (RV) demonstrated a strong preference for the transduction into neuronal cells (reviewed in [3]). However, the most common envelope used to pseudotype viral particles is that of vesicular stomatitis virus protein G (VSV-G). The envelope has been shown to support an extremely broad range of tropism and as such is used for transduction into most cells and tissues (reviewed in [2]). In addition to gag and pol, lentiviruses carry six supplementary genes: rev and tat, involved in viral transcription and export, respectively, and nef, vif, vpr, and vpu, involved in viral entry, assembly, replication, particle formation and release (Figure 1) and [4]. It is important to note that the latter four accessory products are dispensable for vector production, and as such can be omitted from the packaging cassette of the vector (Figure 2A). The removal of these products has been shown to have a positive effect on the vector safety; importantly, their deletion also creates space for cloning larger inserts [5,6,7,8]. Indeed, the second generation of the packaging system carried only the tat and rev genes (Figure 2B) and [9]. The third generation of the packaging system does not have the tat gene, which is in co-incidental with the deletion of the endogenous promoter harboring tat-responsive element, TAR, at the U3′ region of the LTRs (Figure 2C). Instead, full-length RNA of the virus is transcribed from the strong ubiquitous promoter derived from Rous sarcoma virus (RSV) or cytomegalovirus (CMV). Further improvement of virus safety has been achieved with the development of the fourth-generation packaging plasmid, highlighted by the split of the gag/pol and rev sequences into two different cassettes (Figure 2D). In fact, the fourth generation of the packaging systems is the safest to date [7]. It should be noted that rev gene is present in all the packaging systems, as its product REV plays a key role in exporting fully length and partially spliced viral RNA (vRNA) from the nucleus into the cytoplasm [10]. The advanced generations of the packaging plasmids also harbor a strong, heterologous poly-adenylation signal (poly-A), derived from the SV40 virus or bovine/human growth hormone (bGH/hGH). These potent poly-As enable high level of vRNA stability, and as such their inclusion has been found to be advantageous for the packaging and viral titers [7,10]. Moreover, the inclusion of the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) and the central polypurine tract (cPPT) into the viral transfer cassette has been shown to further improve vRNA stability, transcription efficiency, and overall viral titer [11,12]. Importantly, the above modifications greatly reduce the likelihood of the appearance of recombination-competent retroviruses (RCR), which positively impacts viral safety characteristics.
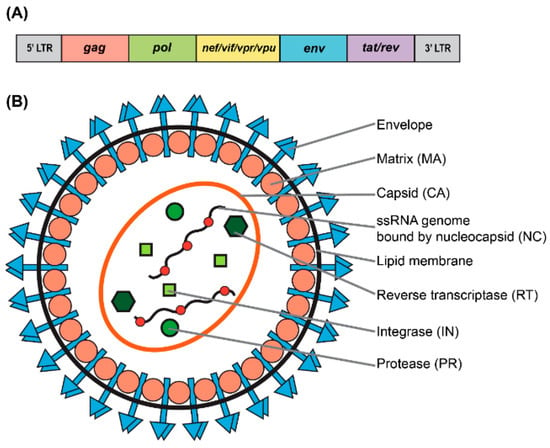
Figure 1.
Genomic and structural organization of lentiviral vector. (A) Simplified schematic of the wild-type human immunodeficiency virus type-1 (HIV-1) genome. (B) Particle structure of a lentivirus.
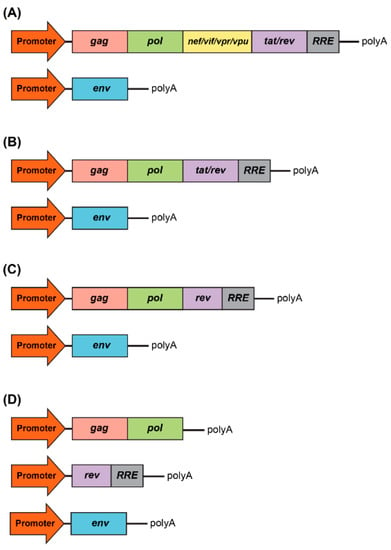
Figure 2.
Development of packaging system for lentiviral production. (A) First generation included all accessory genes, nef, vif, vpr, and vpu, and the regulatory proteins, tat and rev. RRE is the rev response element. (B) Second generation excluded all accessory genes, nef, vif, vpr, and vpu. (C) Third generation excluded the tat regulatory protein. (D) Fours generation is characterized by the split of the gag/pol and rev sequences into two different cassettes, which evidently benefits the vector safety.
2. The Life Cycle of Lentiviral Vectors
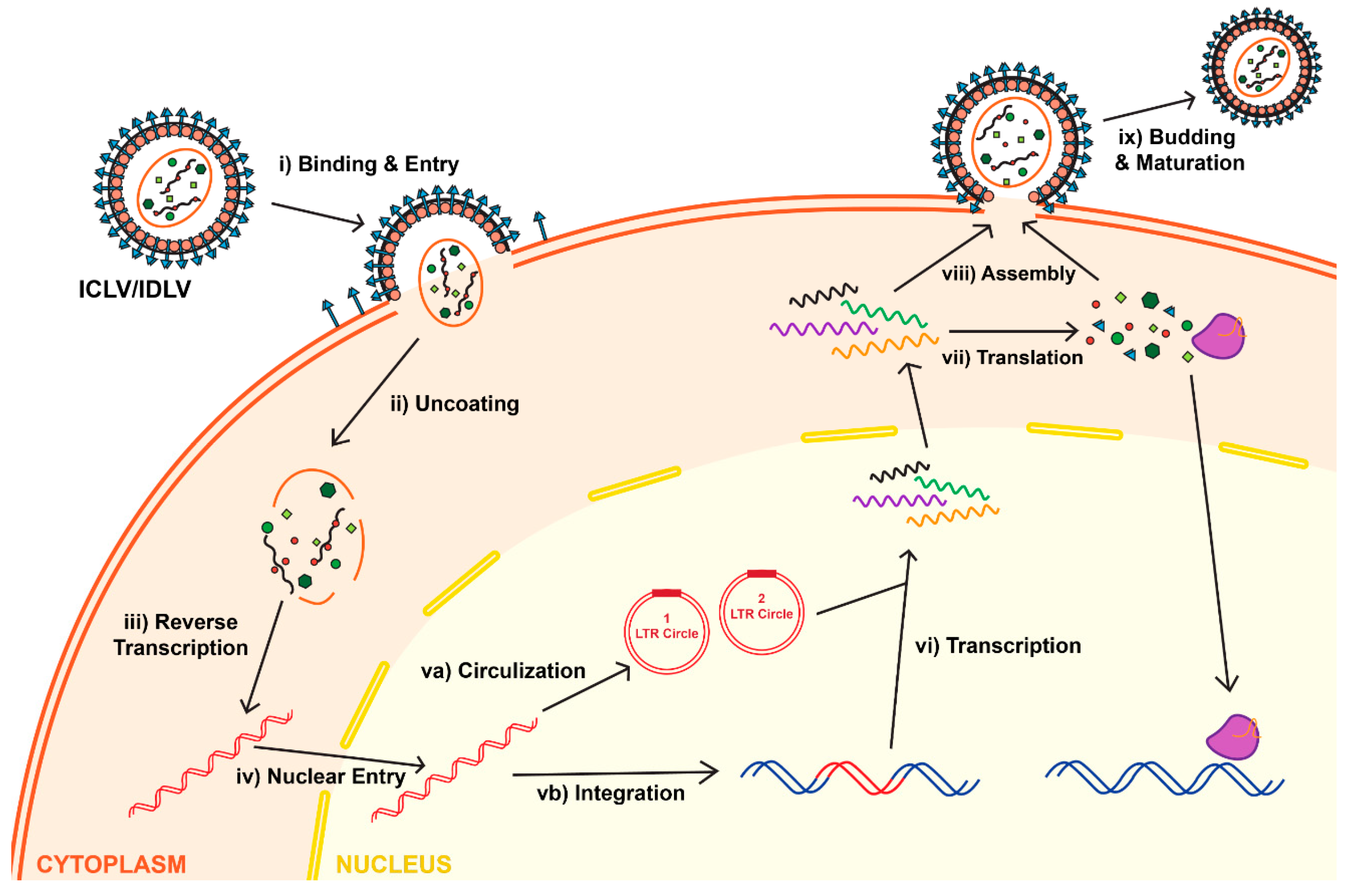
Attachment, binding and entry of the lentivirus is mediated via interaction between the envelope protein and its receptor. Following entry and uncoating, the reverse transcriptase enzyme (RT) is set to start the reverse transcription reaction. The product of the RT reaction is double-stranded (ds) linear DNA. The first step in the RT process depends on two cis-acting elements located within the viral RNA: the primer binding site (PBS) and the polypurine tract (PPT). The PBS is recognized by tRNALys3, which supplies a specific primer for the RT-mediated amplification. The PPT element comprises a purine-rich stretch of nucleotides that are resistant to RNase H-mediated degradation of positive strand of vRNA; the undegraded PPT sequence can then act as a primer towards the positive strand synthesis. As mentioned above, the completion of the RT reaction results in double stranded, linear DNA (reviewed in [2]). It is important to note that the vDNA has the same sequence as the full-length vRNA; the only difference between them is that vDNA carries two U3′, and two U5′ regions placed on both sides of the LTRs; whereas vRNA harbors a single U3′ and U5′ region. The U3′ region on the 5′-LTR carries the viral promoter (RSV or CMV), whereas U5′ of the 3′-LTR harbors the poly-A signal (reviewed in [2]). After the RT reaction is completed, the dsDNA undergoes nuclear import; this process is mediated by host-derived importin complexes. Following nuclear transportation, the vDNA serves as a precursor for viral integration. The int gene encodes the protein integrase (IN), which mediates the integration process by binding to and cleavage within the att sites located on the LTR ends [13,14,15]. Then, vDNA becomes an integral part of the host’s chromosome; it may replicate alongside the host genome, and can be passed on to the cell’s progeny (Figure 3) [16]. The RT enzyme is essential for vector production, whereas a functional lentivirus can be generated without active IN enzyme. We and others have taken an advantage of the fact that IN is not required for viral production and expression to construct an integrase-deficient lentiviral vector (IDLV), which demonstrates some important advantages over the parental integrase-competent lentiviral vectors (ICLV), discussed below.
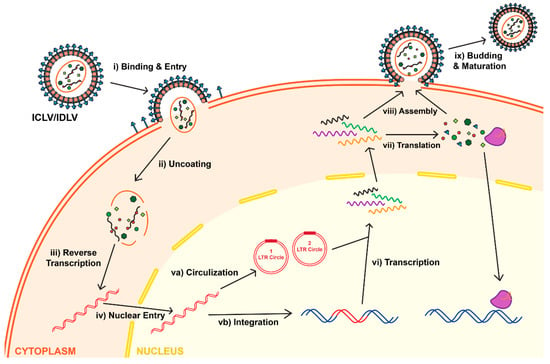
Figure 3.
Life cycle of lentiviruses for both Integrase-Competent Lentivirus (ICLV) and Integrase-Deficient Lentivirus (IDLV). Initially, IDLVs and ICLVs both bind and enter target cells (i). Uncoating of lentiviruses exposes the viral RNA (ii), allowing reverse transcription to occur in the cytoplasm (iii). The dsDNA product is then imported into the nucleus (iv). Some of this dsDNA integrates into the host genome (vb), while the majority recombines into one- or two-LTR circles and remain episomal (va). Due to the differences of the Integrase protein, the rates of integration and circle formation differ between IDLVs and ICLVs. The viral dsDNA undergoes transcription (vi), which is then translated in the cytoplasm into lentiviral structural proteins and gene of interests, such as the CRISPR/Cas system (vii). The viral RNA and structural protein assemble (viii), which then undergoes budding and maturation to form a new lentivirus (ix).
5. Adeno-Associate Vectors (AAVs)
This review is devoted to lentiviral vectors; however, it would be remiss not to mention the most frequently used viral platform for gene therapy, adeno-associated vector (AAV) (reviewed in [2]). AAV is an ideal viral system for several reasons: (i) the vector has no known associated pathologies and causes only a mild immune response in humans; (ii) similarly to IDLV vectors, the AAV genome can be persisted long-termly in episomal forms, and thus presents an opportunity for extended transgene expression in non-dividing cells and tissues [2]. (iii) Lastly, the AAV genomic structure is well-characterized, so the consequences of genome-targeted manipulations can adequately be predicted [2]. For the above considerations, over the last three decades a significant effort has been devoted to developing AAV into one of the gold-standard delivery systems for broad range of gene-therapy applications [40]. Notwithstanding these advances, impressive and rapidly diversifying array of AAV-CRISPR/Cas-derived tools predominantly have been used in vitro (reviewed in [41]). Efficient delivery in vivo using AAV vectors is a significantly more challenging task. In fact, the limited packaging capacity of the AAV genomes is the main bottleneck for its use, in an all-in-one configuration, for delivery of bulky and complex CRISPR/Cas transgenes in vivo. One of the approaches to overcome the significant restraints imposed by AAV’s ~4.7 kb functional packaging capacity is to physically split a CRISPR/Cas transgene into two pieces, which are packaged into separate AAV vectors. The resulting AAVs are then co-delivered, and the complete protein is reassembled in situ by a split intein—a pair of domains which “splice themselves out”, thus re-joining two peptides in the end-to-end configuration [42]. Nevertheless, further improvement of this and other systems (reviewed in [41]) would be necessary to achieve the desired therapeutic efficacy.
6. Overview of CRISPR/Cas9-Based Gene-Editing Systems
The clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein (Cas) system has recently evolved to be a revolutionary platform for both genome- and epigenome-editing manipulations in a broad range of tissues and organs. CRISPR/Cas has tremendously advanced our understanding of hereditary diseases by enabling the rapid generation of novel cellular and animal models. Furthermore, CRISPR/Cas has become a valuable and effective option for the treatment of many diseases and disorders which would otherwise be incurable. In this review, we aim to highlight the ongoing development of innovative gene-editing tools used with LV platforms in a broad range of research and clinical applications.
In nature, CRISPR/Cas systems act as a prokaryotic adaptive immunity mechanism to recognize, target, and destroy the foreign DNA and RNA of phages and viruses [43,44]. The CRISPR/Cas systems are very diverse, composing so far six Cas enzyme types (I–VI), with at least 30 subtypes [45,46]. Despite this diversity, the Cas family’s members share similar components, including a CRISPR-RNA, or guide RNA (gRNA), and in most cases a trans-activating RNA (tracrRNA), [47,48]. The most studied and developed system of CRISPR/Cas is derived from the class II CRISPR-associated enzyme Cas9, which operates as a single effector; in contrast, class I Cas enzymes act as a multi-subunit protein system (reviewed in [49]). In this review, we will focus only on CRISPR/Cas9 systems; for comprehensive reviews on other Cas proteins, we would refer the reader to the following publications [50]. To achieve genome-editing several important steps have to occur, leading to the assembly of both RNAs (gRNA and tracrRNA) into one CRISPR/Cas complex. The combined version of both, namely synthetic guide RNA (sgRNA), greatly simplifies the delivery and expression of CRISPR/Cas systems. Cas9 is capable of binding DNA only in the presence of a specific sequence, known as a protospacer-adjacent motif (PAM). The recognition of the PAM motif enables Watson–Crick RNA-DNA base pairing. Following assembly of the ternary complex between RNA, DNA, and Cas9, the endonuclease domains become active, and cleave within both DNA strands, which results in the formation of double-strand DNA breaks (DSBs). The unprecedented efficiency, accuracy, and specificity of the Cas9 protein has been rapidly recognized and directed towards a wide range of genome-editing applications, from basic science to translational and clinical research and medicine [51].
7. The Use of Active Cas9 for Genome-Editing Applications
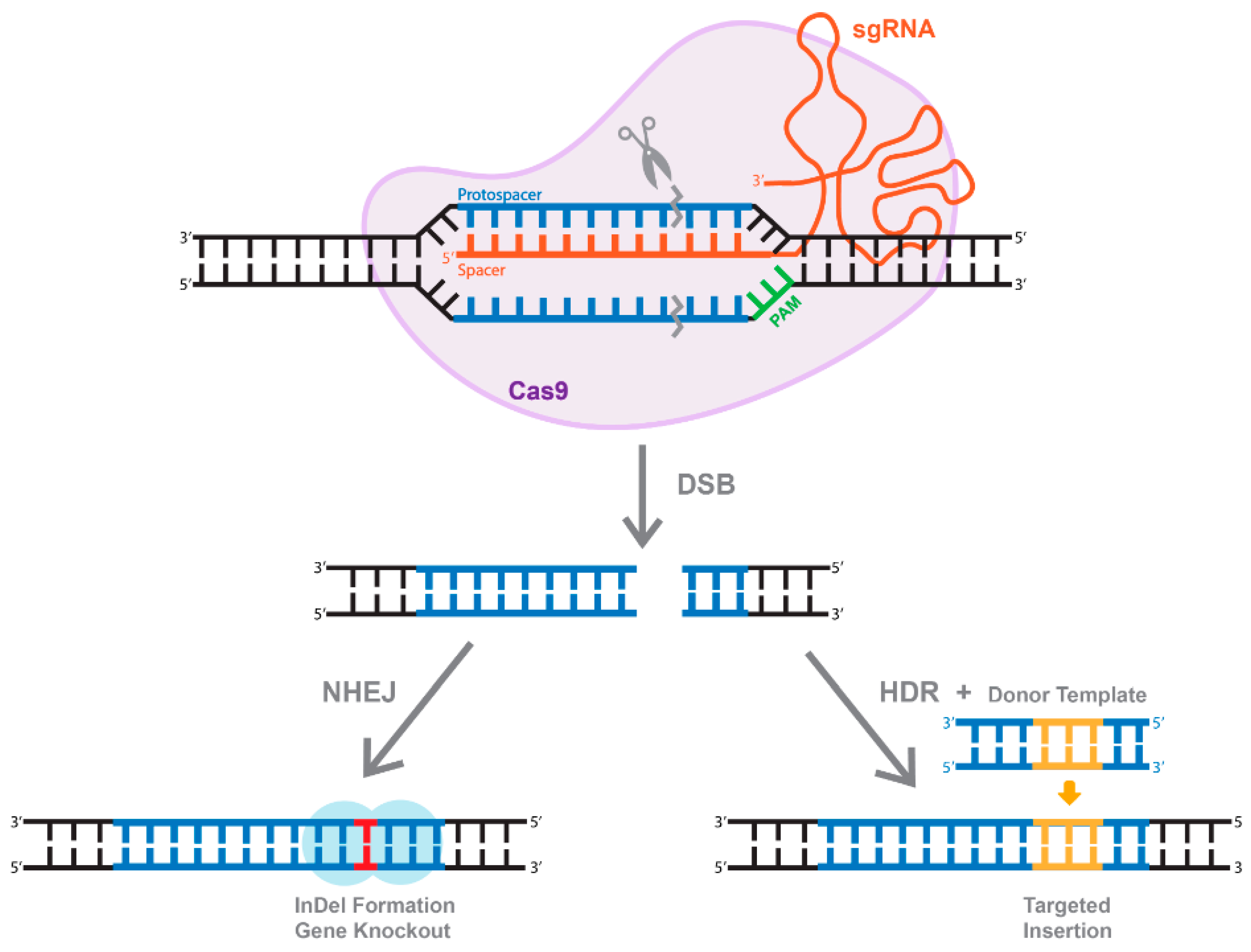
As mentioned above, cleavage mediated by Cas9 results in the generation of DSBs (Figure 4). Eukaryotic organisms mainly repair DSBs through an error-prone non-homologous end joining (NHEJ) mechanism, which leads to the formation of small insertions or deletions (InDels) (Figure 4). Alternatively, if a repair donor template is provided, host-mediated repair machinery can activate the homology-directed repair (HDR) pathway, resulting in error-free replacement of the target sequence (Figure 4). Unfortunately, HDR is typically characterized by very low efficiency [52]. In fact, it is widely accepted that the rate of HDR events range between 0.1 and 1%, depending on the system and the target gene sequence [52]. The rate could be even lower in vivo [53]. Another drawback of the HDR pathway is that it is not active in non-dividing and terminally differentiated cells, including brain neurons. Furthermore, the DSBs needed to trigger more efficient HDR also increase the possibility of non-specific cleavages and recombinations, and even on-target HDR can have highly negative effects on the cells [54,55]. These limitations established a significant need for the development of single-base-pair editing and prime-editing technologies to enable precise genome editing in non-dividing cells and tissues (discussed in detail below and reviewed in [56,57]).
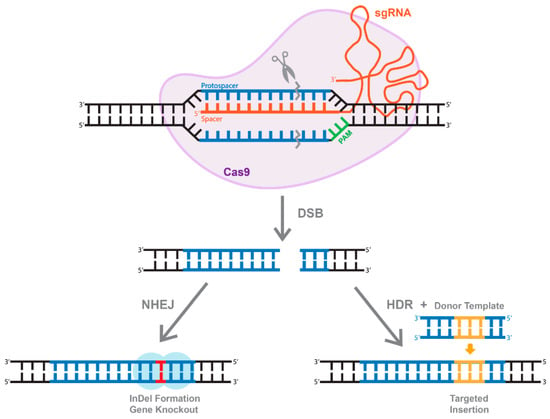
Figure 4.
The overview of CRISPR/Cas system. Active Cas9 enzyme introduces a double-stranded break (DBS). The cell repairs through two methods, one of which is through non-homologous end joining (NHEJ) that create InDels. Alternatively, of a dsDNA donor template is provided, the DSB is repaired through homologous recombination (HDR), resulting in a targeted insertion.
8. Deactivated Cas9 (dCas9) for CRISPRi and CRISPRa Approaches
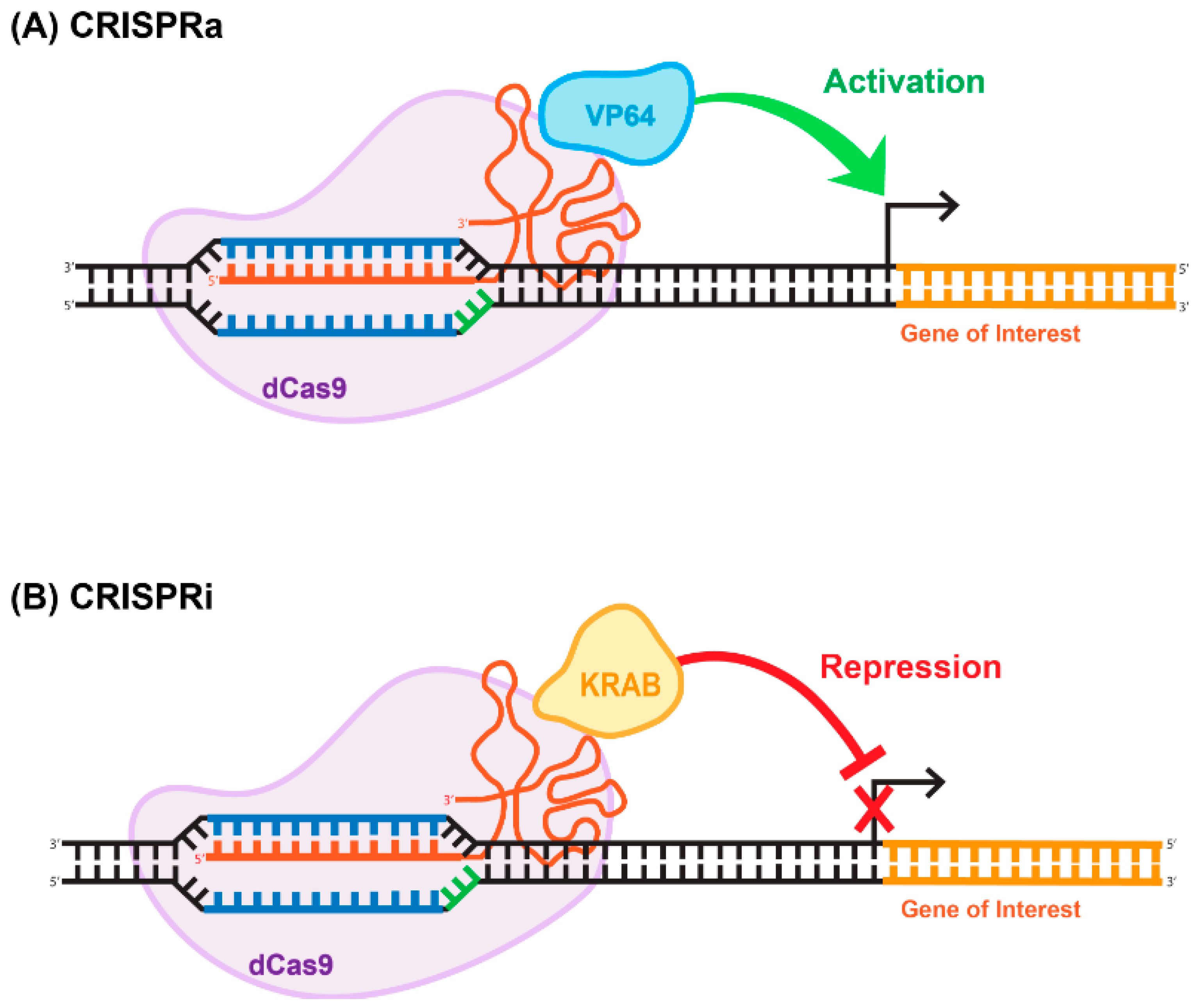
The ability of Cas9 to bind to specific sequences with strong affinity is of immense value in and of itself, independently of its endonuclease-enzymatic activity. Cas9 is a modular enzyme that possesses well-characterized catalytic domains. The amino acid residues that are necessary for the endonucleic activity of the enzyme are located in the RuvC and HNH domains [48,58]. It has been demonstrated that D10A and H840A substitutions in the RuvC and HMH domains, respectively, greatly reduce the catalytic activity of the enzyme without compromising the ability of the deactivated enzyme to bind to the targeted DNA sequence [48,58]. This mutated version of Cas9 is called deactivated or dead; implying that it supports no endonucleatic activity. Deactivated Cas9 has been widely used for transcriptional and epigenomic targeting. For example, dead Cas9 (dCas9) linked to a transcription repressor (e.g., KRAB) can be used for targeted repression of a gene-of-interest. Similarly, gene activation can be achieved using a dCas9- activator fusion variant (reviewed in [59]). The CRISPR/dCas9 systems used for gene activation and repression have been coined CRISPR-activation (CRISPRa) and CRISPR-interference (CRISPRi), (Figure 5). The basic repression system includes a dCas9-KRAB fusion; conversely, activation can be achieved using dCas9 fused to the VP64 effector (Figure 5), (reviewed in [60]). These basic tools paved the way for the development of more robust and efficient activation and repression systems, including SAM and SunTag [61]. The SunTag system, developed by Tanenbaum and colleagues in [61], utilizes the interaction between 10–24 copies of the short epitope GCN4 and its cognate scFV antibody expressed from a separate plasmid [61]. Placing dCas9-GCN4 on one vector and various effectors, activators or repressors fused with the scFV antibody on the other vector, enabled the high-affinity interaction, resulting in functional activation or repression. The reported levels of the activation and repression are significantly higher than those achieved with more basic systems [61]. Alternative to the SunTag system, the human CRISPR/Cas9 synergistic activation mediator (SAM) approach has been developed in Feng Zhang’s laboratory [62]. Similar to SunTag system, SAM uses several strong transcriptional activators linked together to amplify the effect. These activators are VP64, heat-shock factor 1 (HSF-1), and the p65 subunit of NF-kappaB (NF-kB) [63]. It is important to note that CRISPRi- and CRISPRa-mediated outcomes are reversible in general, as both systems target chromatin structure rather than the DNA sequence. Obviously this feature is appealing for many gene therapy applications. We recently developed a novel epigenome-editing platform based on an all-in-one LV for targeted DNA methylation editing within intron 1 of the alpha-synuclein-encoding gene, SNCA. Dysregulation of SNCA expression is one of the causative factors for Parkinson’s disease (PD) [64]. The system consists of dCas9 fused with the catalytic domain of DNA-methyltransferase 3A (DNMT3A). Applying the system to human-induced pluripotent stem cell (hiPSC)-derived dopaminergic neurons from a Parkinson’s disease patient with the SNCA triplication resulted in robust and steady downregulation of SNCA mRNA and protein levels, mediated by targeted DNA methylation at a regulatory sequence located in intron 1 of SNCA. We further demonstrated that this reduction in SNCA levels is capable of rescuing disease-related phenotypes. This developed approach suggests broad potential for this target sequence combined with LV-CRISPR-dCas9 technology as a novel epigenetic-based therapeutic strategy for various cases of PD [65].
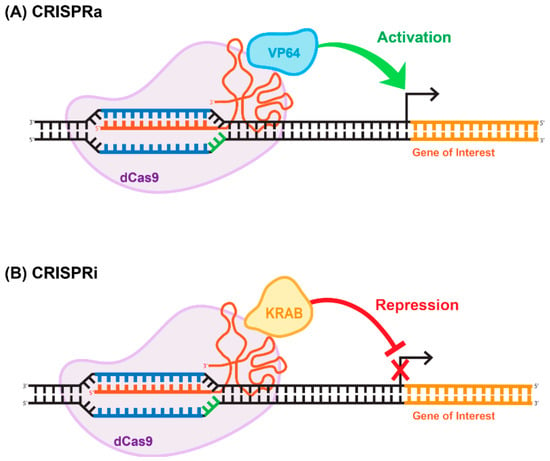
Figure 5.
Repurposing CRISPR/Cas for epigenome-editing applications. (A) CRISPR-activation (CRISPRa) often consists of a dCas9 fused directly to a single transcriptional activator (e.g., VP64), which then activates the gene of interest. (B) Similarly, CRISPR-interference (CRISPRi) often consists of a dCas9 fused directly to a single transcriptional repressor (e.g., KRAB), which then represses the gene of interest.
9. Base-Pair Editing Technology
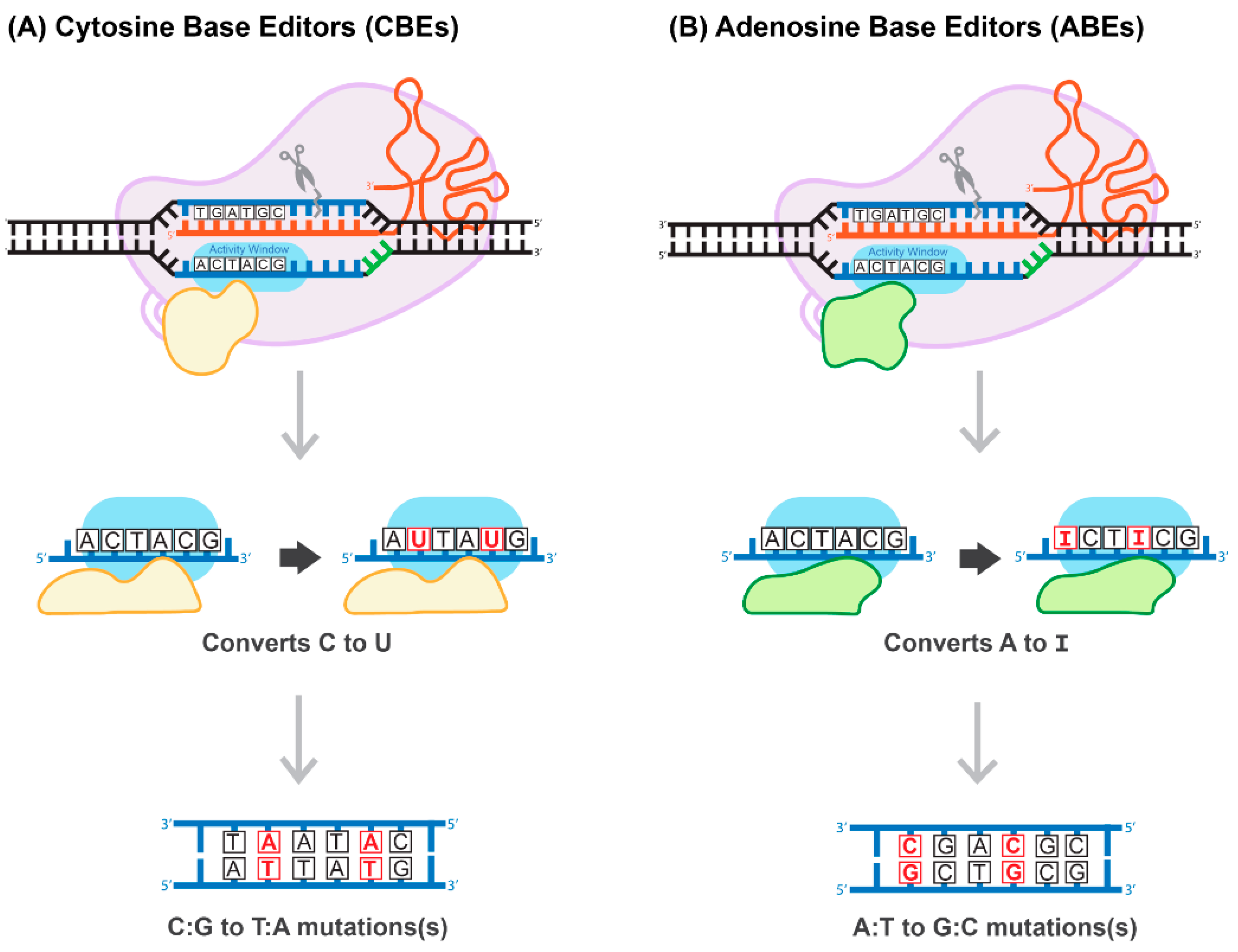
Generally, the most common genetic variants associated with hereditary diseases in humans are point mutations and functional single-nucleotide polymorphisms (SNPs) characterized by high-level penetration [66]. Therefore, gene-editing systems capable of inducing a robust, efficient, and safe conversion on a single nucleotide level have the potential to cure many hereditary diseases. The construction of a cytosine base-editor (CBE) system by David Liu’s group was the first important development towards the establishment of such tools (Figure 6A). In the paper published by Komor and colleagues, dCas9 was fused with rat APOBEC1, a cytosine deaminase enzyme [67]. The resulting system induces conversion of all cytosines to uracils within an approximately six-nucleotide window, counted from the PAM-20 to the left (upstream). The uracil is then read as thymine during replication, completing the C-to-T conversion [67]. To overcome a caveat associated with the activation of base excision repair, catalyzing U-to-C re-conversion, the second generation of CBE carried a uracil glycosylase inhibitor domain to deactivate the base-excision repair pathway. This improvement greatly enhances the efficiency of the base editor. The use of nickase Cas9 (nCas9) to add cuts in non-edited strands further improved CBE efficiency. The new system has been coined BE3 and has been proven to be highly efficient in a variety of human cells, with a correction rate ranging between 25–75%. It is important to note that the nickase approach also results in an unwanted increase in the formation of InDels, from less than 0.1% to approximately 1% [67]. More recently, base-editing systems have been further enhanced. For example, Komor et al. created the BE4-Gam system, carrying a second copy of the uracil glycosylase inhibitor and a bacteriophage protein, Gam. Gam has a high affinity for the free ends of DSBs, thus preventing NHEJ-mediated repair and reducing InDel formation [68]. Koblan et al. inserted two nuclear localization signals (NLS), placing them proximal and distal to nCas9. In addition, they evolved a more efficient deaminase by constructing a codon-optimized version of the APOBEC domain, yielding BE4max and ancBE4max [69]. Other groups have focused on limiting or expanding the cytosine deaminase activity window, allowing for C-to-T conversions within a window as short as three or as long as twelve nucleotides [70].
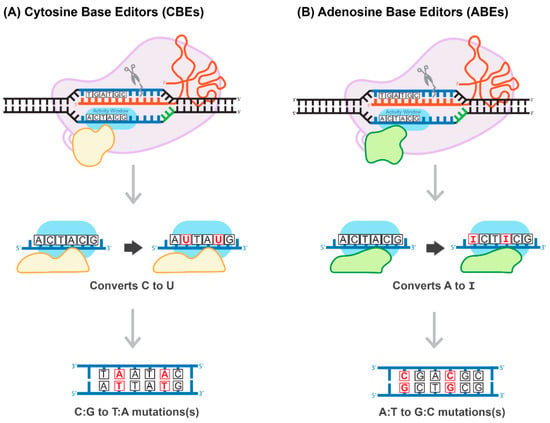
Figure 6.
Proposed mechanism of base-pair editing CRISPR/Cas technology. (A) Cytosine Base Editors (CBEs) catalyze the conversion of all cytosines to uracils within a 5-6 activity window. Uracil is read as thymine during replication, which then converts all C:G to T:A. (B) Adenosine Base Editors (ABEs) catalyze the conversion of all adenosines to inosines within a 5-6 activity window. Inosine is read as guanine during replication, which then converts all A:T to G:C.
The development of an adenosine base editor (ABE) platform supporting A-to-G transitions vastly broadened the applications of base editing (Figure 6B). The original ABE platform was created by Gaudelli and colleagues, who connected nCas9 with an evolved deoxyadenosine deaminase, which catalyzes an adenosine to inosine transition [71]. Like the two-step CBE reaction, the inosine is then read as guanine during replication, completing the A-to-G transition. The efficiency of the ABE7 system engineered by Gaudelli et al. [71] was 50% in HEK293 cells, with an InDel formation rate of less than 0.1%. Notwithstanding this success, ABE7 was found to be incompatible with Cas9 of any species other than Streptococcus pyogenes (SpCas9). The incompatibility is due to the low DNA-bound residence-time of non-SpCas9, coupled with the decelerated enzymatic pace of deoxyadenosine deaminase [72]. To circumvent this bottleneck, Richter and colleagues used phage-assisted-continuous evolution (PACE) and phage-assisted non-continuous evolution (PANCE) methods to evolve the ABE8e system, which shows a greatly accelerated catalytic rate for the deoxyadenosine deaminase process, resulting in substantially higher efficiency [72]. ABE8e also displays increased processivity, which could be particularly advantageous for the multiplexed perturbations. Further improvement of the ABE system by Gaudelli and colleagues created an array of new eighth-generation ABEs, characterized by increased activity and editing efficiency and a broader window of editing [73]. This novel system has shown success in an adult mouse model of Duchenne muscular dystrophy, by enabling correction of the DMB gene in 17% of myofibers, with no measurable InDels or other off-target effects. It has to be pointed out that this rate of gene correction is therapeutic, considering the threshold level of 4% of the gene expression that is needed to correct muscle function [73]. Most recently, three different groups reported the development of CRISPR-Cas9-based dual programmable adenine and cytosine editors, capable of introducing A-to-G and C-to-T substitutions at the same time, with minimal off-target edits [74]. These dual CBE/ABE systems expand the range of possible DNA sequence alterations, broadening the research applications of CRISPR base editors.
10. Prime-Editing Technology
The majority of mutations linked to human genetic diseases are transitions; nevertheless, according with ClinVar database, correcting about 40% of the hereditary diseases would require transforming a purine-base nucleotide into pyrimidine-base nucleotide or vice versa. Thus, the bottleneck of the base-editing technology was recently addressed by David Liu’s group, who found a very elegant way to address this problem. In their recent publication, entitled “Search-and-replace genome editing without double-strand breaks or donor DNA,” Anzalone et al. described a new method of genome editing based on the rewriting genetic information into a specified DNA site using a dCas9 or nCas9 fused to an engineered viral reverse transcriptase (RT), paired with a prime editing guide RNA (pegRNA) that both specifies the target sequence and encodes the edit-of-interest [57] and (Figure 7). The authors validated these “prime editing” tools in human cells by correcting mutations in sickle cell disease and Tay-Sachs disease (efficiently and with few byproducts), generating a protective transversion in PRNP, and introducing various epitopes and tags into targets-of-interest [57]. Prime editing has demonstrated comparable or higher efficiency than the HDR-based equivalent and has had complementary strengths and weaknesses compared to base editing [57]. Consistent with these observations, we demonstrated that prime-editing system delivered by lentiviral vector is capable to correct any mutations precisely and specifically, albeit with low efficiency (personal communications). Nevertheless, in our hands, the efficacy of the prime editing was found to be significantly higher than that of the HDR. In fact, we found that the rate of the mutations introduced using the prime-editing approach ranges between 5 and 15% depending on the used system (personal communications). We believe that the low editing capacity of the prime-editing technology could stem from the fact that the expression level of Cas9-RT is fairly reduced, similar to the expression of base-editing tools discussed above (personal communications). As such, further evolution of the prime editing system will be necessary to address its shortcomings. Overall, prime editing substantially expands the scope and capabilities of genome editing, and in principle could correct up to 89% of known genetic variants associated with human diseases [57]. A good summary expanding on the diversity of dCas9-effector tools and systems could be also found in Rittiner et al. [41].
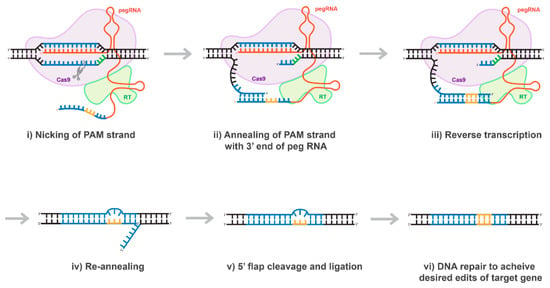
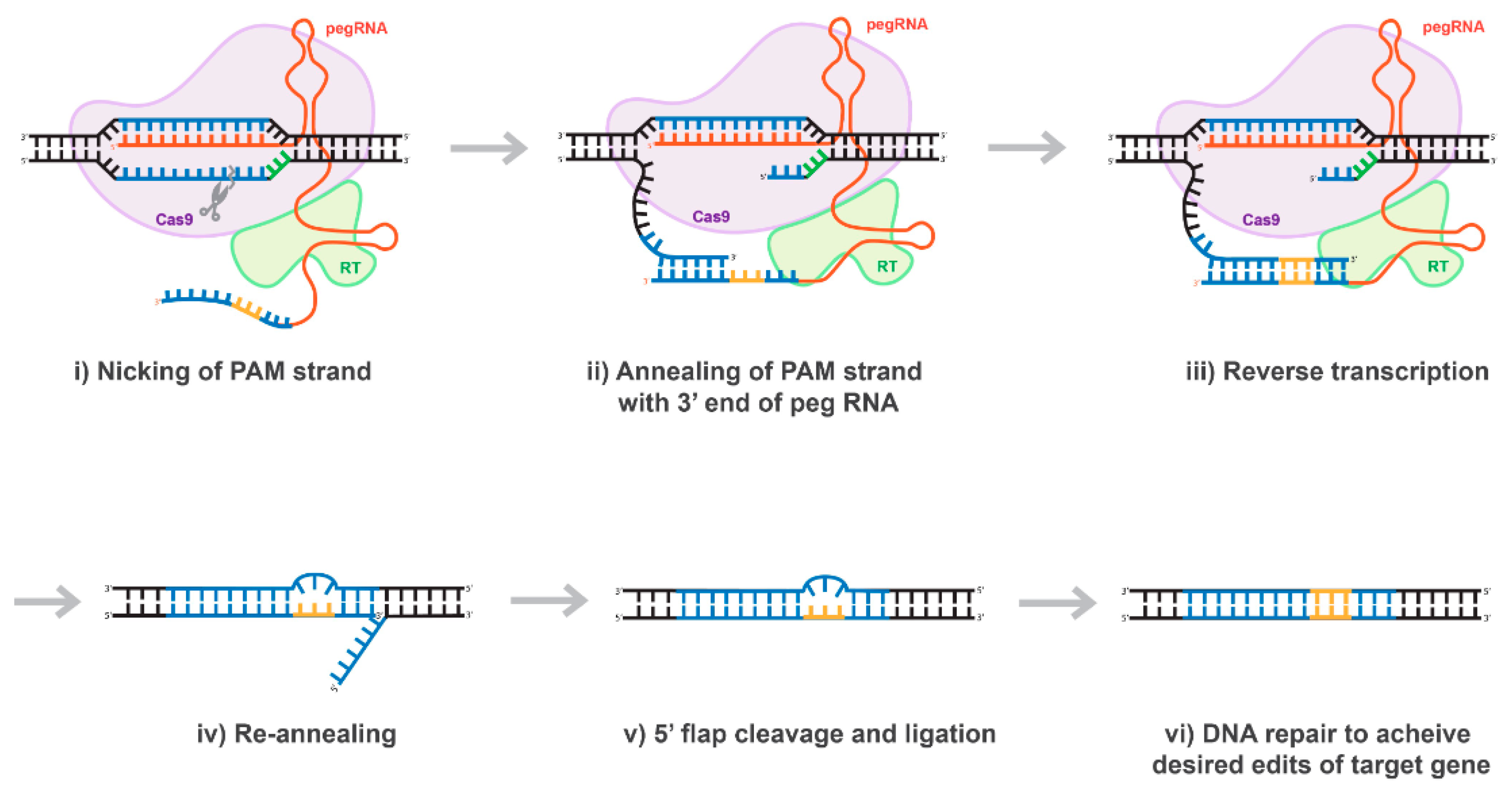
Figure 7.
Proposed mechanism of prime editing CRISPR/Cas technology. Once the 5’ end of the pegRNA (spacer) binds to the protospacer of the target DNA, the protospacer-adjacent motif (PAM) strand of the target DNA is nicked (i). The nicked PAM strand hybridizes with the primer binding site (PBS) on the 3’ end of the pegRNA (ii). The pegRNA serves as a template as the reverse transcriptase (RT) fused to the Cas9 enzyme extends the 3’ end of the nicked PAM strand (iii). The prime editing CRISPR/Cas system disengages and the target site is left with a 3’ flap (the edited PAM strand) and a 5’ flap (the original PAM strand) (iv). The 5’ flap is preferentially degraded by cellular endonucleases and the edited PAM strand ligases and hybridizes with the non-PAM strand (v). Finally, through DNA repair mechanisms, the desired edits of the target gene are transferred to the non-PAM strand (vi).
12. Conclusions
CRISPR/Cas technology has revolutionized all aspects of modern life science research, ranging from basic science to clinical research. Importantly, recent progress in lentiviral vectorology has fully supported and enabled robust and efficient delivery of CRISPR/Cas tools into tissues- and organs-of-interest in vivo, opening doors for novel approaches and strategies previously unimaginable. While these innovative technologies and tools continue to progress, several key issues still need to be solved. One of the issues is the relatively high levels of undesirable off-target effects induced as the consequence of permanent expression of CRISPR/Cas9 tools delivered by LVs. In addition to causing undesirable effects outside of the targeted gene or region, integrating lentivirus (mostly meaning simple retroviruses, e.g., γ-retrovirus) is capable of inducing measurable levels of toxicity and pathogenicity, stemming mostly from its oncogenic potential. Unfortunately, other viral delivery platforms are not fully detached from the adverse effects generally associated with viruses and may cause tragic outcomes. [71,72,73]. Adverse reactions resulting from the integrating nature of WT-LVs could be minimized with transient, non-integrating vectors, e.g., IDLVs. Indeed, we recently reported that IDLVs demonstrate significantly lower levels of InDel formation and other off-target effects compared to their integrase-competent counterparts, both in vitro and in vivo [37,39]. Another potential issue is that permanently expressed, host genome-integrated lentivirus may be a target for mobilization by incoming non-infectious (vector) or infectious (HIV-1) viral particles in the case of superinfection. Vector mobilization may lead to even higher levels of integration events, and eventually to the insertional mutagenesis, oncogenic activation, and other off-target perturbations mentioned above. From this perspective, IDLVs would also be preferred over WT-LVs, as IDLVs have demonstrated lower levels of vector-with-vector and vector-with-virus mobilization events [21]. However, further improvement in lentivirus design aimed to create a recombinogenic-less vector will be needed to fully address this concern [17]. Lastly, as disease progression is often dynamic over time, it will be important to improve spatial- and time-regulation of target gene expression to achieve more accurate and fine-tuned treatment.
Author Contributions
W.D.; Writing, and editing, figures preparation. B.K.; Conceptualization, Writing—Original draft, Writing—Review and Editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We thank Joseph Rittiner and Benjamin Johnson helpful discussion and editing of the manuscript. We thank Seelos Therapeutics Inc for supporting the study.
Conflicts of Interest
Boris Kantor is the owner of CLAIRIgene LLC and consultant for Seelos Therapeutics, Inc.
References
- Lewis, P.F.; Emerman, M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 1994, 68, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Kantor, B.; Bailey, R.M.; Wimberly, K.; Kalburgi, S.N.; Gray, S.J. Methods for Gene Transfer to the Central Nervous System. Agric. Food Prod. 2014, 87, 125–197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Y.; Reiser, J. Altering the Tropism of Lentiviral Vectors through Pseudotyping. Curr. Gene Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef]
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. The Interactions of Retroviruses and their Hosts. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1997. [Google Scholar]
- Naldini, L.; Blomer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomer, U.; Naldini, L.; Kafri, T.; Trono, D.; Verma, I.M.; Gage, F.H. Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J. Virol. 1997, 71, 6641–6649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafri, T.; Blömer, U.; Peterson, D.A.; Gage, F.H.; Verma, I.M. Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nat. Genet. 1997, 17, 314–317. [Google Scholar] [CrossRef]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Cockrell, A.S.; Ma, H.; Fu, K.; McCown, T.J.; Kafri, T. A trans-lentiviral packaging cell line for high-titer conditional self-inactivating HIV-1 vectors. Mol. Ther. 2006, 14, 276–284. [Google Scholar] [CrossRef]
- Zufferey, R.; Donello, J.E.; Trono, D.; Hope, T.J. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol. 1999, 73, 2886–2892. [Google Scholar] [CrossRef] [Green Version]
- Zennou, V.; Petit, C.; Guetard, D.; Nerhbass, U.; Montagnier, L.; Charneau, P. HIV-1 Genome Nuclear Import Is Mediated by a Central DNA Flap. Cell 2000, 101, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Colicelli, J.; Goff, S.P. Mutants and pseudorevertants of moloney murine leukemia virus with alterations at the integration site. Cell 1985, 42, 573–580. [Google Scholar] [CrossRef]
- Craigie, R.; Fujiwara, T.; Bushman, F. The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell 1990, 62, 829–837. [Google Scholar] [CrossRef]
- Leavitt, A.D.; Rose, R.B.; Varmus, H.E. Both substrate and target oligonucleotide sequences affect in vitro integration mediated by human immunodeficiency virus type 1 integrase protein produced in Saccharomyces cerevisiae. J. Virol. 1992, 66, 2359–2368. [Google Scholar] [CrossRef] [Green Version]
- Buchow, H.D.; Tschachler, E.; Gallo, R.C.; Reitz, M. HIV-I Replication Requires an Intact Integrase Reading Frame. Acute Leuk. 1989, 32, 402–405. [Google Scholar] [CrossRef] [Green Version]
- Kim, V.N.; Mitrophanous, K.; Kingsman, S.M.; Kingsman, A.J. Minimal requirement for a lentivirus vector based on human immunodeficiency virus type 1. J. Virol. 1998, 72, 811–816. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Blomer, U.; Takahashi, M.; Gage, F.H.; Verma, I.M. Development of a self-inactivating lentivirus vector. J. Virol. 1998, 72, 8150–8157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnell, T.; Foley, P.; Wirth, M.; Munch, J.; Uberla, K. Development of a Self-Inactivating, Minimal Lentivirus Vector Based on Simian Immunodeficiency Virus. Hum. Gene Ther. 2000, 11, 439–447. [Google Scholar] [CrossRef]
- Iwakuma, T.; Cui, Y.; Chang, L.-J. Self-Inactivating Lentiviral Vectors with U3 and U5 Modifications. Virology 1999, 261, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Kantor, B.; Ma, H.; Webster-Cyriaque, J.; Monahan, P.E.; Kafri, T. Epigenetic activation of unintegrated HIV-1 genomes by gut-associated short chain fatty acids and its implications for HIV infection. Proc. Natl. Acad. Sci. USA 2009, 106, 18786–18791. [Google Scholar] [CrossRef] [Green Version]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; Basile, G.D.S.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.-L.; et al. Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi, L.S.; Benedicenti, F.; Ambrosi, A.; DI Serio, M.S.; et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Themis, M.; Waddington, S.N.; Schmidt, M.; von Kalle, C.; Wang, Y.; Al-Allaf, F.; Gregory, L.G.; Nivsarkar, M.; Themis, M.; Holder, M.V.; et al. Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol. Ther. 2005, 12, 763–771. [Google Scholar] [CrossRef]
- Tucci, F.; Scaramuzza, S.; Aiuti, A.; Mortellaro, A. Update on Clinical Ex Vivo Hematopoietic Stem Cell Gene Therapy for Inherited Monogenic Diseases. Mol. Ther. 2021, 29, 489–504. [Google Scholar] [CrossRef]
- Aiuti, A.; Roncarolo, M.G.; Naldini, L. Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: Paving the road for the next generation of advanced therapy medicinal products. EMBO Mol. Med. 2017, 9, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Schuessler-Lenz, M.; Enzmann, H.; Vamvakas, S. Regulators’ Advice Can Make a Difference: European Medicines Agency Approval of Zynteglo for Beta Thalassemia. Clin. Pharmacol. Ther. 2020, 107, 492–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Englund, G.; Orenstein, J.M.; Martin, M.A.; Craigie, R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 1995, 69, 2729–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, M.; Kantor, B.; Cockrell, A.; Ma, H.; Zeithaml, B.; Li, X.; McCown, T.; Kafri, T. A Large U3 Deletion Causes Increased In Vivo Expression From a Nonintegrating Lentiviral Vector. Mol. Ther. 2008, 16, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Muñoz, R.J.; Balaggan, K.S.; MacNeil, A.; Howe, S.J.; Schmidt, M.; Smith, A.J.; Buch, P.; MacLaren, R.; Anderson, P.N.; Barker, S.E.; et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 2006, 12, 348–353. [Google Scholar] [CrossRef]
- Philippe, S.; Sarkis, C.; Barkats, M.; Mammeri, H.; Ladroue, C.; Petit, C.; Mallet, J.; Serguera, C. Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 17684–17689. [Google Scholar] [CrossRef] [Green Version]
- Kantor, B.; Bayer, M.; Ma, H.; Samulski, J.; Li, C.; McCown, T.J.; Kafri, T. Notable Reduction in Illegitimate Integration Mediated by a PPT-deleted, Nonintegrating Lentiviral Vector. Mol. Ther. 2011, 19, 547–556. [Google Scholar] [CrossRef]
- Ortinski, P.I.; O’Donovan, B.; Dong, X.; Kantor, B. Integrase-Deficient Lentiviral Vector as an All-in-One Platform for Highly Efficient CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. - Methods Clin. Dev. 2017, 5, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Saida, H.; Matsuzaki, Y.; Takayama, K.; Iizuka, A.; Konno, A.; Yanagi, S.; Hirai, H. One-year follow-up of transgene expression by integrase-defective lentiviral vectors and their therapeutic potential in spinocerebellar ataxia model mice. Gene Ther. 2014, 21, 820–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VijayRaghavan, S.; Kantor, B. A Protocol for the Production of Integrase-deficient Lentiviral Vectors for CRISPR/Cas9-mediated Gene Knockout in Dividing Cells. J. Vis. Exp. 2017, e56915. [Google Scholar] [CrossRef]
- Kantor, B.; McCown, T.; Leone, P.; Gray, S.J. Clinical Applications Involving CNS Gene Transfer. Adv. Genet. 2014, 87, 71–124. [Google Scholar] [CrossRef] [Green Version]
- Rittiner, J.E.; Moncalvo, M.; Chiba-Falek, O.; Kantor, B. Gene-Editing Technologies Paired With Viral Vectors for Translational Research Into Neurodegenerative Diseases. Front. Mol. Neurosci. 2020, 13, 148. [Google Scholar] [CrossRef]
- Lim, C.K.; Gapinske, M.; Brooks, A.K.; Woods, W.S.; Powell, J.E.; C., M.A.Z.; Winter, J.; Perez-Pinera, P.; Gaj, T. Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol. Ther. 2020, 28, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Sorek, R.; Kunin, V.; Hugenholtz, P. CRISPR — A widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Genet. 2008, 6, 181–186. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Annotation and Classification of CRISPR-Cas Systems. Methods Mol. Biol. 2015, 1311, 47–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, K.S.; Wolf, Y.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.; Barrangou, R.; Brouns, S.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Genet. 2015, 13, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nat. Cell Biol. 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef]
- Hsu, P.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.D.; Ray, G.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Rosenblum, D.; Gutkin, A.; Dammes, N.; Peer, D. Progress and challenges towards CRISPR/Cas clinical translation. Adv. Drug Deliv. Rev. 2020, 154-155, 176–186. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Badran, A.H.; Liu, D.R. Editing the Genome Without Double-Stranded DNA Breaks. ACS Chem. Biol. 2018, 13, 383–388. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.; Levy, J.M.; Chen, P.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nat. Cell Biol. 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Thakore, P.I.; Black, J.B.; Hilton, I.B.; Gersbach, C.A. Editing the epigenome: Technologies for programmable transcription and epigenetic modulation. Nat. Methods 2016, 13, 127–137. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.; Habib, N.; Gootenberg, J.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nat. Cell Biol. 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, A.; Scheiman, J.; Vora, S.D.; Pruitt, B.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA Expression by Targeted Editing of DNA Methylation: A Potential Strategy for Precision Therapy in PD. Mol. Ther. 2018, 26, 2638–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.; Moncalvo, M.; Tringali, D.; Tagliafierro, L.; Shriskanda, A.; Ilich, E.; Dong, W.; Kantor, B.; Chiba-Falek, O. The mechanistic role of alpha-synuclein in the nucleus: Impaired nuclear function caused by familial Parkinson’s disease SNCA mutations. Hum. Mol. Genet. 2020, 29, 3107–3121. [Google Scholar] [CrossRef] [PubMed]
- Veltman, J.A.; Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nat. Cell Biol. 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koblan, L.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nat. Cell Biol. 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.-M.; Koo, T.; Kim, K.; Lim, K.; Baek, G.; Kim, S.-T.; Kim, H.S.; Kim, D.-E.; Lee, H.; Chung, E.; et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018, 36, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, J.; Zhou, R.; Lareau, C.A.; Garcia, S.P.; Iyer, S.; Miller, B.R.; Langner, L.M.; Hsu, J.Y.; Aryee, M.J.; Joung, J.K. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol. 2020, 38, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjana, N.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; McConkey, M.E.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.; Garcia, S.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nat. Cell Biol. 2015, 527, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.G.; Dang, Y.; Abraham, S.; Ma, H.; Zhang, J.; Guo, H.; Cai, Y.; Mikkelsen, J.G.; Wu, H.; Shankar, P.; et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene. Ther. 2016, 23, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, A.V.; Hollis, R.P.; Kuftinec, G.; Senadheera, S.; Chan, R.; Kohn, D.B. Integrase-defective Lentiviral Vectors as a Delivery Platform for Targeted Modification of Adenosine Deaminase Locus. Mol. Ther. 2013, 21, 1705–1717. [Google Scholar] [CrossRef] [Green Version]
- Tagliafierro, L.; Ilich, E.; Moncalvo, M.; Gu, J.; Sriskanda, A.; Grenier, C.; Murphy, S.K.; Chiba-Falek, O.; Kantor, B. Lentiviral Vector Platform for the Efficient Delivery of Epigenome-editing Tools into Human Induced Pluripotent Stem Cell-derived Disease Models. J. Vis. Exp. 2019, e59241. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).