
Hepatitis B Virus Pre-S Gene Deletions and Pre-S Deleted Proteins: Clinical and Molecular Implications in Hepatocellular Carcinoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
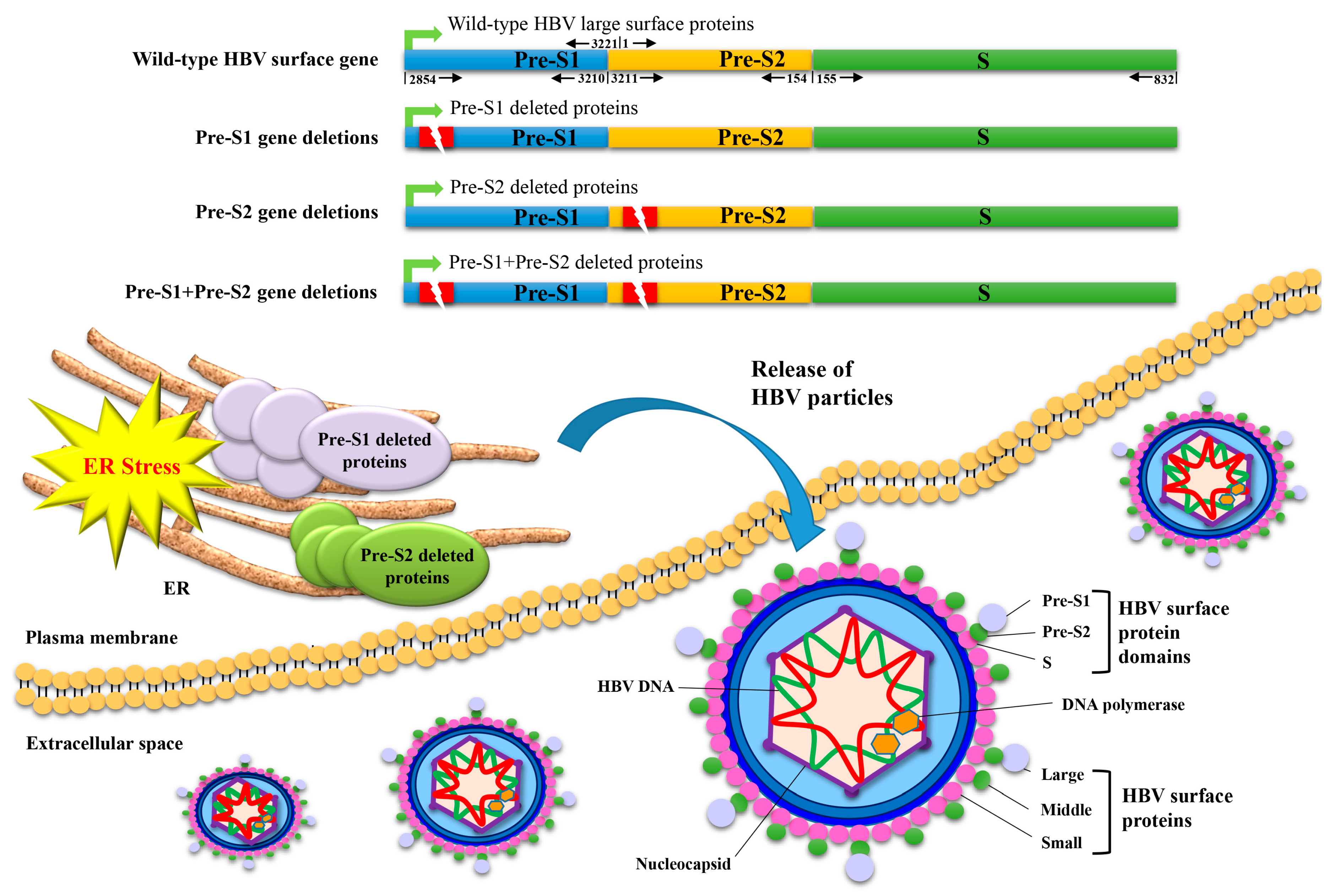
1. Introduction
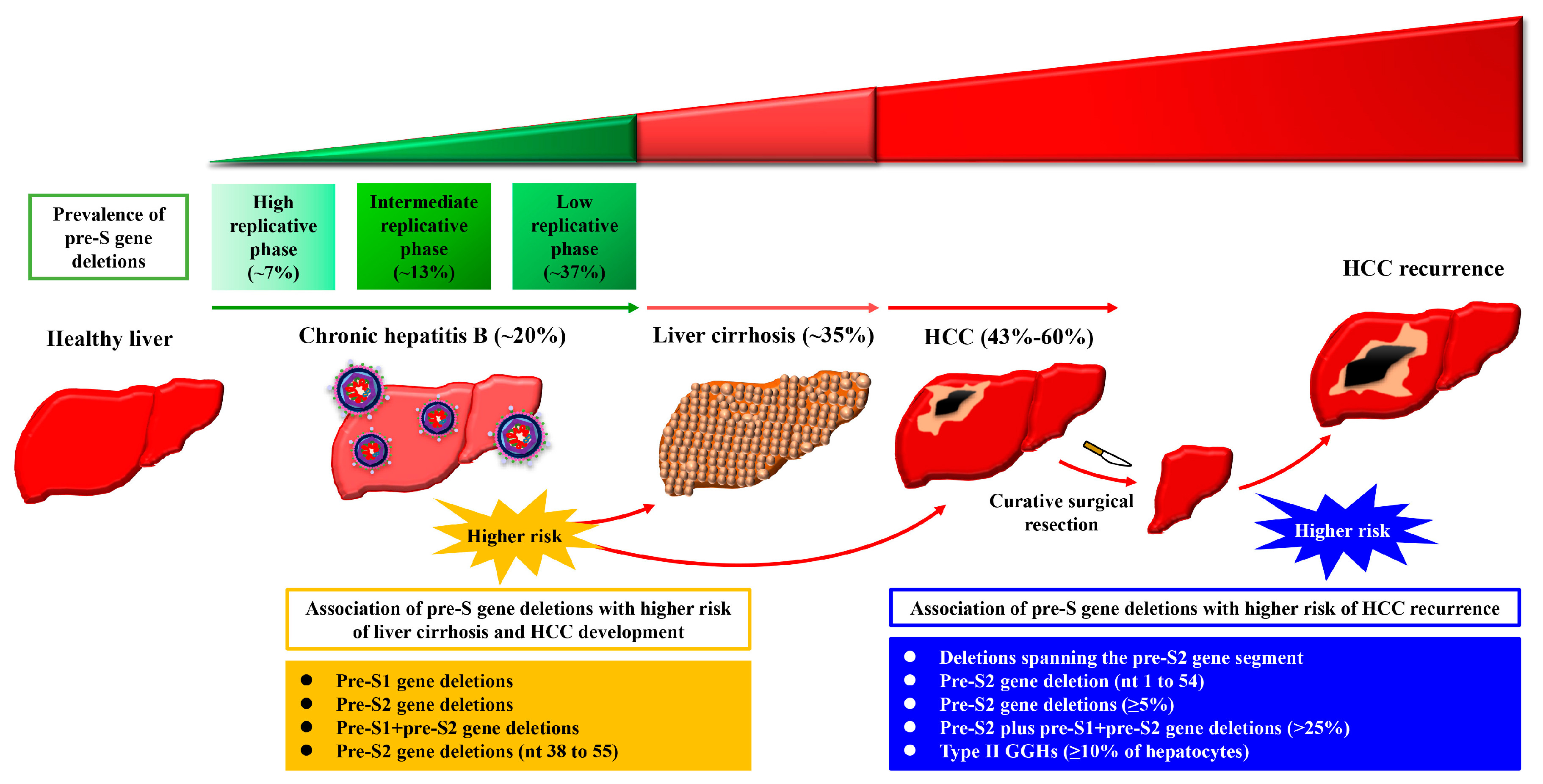
2. The Presence of HBV Pre-S Gene Deletions in the Blood of Patients with Chronic Hepatitis B Is Associated with Liver Disease Progression and Higher Incidence of Liver Cirrhosis and HCC Development
3. The Presence of HBV Pre-S Gene Deletions in the Blood of Patients with HCC Is Associated with Higher Risk of HCC Recurrence after Curative Surgical Resection
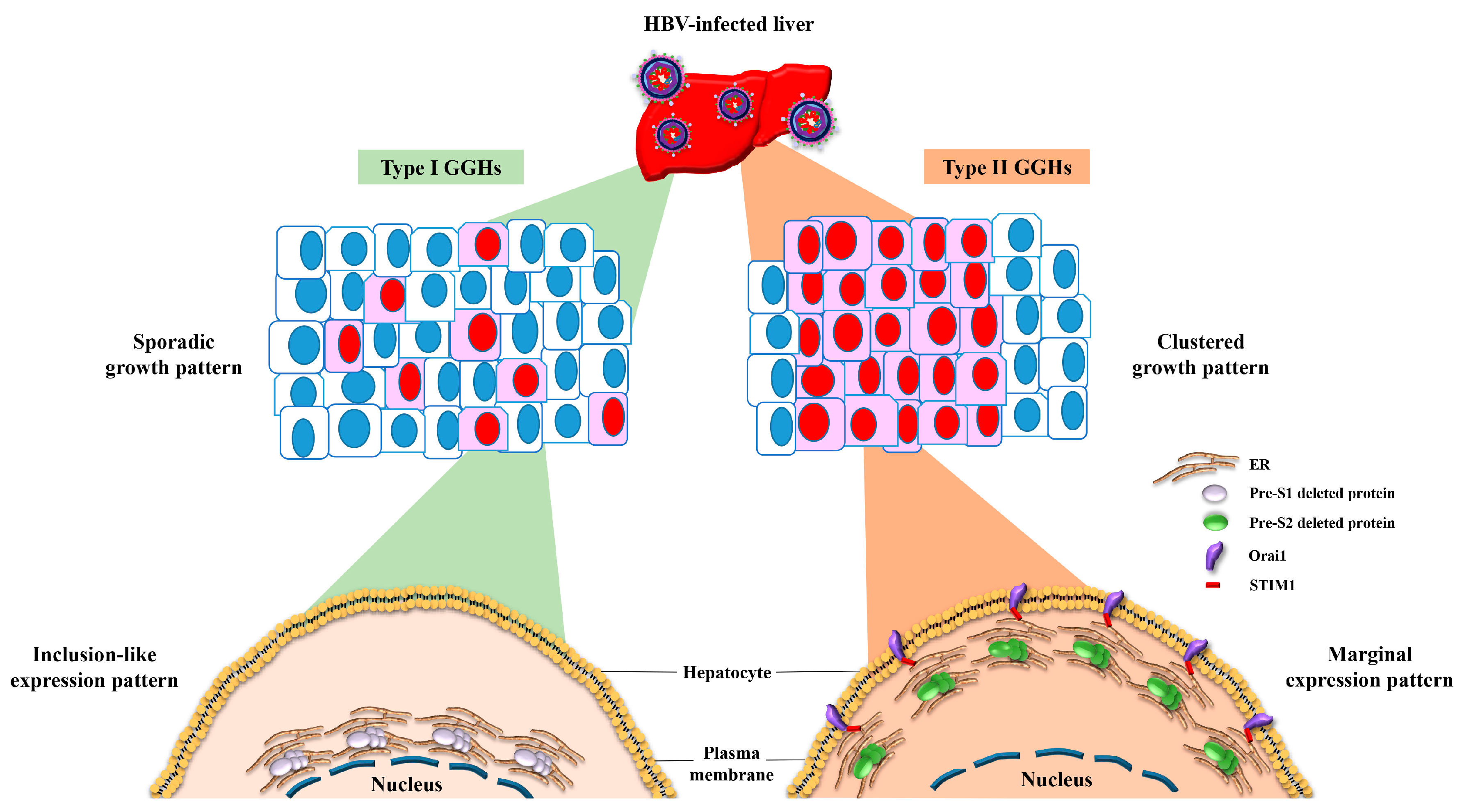
4. The Expression of Pre-S Deleted Proteins in the Liver Tissues of Patients with HCC Is Associated with Higher Risk of HCC Recurrence after Curative Surgical Resection
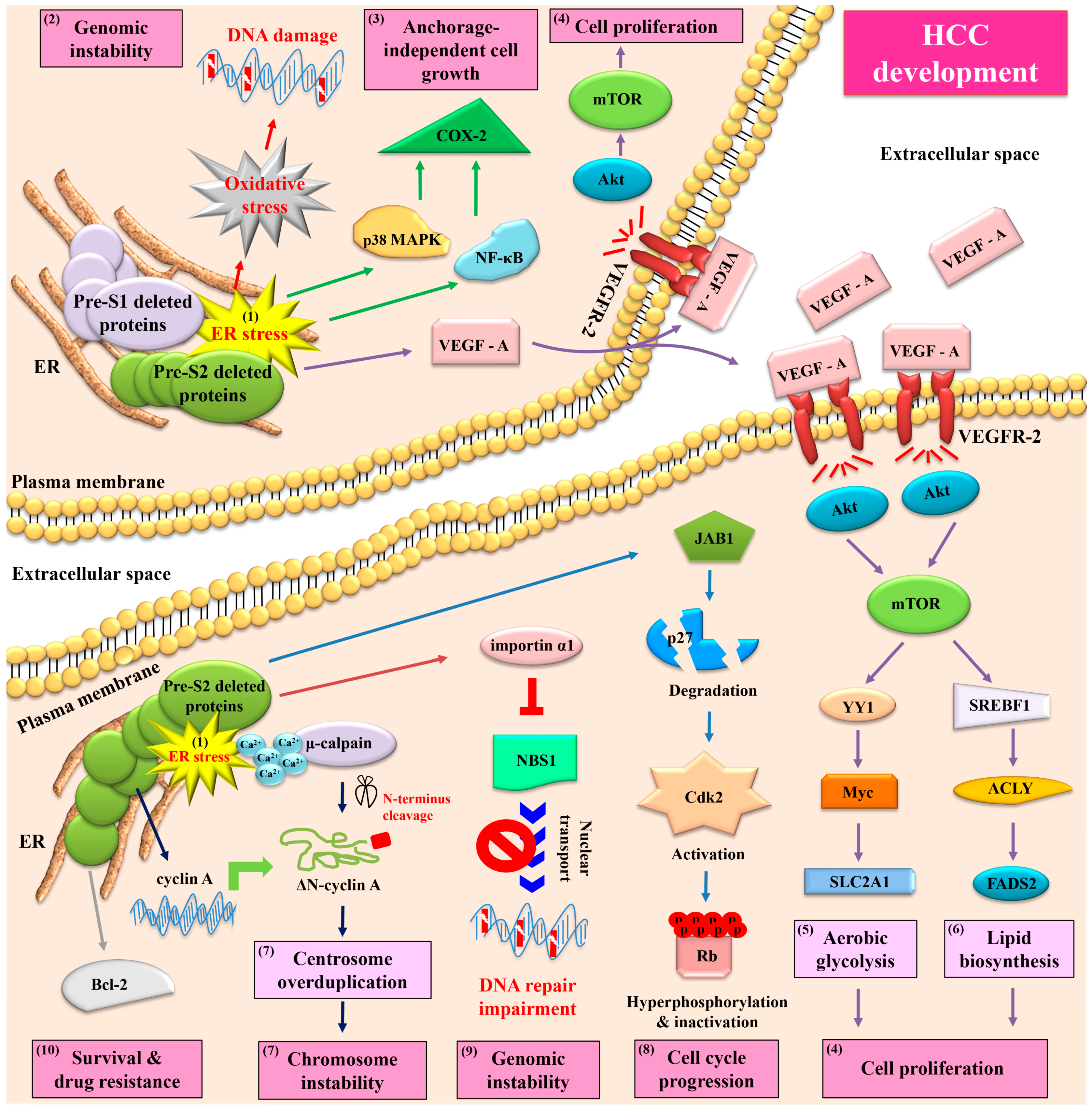
5. Both HBV Pre-S1 and Pre-S2 Deleted Proteins Activate Endoplasmic Reticulum (ER) Stress-Dependent Signaling Pathways to Induce DNA Damage and to Promote Growth and Proliferation in Hepatocytes In Vitro and In Vivo
6. HBV Pre-S2 Deleted Proteins Additionally Activate ER Stress-Dependent or Independent Signaling Pathways to Induce Centrosome Overduplication, Promote Cell Cycle Progression, Inhibit DNA Repair, and Enhance Survival and Drug Resistance in Hepatocytes In Vitro and In Vivo
7. HBV Pre-S2 Deleted Proteins Induce Malignant Transformation of Hepatocytes and Trigger HCC Development in a Transgenic Mouse Model
8. Inhibition of Signaling Pathways Activated by HBV Pre-S Deleted Proteins Exhibits a Preventive Effect on Liver Pathology and HCC Development in a Transgenic Mouse Model
9. Discussion
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Cheng, K.-C.; Lin, W.-Y.; Liu, C.-S.; Lin, C.-C.; Lai, H.-C.; Lai, S.-W. Association of different types of liver disease with demographic and clinical factors. Biomedicine 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kulik, L.; El-Serag, H.B. Epidemiology and management of hepatocellular carcinoma. Gastroenterology 2019, 156, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 68, 394–424. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, S.-B.; Chen, S.-G.; Qu, Q.; Rui, J.-A. Risk factors of recurrence and poor survival in curatively resected hepatocellular carcinoma with microvascular invasion. Adv. Clin. Exp. Med. 2020, 29, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Tampaki, M.; Papatheodoridis, G.V.; Cholongitas, E. Intrahepatic recurrence of hepatocellular carcinoma after resection: An update. Clin. J. Gastroenterol. 2021. [Google Scholar] [CrossRef]
- Maucort-Boulch, D.; de Martel, C.; Franceschi, S.; Plummer, M. Fraction and incidence of liver cancer attributable to hepatitis B and C viruses worldwide: Liver cancer attributable to hepatitis viruses worldwide. Int. J. Cancer 2003, 142, 2471–2477. [Google Scholar] [CrossRef]
- Llovet, J.M.; Burroughs, A.; Bruix, J. Hepatocellular carcinoma. Lancet 2003, 362, 1907–1917. [Google Scholar] [CrossRef]
- Tarocchi, M.; Polvani, S.; Marroncini, G.; Galli, A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J. Gastroenterol. 2014, 20, 11630–11640. [Google Scholar] [CrossRef]
- Mani, S.K.K.; Andrisani, O. Hepatitis B Virus-Associated Hepatocellular Carcinoma and Hepatic Cancer Stem Cells. Genes 2018, 9, 137. [Google Scholar] [CrossRef]
- Robinson, W.S. The Genome of Hepatitis B Virus. Annu. Rev. Microbiol. 1977, 31, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Hepatitis B Virus Infection. N. Engl. J. Med. 1997, 337, 1733–1745. [Google Scholar] [CrossRef]
- Wang, H.-C.; Wu, H.-C.; Chen, C.-F.; Fausto, N.; Lei, H.-Y.; Su, I.-J. Different Types of Ground Glass Hepatocytes in Chronic Hepatitis B Virus Infection Contain Specific Pre-S Mutants that May Induce Endoplasmic Reticulum Stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef]
- Su, I.J.; Wang, H.C.; Wu, H.C.; Huang, W.Y. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J. Gastroenterol. Hepatol. 2008, 23, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Su, I.-J.; Wang, L.H.-C.; Hsieh, W.-C.; Wu, H.-C.; Teng, C.-F.; Tsai, H.-W.; Huang, W. The emerging role of hepatitis B virus Pre-S2 deletion mutant proteins in HBV tumorigenesis. J. Biomed. Sci. 2014, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.-F.; Wu, H.-C.; Su, I.-J.; Jeng, L.-B. Hepatitis B Virus Pre-S Mutants as Biomarkers and Targets for the Development and Recurrence of Hepatocellular Carcinoma. Viruses 2020, 12, 945. [Google Scholar] [CrossRef]
- Teng, C.F.; Wu, H.C.; Shyu, W.C.; Jeng, L.B.; Su, I.J. Pre-S2 Mutant-Induced Mammalian Target of Rapamycin Signal Pathways as Potential Therapeutic Targets for Hepatitis B Virus-Associated Hepatocellular Carcinoma. Cell Transplant. 2017, 26, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-L.; Hung, J.-H.; Huang, W. Association of the Hepatitis B Virus Large Surface Protein with Viral Infectivity and Endoplasmic Reticulum Stress-mediated Liver Carcinogenesis. Cells 2020, 9, 2052. [Google Scholar] [CrossRef]
- Fan, Y.; Lu, C.; Chen, W.; Yao, W.; Wang, H.; Chang, T.; Lei, H.; Shiau, A.; Su, I. Prevalence and significance of hepatitis B virus (HBV) pre-S mutants in serum and liver at different replicative stages of chronic HBV infection. Hepatology 2001, 33, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.C.; Su, I.J.; Wu, H.C.; Hsieh, Y.H.; Yao, W.J.; Young, K.C.; Chang, T.C.; Hsieh, H.C.; Tsai, H.N.; Huang, W. A pre-S gene chip to detect pre-S deletions in hepatitis B virus large surface antigen as a predictive marker for hepatoma risk in chronic hepatitis B virus carriers. J. Biomed. Sci. 2009, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Kim, D.Y.; Lee, D.H.; Lee, J.H.; Koh, K.C.; Paik, S.W.; Rhee, J.C.; Yoo, B.C. Clinical significance of pre-S mutations in patients with genotype C hepatitis B virus infection. J. Viral Hepat. 2006, 14, 161–168. [Google Scholar] [CrossRef]
- Li, X.; Qin, Y.; Liu, Y.; Li, F.; Liao, H.; Lu, S.; Qiao, Y.; Xu, D.; Li, J. PreS deletion profiles of hepatitis B virus (HBV) are associated with clinical presentations of chronic HBV infection. J. Clin. Virol. 2016, 82, 27–32. [Google Scholar] [CrossRef]
- Zhao, Z.-M.; Jin, Y.; Gan, Y.; Zhu, Y.; Chen, T.-Y.; Wang, J.-B.; Sun, Y.; Cao, Z.-G.; Qian, G.-S.; Tu, H. Novel approach to identifying the hepatitis B virus pre-S deletions associated with hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 13573–13581. [Google Scholar] [CrossRef]
- Jia, J.; Liang, X.; Chen, S.; Wang, H.; Li, H.; Fang, M.; Bai, X.; Wang, Z.; Wang, M.; Zhu, S.; et al. Next-generation sequencing revealed divergence in deletions of the preS region in the HBV genome between different HBV-related liver diseases. J. Gen. Virol. 2017, 98, 2748–2758. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Hung, C.H.; Lee, C.M.; Hu, T.H.; Wang, J.H.; Wang, J.C.; Lu, S.N.; Changchien, C.S. Pre-S deletion and complex mutations of hepatitis B virus related to advanced liver disease in HBeAg-negative patients. Gastroenterology 2007, 133, 1466–1474. [Google Scholar] [CrossRef]
- Sinn, D.H.; Choi, M.S.; Gwak, G.-Y.; Paik, Y.-H.; Lee, J.H.; Koh, K.C.; Paik, S.W.; Yoo, B.C. Pre-S Mutation Is a Significant Risk Factor for Hepatocellular Carcinoma Development: A Long-Term Retrospective Cohort Study. Dig. Dis. Sci. 2012, 58, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Ghosh, S.; Shimakawa, Y.; Ramou, N.; Garcia, P.S.; Dubois, A.; Guillot, C.; Deluce, N.K.-N.; Tilloy, V.; Durand, G.; et al. Hepatitis B virus preS2Δ38–55 variants: A newly identified risk factor for hepatocellular carcinoma. JHEP Rep. 2020, 2, 100144. [Google Scholar] [CrossRef]
- Li-Shuai, Q.; Yu-Yan, C.; Hai-Feng, Z.; Jin-Xia, L.; Cui-Hua, L. Pre-S deletions of hepatitis B virus predict recurrence of hepatocellular carcinoma after curative resection. Medicine 2017, 96, e8311. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.J.; Ai, Y.L.; Tsai, H.W.; Chan, S.H.; Yen, C.S.; Cheng, K.H.; Lee, Y.P.; Kao, C.W.; Wang, Y.C.; Chen, Y.L.; et al. Hepatitis B virus surface gene pre-S2 mutant as a high-risk serum marker for hepatoma recurrence after curative hepatic resection. Hepatology 2018, 68, 815–826. [Google Scholar] [CrossRef]
- Teng, C.F.; Huang, H.Y.; Li, T.C.; Shyu, W.C.; Wu, H.C.; Lin, C.Y.; Su, I.J.; Jeng, L.B. A Next-Generation Sequencing-Based Platform for Quantitative Detection of Hepatitis B Virus Pre-S Mutants in Plasma of Hepatocellular Carcinoma Patients. Sci. Rep. 2018, 8, 14816. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.-F.; Tsai, H.-W.; Li, T.-C.; Wang, T.; Wang, J.; Shyu, W.-C.; Wu, H.-C.; Su, I.-J.; Jeng, L.-B. Detection of hepatitis B virus pre-S mutants in plasma by a next-generation sequencing-based platform determines their patterns in liver tissues. PLoS ONE 2020, 15, e0234773. [Google Scholar] [CrossRef]
- Teng, C.F.; Li, T.C.; Huang, H.Y.; Lin, J.H.; Chen, W.S.; Shyu, W.C.; Wu, H.C.; Peng, C.Y.; Su, I.J.; Jeng, L.B. Next-generation sequencing-based quantitative detection of hepatitis B virus pre-S mutants in plasma predicts hepatocellular carcinoma recurrence. Viruses 2020, 12, 796. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.F.; Li, T.C.; Huang, H.Y.; Chan, W.L.; Wu, H.C.; Shyu, W.C.; Su, I.J.; Jeng, L.B. Hepatitis B virus pre-S2 deletion (nucleotide 1 to 54) in plasma predicts recurrence of hepatocellular carcinoma after curative surgical resection. PLoS ONE 2020, 15, e0242748. [Google Scholar] [CrossRef]
- Thedja, M.D.; Muljono, D.H.; Ie, S.I.; Sidarta, E.; Verhoef, J.; Marzuki, S. Genogeography and Immune Epitope Characteristics of Hepatitis B Virus Genotype C Reveals Two Distinct Types: Asian and Papua-Pacific. PLoS ONE 2015, 10, e0132533. [Google Scholar] [CrossRef]
- Hatazawa, Y.; Yano, Y.; Okada, R.; Tanahashi, T.; Hayashi, H.; Hirano, H.; Minami, A.; Kawano, Y.; Tanaka, M.; Fukumoto, T.; et al. Quasispecies variant of pre-S/S gene in HBV-related hepatocellular carcinoma with HBs antigen positive and occult infection. Infect. Agents Cancer 2018, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-W.; Lin, Y.-J.; Lin, P.-W.; Wu, H.-C.; Hsu, K.-H.; Yen, C.-J.; Chan, S.-H.; Huang, W.; Su, I.-J. A clustered ground-glass hepatocyte pattern represents a new prognostic marker for the recurrence of hepatocellular carcinoma after surgery. Cancer 2011, 117, 2951–2960. [Google Scholar] [CrossRef]
- Tsai, H.-W.; Lin, Y.-J.; Wu, H.-C.; Chang, T.-T.; Wu, I.-C.; Cheng, P.-N.; Yen, C.-J.; Chan, S.-H.; Huang, W.; Su, I.-J. Resistance of ground glass hepatocytes to oral antivirals in chronic hepatitis B patients and implication for the development of hepatocellular carcinoma. Oncotarget 2016, 7, 27724–27734. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-H.; Su, I.-J.; Wang, H.-C.; Chang, W.-T.; Lei, H.-Y.; Lai, M.-D.; Huang, W. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004, 25, 2023–2032. [Google Scholar] [CrossRef]
- Hung, J.H.; Su, I.J.; Lei, H.Y.; Wang, H.C.; Lin, W.C.; Chang, W.T.; Huang, W.; Chang, W.C.; Chang, Y.S.; Chen, C.C.; et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J. Biol. Chem. 2004, 279, 46384–46392. [Google Scholar] [CrossRef]
- Yang, J.C.; Teng, C.F.; Wu, H.C.; Tsai, H.W.; Chuang, H.C.; Tsai, T.F.; Hsu, Y.H.; Huang, W.; Wu, L.W.; Su, I.J. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology 2009, 49, 1962–1971. [Google Scholar] [CrossRef]
- Teng, C.-F.; Hsieh, W.-C.; Wu, H.-C.; Lin, Y.-J.; Tsai, H.-W.; Huang, W.; Su, I.-J. Hepatitis B Virus Pre-S2 Mutant Induces Aerobic Glycolysis through Mammalian Target of Rapamycin Signal Cascade. PLoS ONE 2015, 10, e0122373. [Google Scholar] [CrossRef]
- Teng, C.F.; Wu, H.C.; Hsieh, W.C.; Tsai, H.W.; Su, I.J. Activation of ATP citrate lyase by mTOR signal induces disturbed lipid metabolism in hepatitis B virus pre-S2 mutant tumorigenesis. J. Virol. 2015, 89, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-C.; Chang, W.-T.; Wu, H.-C.; Huang, W.; Lei, H.-Y.; Lai, M.-D.; Fausto, N.; Su, I.-J.; Chang, W.-W. Hepatitis B virus pre-S2 mutant upregulates cyclin A expression and induces nodular proliferation of hepatocytes. Hepatology 2005, 41, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.-C.; Huang, W.; Lai, M.-D.; Su, I.-J. Aberrant cyclin A expression and centrosome overduplication induced by hepatitis B virus Pre-S2 mutants and its implication in hepatocarcinogenesis. Carcinogenesis 2011, 33, 466–472. [Google Scholar] [CrossRef]
- Yen, T.T.-C.; Yang, A.; Chiu, W.-T.; Li, T.-N.; Wang, L.-H.; Wu, Y.-H.; Wang, H.-C.; Chen, L.; Wang, W.-C.; Huang, W.; et al. Hepatitis B virus PreS2-mutant large surface antigen activates store-operated calcium entry and promotes chromosome instability. Oncotarget 2016, 7, 23346–23360. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Su, I.J.; Wang, H.C.; Tsai, J.H.; Huang, Y.J.; Chang, W.W.; Lai, M.D.; Lei, H.Y.; Huang, W. Hepatitis B virus pre-S2 mutant sur-face antigen induces degradation of cyclin-dependent kinase inhibitor p27Kip1 through c-Jun activation do-main-binding protein 1. Mol. Cancer Res. 2007, 5, 1063–1072. [Google Scholar] [CrossRef]
- Hsu, J.-L.; Chuang, W.-J.; Su, I.-J.; Gui, W.-J.; Chang, Y.-Y.; Lee, Y.-P.; Ai, Y.-L.; Chuang, D.T.; Huang, W. Zinc-Dependent Interaction between JAB1 and Pre-S2Mutant Large Surface Antigen of Hepatitis B Virus and Its Implications for Viral Hepatocarcinogenesis. J. Virol. 2013, 87, 12675–12684. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hsieh, Y.-H.; Chang, Y.-Y.; Su, I.-J.; Yen, C.-J.; Liu, Y.-R.; Liu, R.-J.; Hsieh, W.-C.; Tsai, H.-W.; Wang, L.H.-C.; Huang, W. Hepatitis B virus pre-S2 mutant large surface protein inhibits DNA double-strand break repair and leads to genome instability in hepatocarcinogenesis. J. Pathol. 2015, 236, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.H.; Teng, Y.N.; Wang, L.H.C.; Su, I.J.; Wang, C.C.; Huang, W.; Lee, K.H.; Lu, K.Y.; Wang, L.H. Induction of Bcl-2 expression by hepatitis B virus pre-S2 mutant large surface protein resistance to 5-fluorouracil treatment in Huh-7 cells. PLoS ONE 2011, 6, e28977. [Google Scholar] [CrossRef]
- Teng, C.-F.; Li, T.-C.; Wang, T.; Wu, T.-H.; Wang, J.; Wu, H.-C.; Shyu, W.-C.; Su, I.-J.; Jeng, L.-B. Increased Expression of Programmed Death Ligand 1 in Hepatocellular Carcinoma of Patients with Hepatitis B Virus Pre-S2 Mutant. J. Hepatocell. Carcinoma 2020, 7, 385–401. [Google Scholar] [CrossRef]
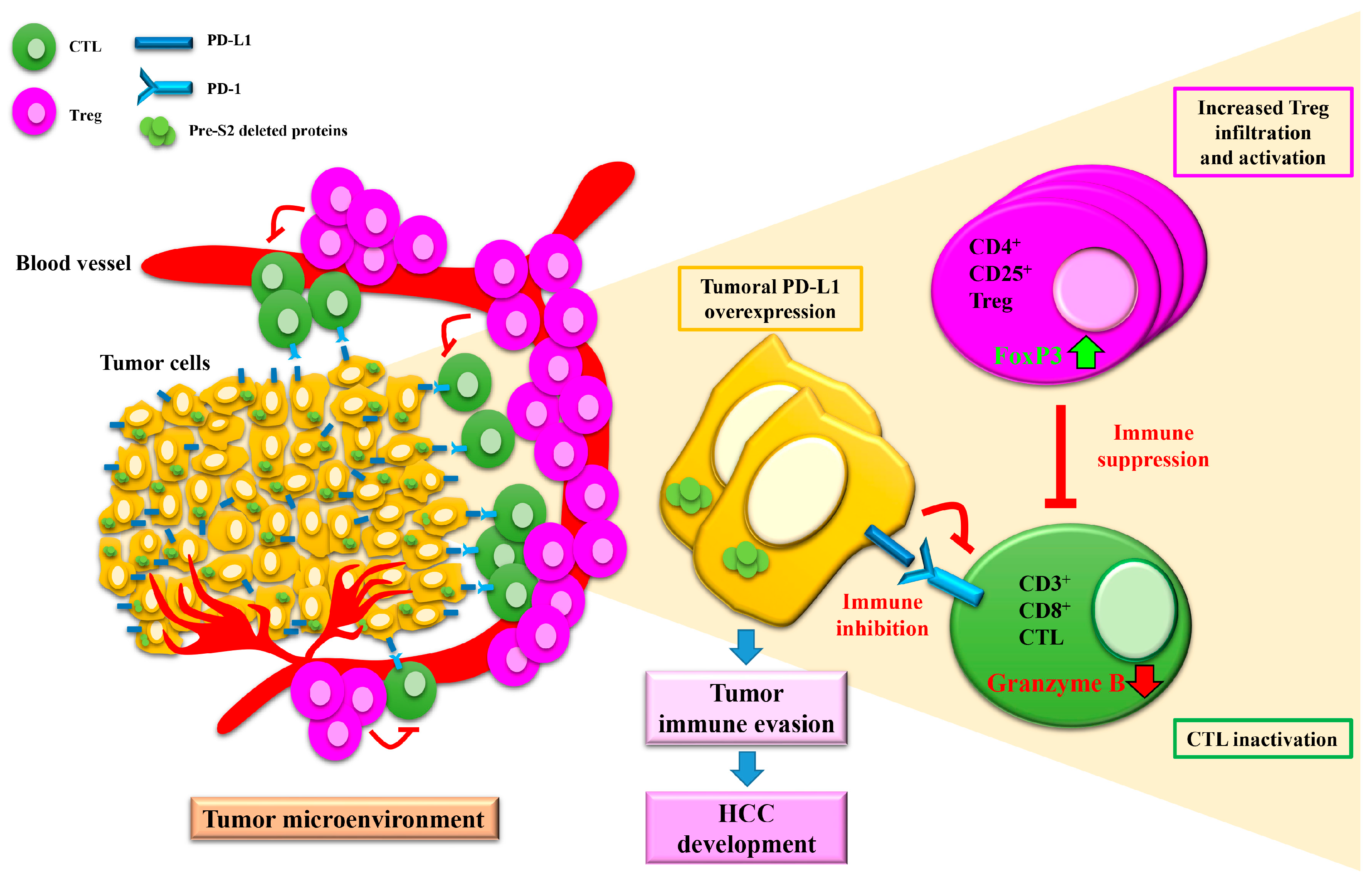
- Teng, C.-F.; Li, T.-C.; Wang, T.; Liao, D.-C.; Wen, Y.-H.; Wu, T.-H.; Wang, J.; Wu, H.-C.; Shyu, W.-C.; Su, I.-J.; et al. Increased infiltration of regulatory T cells in hepatocellular carcinoma of patients with hepatitis B virus pre-S2 mutant. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, X.-Y.; Qiu, S.-J.; Yamato, I.; Sho, M.; Nakajima, Y.; Zhou, J.; Li, B.-Z.; Shi, Y.-H.; Xiao, Y.-S.; et al. Overexpression of PD-L1 Significantly Associates with Tumor Aggressiveness and Postoperative Recurrence in Human Hepatocellular Carcinoma. Clin. Cancer Res. 2009, 15, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-S.; Gu, X.; Xiong, W.; Guo, W.; Han, L.; Bai, Y.; Peng, C.; Cui, M.; Xie, M. Increased programmed death ligand-1 expression predicts poor prognosis in hepatocellular carcinoma patients. OncoTargets Ther. 2016, 9, 4805–4813. [Google Scholar] [CrossRef]
- Jung, H.I.; Jeong, D.; Ji, S.; Ahn, T.S.; Bae, S.H.; Chin, S.; Chung, J.C.; Kim, H.C.; Lee, M.S.; Baek, M.-J. Overexpression of PD-L1 and PD-L2 Is Associated with Poor Prognosis in Patients with Hepatocellular Carcinoma. Cancer Res. Treat. 2017, 49, 246–254. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Shevach, E.M.; McHugh, R.S.; Piccirillo, C.A.; Thornton, A.M. Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol. Rev. 2001, 182, 58–67. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Teng, Y.C.; Neo, J.C.; Wu, J.C.; Chen, Y.F.; Kao, C.H.; Tsai, T.F. Expression of a hepatitis B virus pre-S2 deletion mutant in the liver results in hepatomegaly and hepatocellular carcinoma in mice. J. Pathol. 2017, 241, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-C.; Tsai, H.-W.; Teng, C.-F.; Hsieh, W.-C.; Lin, Y.-J.; Wang, L.H.-C.; Yuan, Q.; Su, I.-J. Ground-glass hepatocytes co-expressing hepatitis B virus X protein and surface antigens exhibit enhanced oncogenic effects and tumorigenesis. Hum. Pathol. 2014, 45, 1294–1301. [Google Scholar] [CrossRef]
- Teng, C.-F.; Yu, C.-H.; Chang, H.-Y.; Hsieh, W.-C.; Wu, T.-H.; Lin, J.-H.; Wu, H.-C.; Jeng, L.-B.; Su, I.-J. Chemopreventive Effect of Phytosomal Curcumin on Hepatitis B Virus-Related Hepatocellular Carcinoma in A Transgenic Mouse Model. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-H.; Su, I.-J.; Yen, C.-J.; Tsai, T.-F.; Tsai, H.-W.; Tsai, H.-N.; Huang, Y.-J.; Chen, Y.-Y.; Ai, Y.-L.; Kao, L.-Y.; et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid suppresses the pro-oncogenic effects induced by hepatitis B virus pre-S 2 mutant oncoprotein and represents a potential chemopreventive agent in high-risk chronic HBV patients. Carcinogenesis 2012, 34, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.N.; Chisari, F.V. Strong, sustained hepatocellular proliferation precedes hepatocarcinogenesis in hepatitis B surface antigen transgenic mice. Hepatology 1995, 21, 620–626. [Google Scholar] [PubMed]
- Xu, Z.; Jensen, G.; Yen, T.S. Activation of hepatitis B virus S promoter by the viral large surface protein via induction of stress in the endoplasmic reticulum. J. Virol. 1997, 71, 7387–7392. [Google Scholar] [CrossRef] [PubMed]
- Li, T.N.; Wu, Y.J.; Tsai, H.W.; Sun, C.P.; Wu, Y.H.; Wu, H.L.; Pei, Y.N.; Lu, K.Y.; Yen, T.T.C.; Chang, C.W.; et al. Intrahepatic hepatitis B virus large surface an-tigen induces hepatocyte hyperploidy via failure of cytokinesis. J. Pathol. 2018, 245, 502–513. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-T.; Jeng, L.-B.; Chan, W.-L.; Su, I.-J.; Teng, C.-F. Hepatitis B Virus Pre-S Gene Deletions and Pre-S Deleted Proteins: Clinical and Molecular Implications in Hepatocellular Carcinoma. Viruses 2021, 13, 862. https://doi.org/10.3390/v13050862
Lin Y-T, Jeng L-B, Chan W-L, Su I-J, Teng C-F. Hepatitis B Virus Pre-S Gene Deletions and Pre-S Deleted Proteins: Clinical and Molecular Implications in Hepatocellular Carcinoma. Viruses. 2021; 13(5):862. https://doi.org/10.3390/v13050862
Chicago/Turabian StyleLin, Yueh-Te, Long-Bin Jeng, Wen-Ling Chan, Ih-Jen Su, and Chiao-Fang Teng. 2021. "Hepatitis B Virus Pre-S Gene Deletions and Pre-S Deleted Proteins: Clinical and Molecular Implications in Hepatocellular Carcinoma" Viruses 13, no. 5: 862. https://doi.org/10.3390/v13050862
APA StyleLin, Y.-T., Jeng, L.-B., Chan, W.-L., Su, I.-J., & Teng, C.-F. (2021). Hepatitis B Virus Pre-S Gene Deletions and Pre-S Deleted Proteins: Clinical and Molecular Implications in Hepatocellular Carcinoma. Viruses, 13(5), 862. https://doi.org/10.3390/v13050862