
A New Generation of Functional Tagged Proteins for HIV Fluorescence Imaging


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids and Vector Cloning
2.1.1. pCMV-optiGag-FluoIN
2.1.2. psPAX2-FluoIN
2.2. Cells, Virus Production, and Quantification
2.3. Virus on Glass Experiments
2.4. Immunofluorescence Imaging
2.5. Particle Detection and Quantification
2.6. Infection Analysis
3. Results
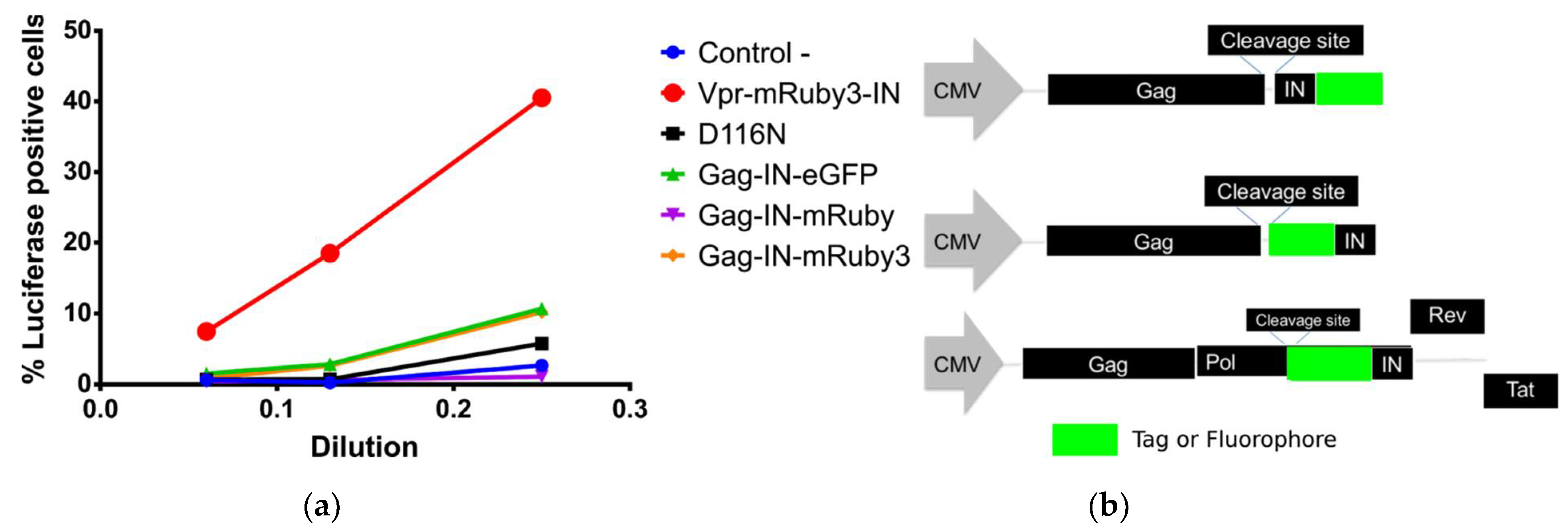
3.1. Development of Codon Optimized Gag-FluorophoreIN Constructs
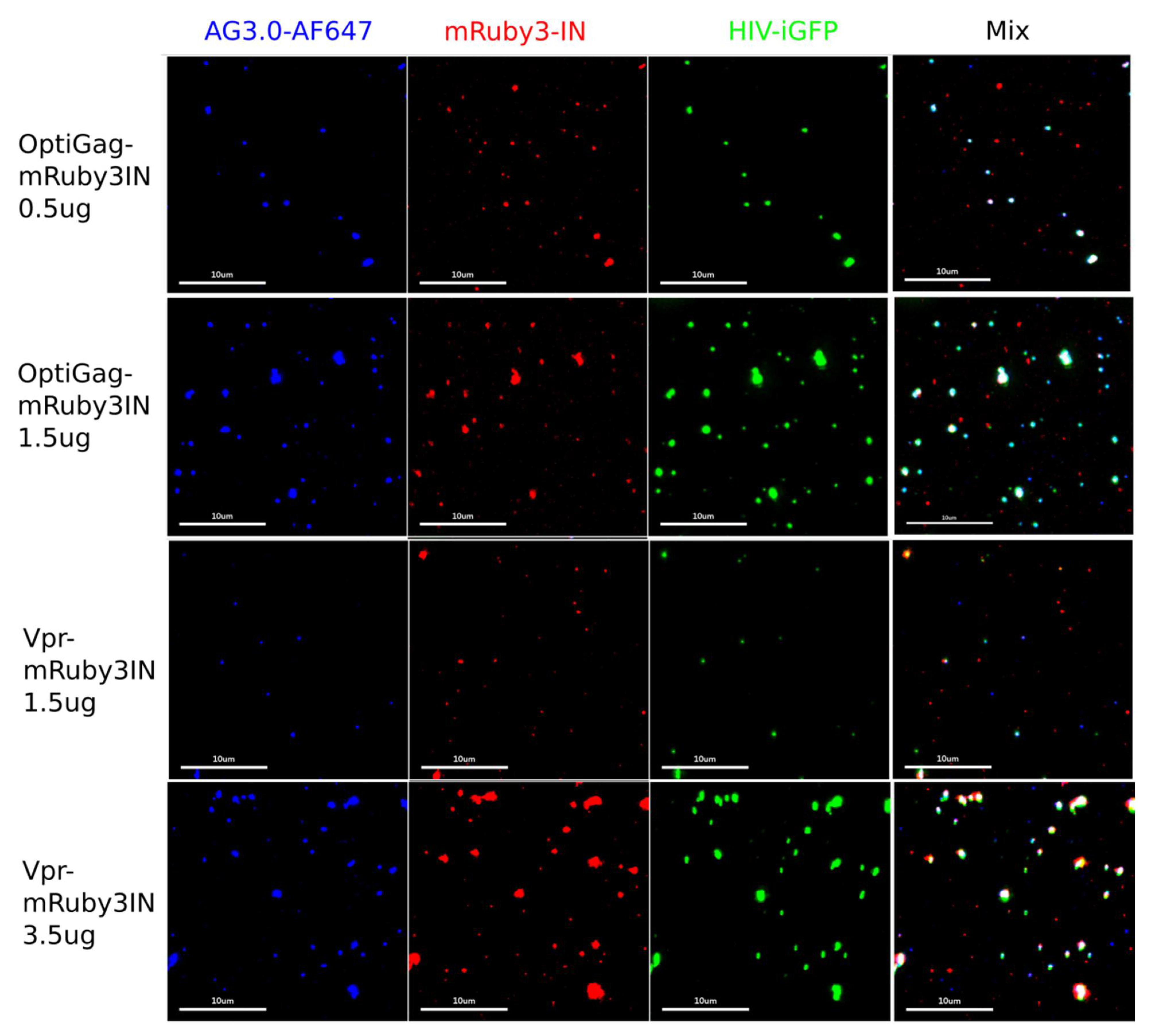
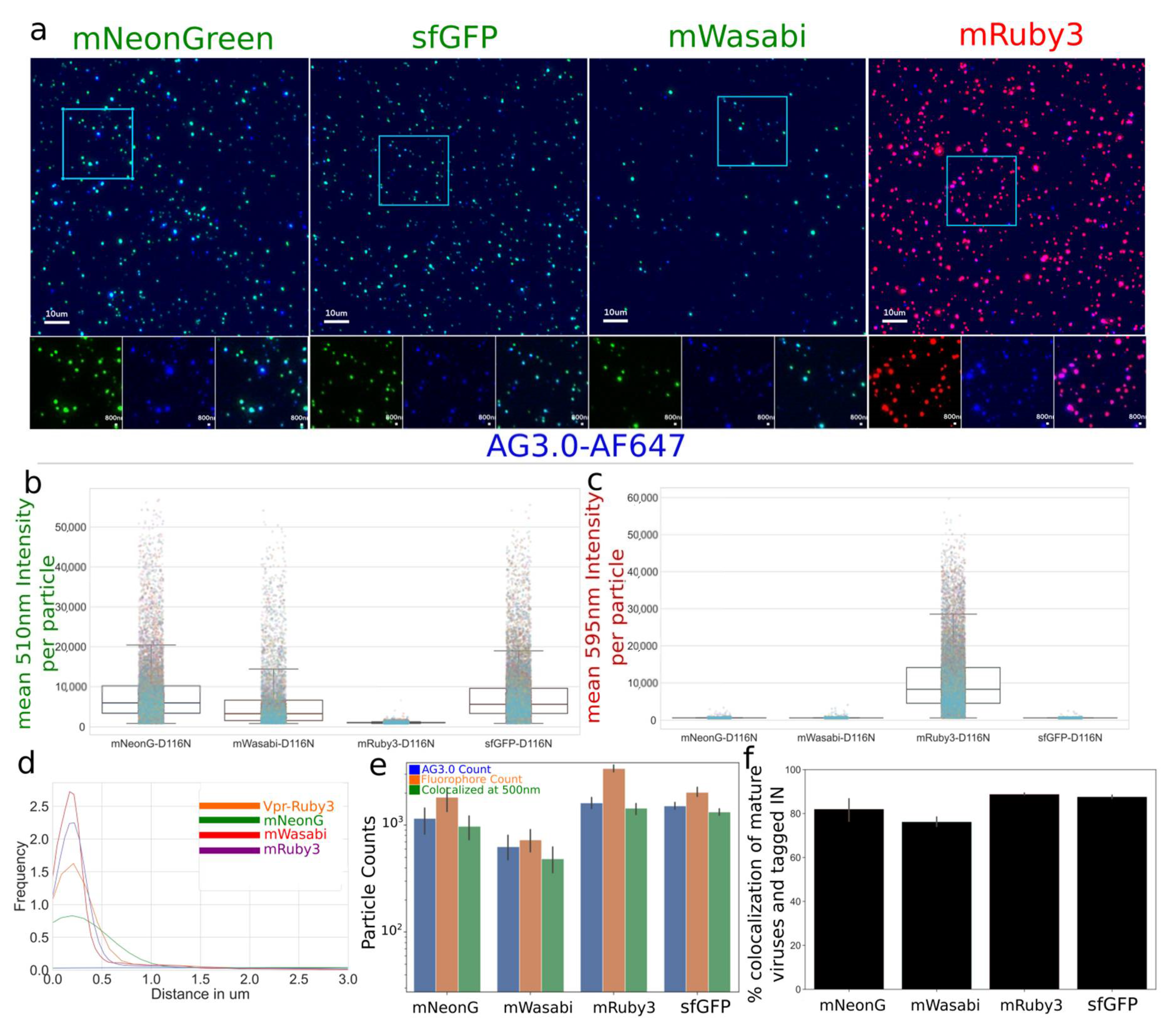
3.1.1. Characterization and Quantification of Labeled IN Incorporation into Viral Particles
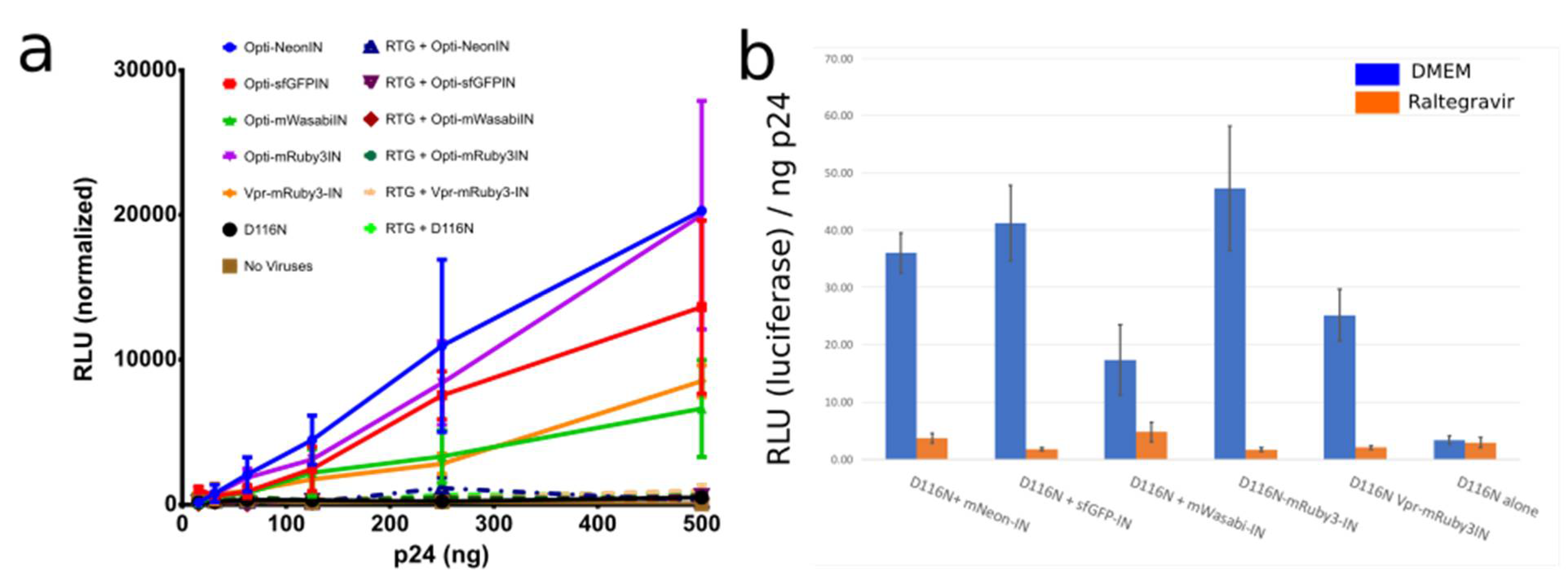
3.1.2. OptiGag-FluoIN Restores Infectivity of Catalytically Inactive D116N Integrase
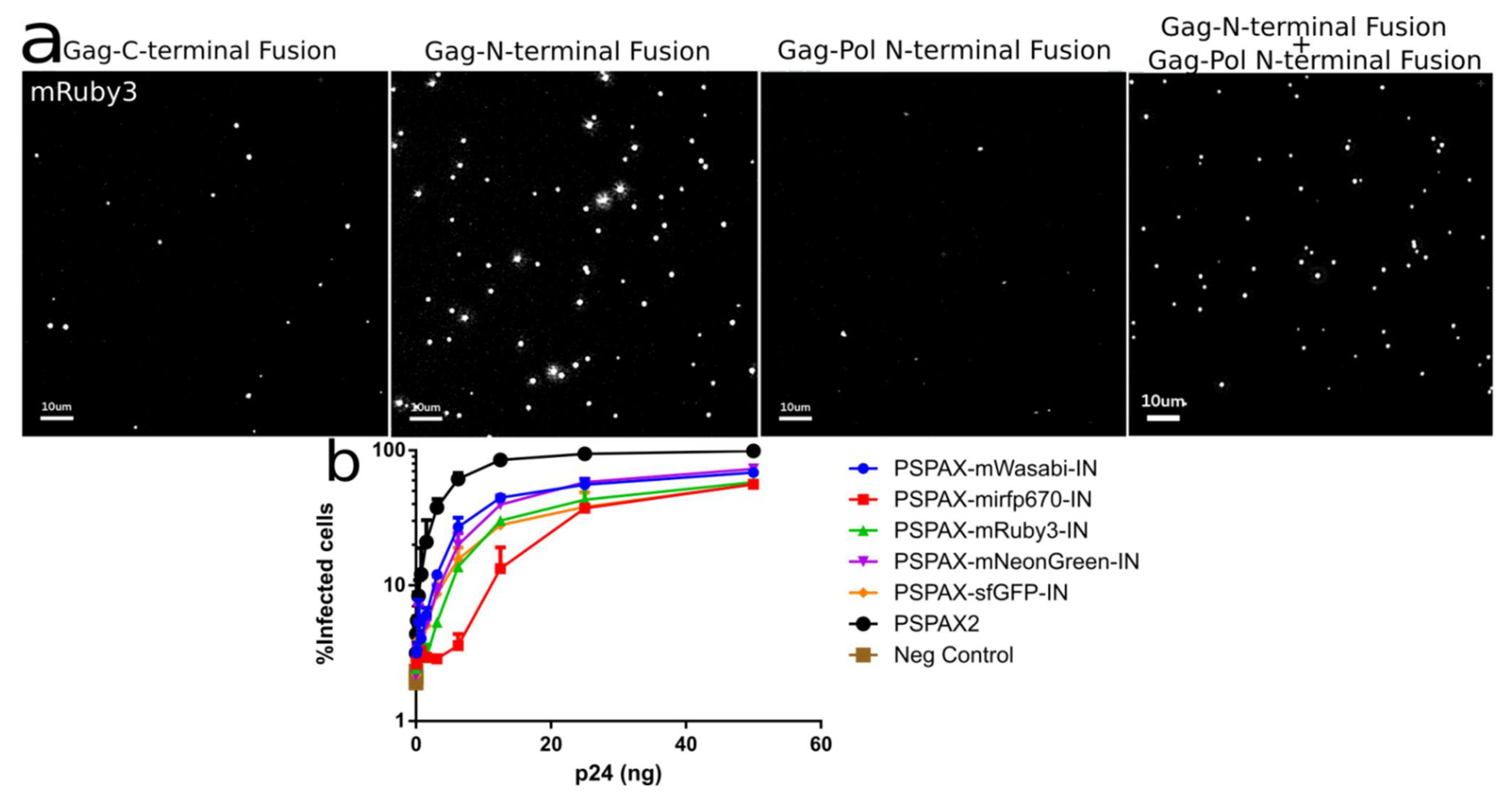
3.2. Development of Gag-Pol Constructs Carrying IN with N-Terminally Fused Fluorescent Proteins
Characterization and Quantification of Fully Labeled Fluorescent IN Labeled Viral Particles that Are Infectious
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barré-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-Lymphotropic Retrovirus from a Patient at Risk for Acquired Immune Deficiency Syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef]
- Popovic, M.; Sarngadharan, M.G.; Read, E.; Gallo, R.C. Detection, Isolation, and Continuous Production of Cytopathic Retroviruses (HTLV-III) from Patients with AIDS and Pre-AIDS. Science 1984, 224, 497–500. [Google Scholar] [CrossRef]
- Campbell, E.M.; Hope, T.J. HIV-1 Capsid: The Multifaceted Key Player in HIV-1 Infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T.J. Early Cytoplasmic Uncoating Is Associated with Infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, E7169–E7178. [Google Scholar] [CrossRef]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the Intracellular Behavior of HIV in Living Cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Di Nunzio, F.; Zimmer, C. FlAsH-PALM: Super-Resolution Pointillist Imaging with FlAsH-Tetracysteine Labeling. Methods Mol. Biol. Clifton NJ 2014, 1174, 183–193. [Google Scholar] [CrossRef]
- Lelek, M.; Nunzio, F.D.; Henriques, R.; Charneau, P.; Arhel, N.; Zimmer, C. Superresolution Imaging of HIV in Infected Cells with FlAsH-PALM. Proc. Natl. Acad. Sci. USA 2012, 109, 8564–8569. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Hope, T.J. Live Cell Imaging of the HIV-1 Life Cycle. Trends Microbiol. 2008, 16, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Marin, M.; Shi, J.; Aiken, C.; Melikyan, G.B. Time-Resolved Imaging of Single HIV-1 Uncoating In Vitro and in Living Cells. PLoS Pathog. 2016, 12, e1005709. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Di Primio, C.; Quercioli, V.; Valentini, P.; Boll, A.; Girelli, G.; Demichelis, F.; Arosio, D.; Cereseto, A. Second Generation Imaging of Nuclear/Cytoplasmic HIV-1 Complexes. AIDS Res. Hum. Retrovir. 2014, 30, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Zhong, Z.; Fischer, D.K.; Harris, G.; Watkins, S.C.; Ambrose, Z.; Zhang, P. Truncated CPSF6 Forms Higher-Order Complexes That Bind and Disrupt HIV-1 Capsid. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Hübner, W.; Chen, P.; Del Portillo, A.; Liu, Y.; Gordon, R.E.; Chen, B.K. Sequence of Human Immunodeficiency Virus Type 1 (HIV-1) Gag Localization and Oligomerization Monitored with Live Confocal Imaging of a Replication-Competent, Fluorescently Tagged HIV-1. J. Virol. 2007, 81, 12596–12607. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Parra, S.; Marin, M.; Gahlaut, N.; Suter, R.; Kondo, N.; Melikyan, G.B. Fusion of Mature HIV-1 Particles Leads to Complete Release of a Gag-GFP-Based Content Marker and Raises the Intraviral PH. PLoS ONE 2013, 8, e71002. [Google Scholar] [CrossRef] [PubMed]
- Márquez, C.L.; Lau, D.; Walsh, J.; Shah, V.; McGuinness, C.; Wong, A.; Aggarwal, A.; Parker, M.W.; Jacques, D.A.; Turville, S.; et al. Kinetics of HIV-1 Capsid Uncoating Revealed by Single-Molecule Analysis. eLife 2018, 7, e34772. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Melikyan, G.B. Single HIV-1 Imaging Reveals Progression of Infection through CA-Dependent Steps of Docking at the Nuclear Pore, Uncoating, and Nuclear Transport. Cell Host Microbe 2018, 23, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.O.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 Uncoats in the Nucleus near Sites of Integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef] [PubMed]
- Bönisch, I.Z.; Dirix, L.; Lemmens, V.; Borrenberghs, D.; Wit, F.D.; Vernaillen, F.; Rocha, S.; Christ, F.; Hendrix, J.; Hofkens, J.; et al. Capsid-Labelled HIV To Investigate the Role of Capsid during Nuclear Import and Integration. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Hulme, A.E.; Kelley, Z.; Foley, D.; Hope, T.J. Complementary Assays Reveal a Low Level of CA Associated with Viral Complexes in the Nuclei of HIV-1-Infected Cells. J. Virol. 2015, 89, 5350–5361. [Google Scholar] [CrossRef]
- Dharan, A.; Talley, S.; Tripathi, A.; Mamede, J.I.; Majetschak, M.; Hope, T.J.; Campbell, E.M. KIF5B and Nup358 Cooperatively Mediate the Nuclear Import of HIV-1 during Infection. PLoS Pathog. 2016, 12, e1005700. [Google Scholar] [CrossRef] [PubMed]
- Ganser-Pornillos, B.K.; Yeager, M.; Pornillos, O. Assembly and Architecture of HIV. Adv. Exp. Med. Biol. 2012, 726, 441–465. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K.; Yeager, M. Atomic-Level Modelling of the HIV Capsid. Nature 2011, 469, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Englund, G.; Orenstein, J.M.; Martin, M.A.; Craigie, R. Multiple Effects of Mutations in Human Immunodeficiency Virus Type 1 Integrase on Viral Replication. J. Virol. 1995, 69, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, H.; Xiao, H.; Conway, J.A.; Hunter, E.; Kappes, J.C. Functional RT and IN Incorporated into HIV-1 Particles Independently of the Gag/Pol Precursor Protein. EMBO J. 1997, 16, 5113–5122. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shehu-Xhilaga, M.; Crowe, S.M.; Mak, J. Maintenance of the Gag/Gag-Pol Ratio Is Important for Human Immunodeficiency Virus Type 1 RNA Dimerization and Viral Infectivity. J. Virol. 2001, 75, 1834–1841. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Cullen, B.R. HIV-1 Structural Gene Expression Requires the Binding of Multiple Rev Monomers to the Viral RRE: Implications for HIV-1 Latency. Cell 1991, 65, 241–248. [Google Scholar] [CrossRef]
- Sherer, N.M.; Swanson, C.M.; Papaioannou, S.; Malim, M.H. Matrix Mediates the Functional Link between Human Immunodeficiency Virus Type 1 RNA Nuclear Export Elements and the Assembly Competency of Gag in Murine Cells. J. Virol. 2009, 83, 8525–8535. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.J.; Hope, T.J.; Bond, B.L.; McDonald, D.; Grahl, K.; Parslow, T.G. Minimal Rev-Response Element for Type 1 Human Immunodeficiency Virus. J. Virol. 1991, 65, 2131–2134. [Google Scholar] [CrossRef] [PubMed]
- Smulevitch, S.; Bear, J.; Alicea, C.; Rosati, M.; Jalah, R.; Zolotukhin, A.S.; von Gegerfelt, A.; Michalowski, D.; Moroni, C.; Pavlakis, G.N.; et al. RTE and CTE MRNA Export Elements Synergistically Increase Expression of Unstable, Rev-Dependent HIV and SIV MRNAs. Retrovirology 2006, 3, 6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Malim, M.H.; Hauber, J.; Le, S.-Y.; Maizel, J.V.; Cullen, B.R. The HIV-1 Rev Trans -Activator Acts through a Structured Target Sequence to Activate Nuclear Export of Unspliced Viral MRNA. Nature 1989, 338, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, J.; Ganser-Pornillos, B.K.; Tivol, W.F.; Sundquist, W.I.; Jensen, G.J. Three-Dimensional Structure of HIV-1 Virus-like Particles by Electron Cryotomography. J. Mol. Biol. 2005, 346, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Mamede, J.I.; Hope, T.J. Detection and Tracking of Dual-Labeled HIV Particles Using Wide-Field Live Cell Imaging to Follow Viral Core Integrity. In HIV Protocols; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; pp. 49–59. ISBN 978-1-4939-3045-6. [Google Scholar]
- Burdick, R.C.; Delviks-Frankenberry, K.A.; Chen, J.; Janaka, S.K.; Sastri, J.; Hu, W.-S.; Pathak, V.K. Dynamics and Regulation of Nuclear Import and Nuclear Movements of HIV-1 Complexes. PLoS Pathog. 2017, 13, e1006570. [Google Scholar] [CrossRef] [PubMed]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A Third-Generation Lentivirus Vector with a Conditional Packaging System. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Ott, D.E.; Gorelick, R.J. Efficiency of Human Immunodeficiency Virus Type 1 Postentry Infection Processes: Evidence against Disproportionate Numbers of Defective Virions. J. Virol. 2007, 81, 4367–4370. [Google Scholar] [CrossRef] [PubMed]
- Butler, S.L.; Hansen, M.S.; Bushman, F.D. A Quantitative Assay for HIV DNA Integration in Vivo. Nat. Med. 2001, 7, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear Pore Blockade Reveals That HIV-1 Completes Reverse Transcription and Uncoating in the Nucleus. Nat. Microbiol. 2020, 5, 1088–1095. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamede, J.I.; Griffin, J.; Gambut, S.; Hope, T.J. A New Generation of Functional Tagged Proteins for HIV Fluorescence Imaging. Viruses 2021, 13, 386. https://doi.org/10.3390/v13030386
Mamede JI, Griffin J, Gambut S, Hope TJ. A New Generation of Functional Tagged Proteins for HIV Fluorescence Imaging. Viruses. 2021; 13(3):386. https://doi.org/10.3390/v13030386
Chicago/Turabian StyleMamede, João I., Joseph Griffin, Stéphanie Gambut, and Thomas J. Hope. 2021. "A New Generation of Functional Tagged Proteins for HIV Fluorescence Imaging" Viruses 13, no. 3: 386. https://doi.org/10.3390/v13030386
APA StyleMamede, J. I., Griffin, J., Gambut, S., & Hope, T. J. (2021). A New Generation of Functional Tagged Proteins for HIV Fluorescence Imaging. Viruses, 13(3), 386. https://doi.org/10.3390/v13030386