
The Valproic Acid Derivative Valpromide Inhibits Pseudorabies Virus Infection in Swine Epithelial and Mouse Neuroblastoma Cell Lines

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Viruses
2.3. Antibodies and Reagents
2.4. Viral Infections
2.5. Endpoint Dilution Assay
2.6. Immunofluorescence Microscopy
2.7. Cell Viability Assay
2.8. Virucidal Effect of VPD against PRV
2.9. Antiviral Effect of ACV and VPD in PK15 Cells Infected with PRV-XGF
2.10. Immunoblot
2.11. Statistics
3. Results
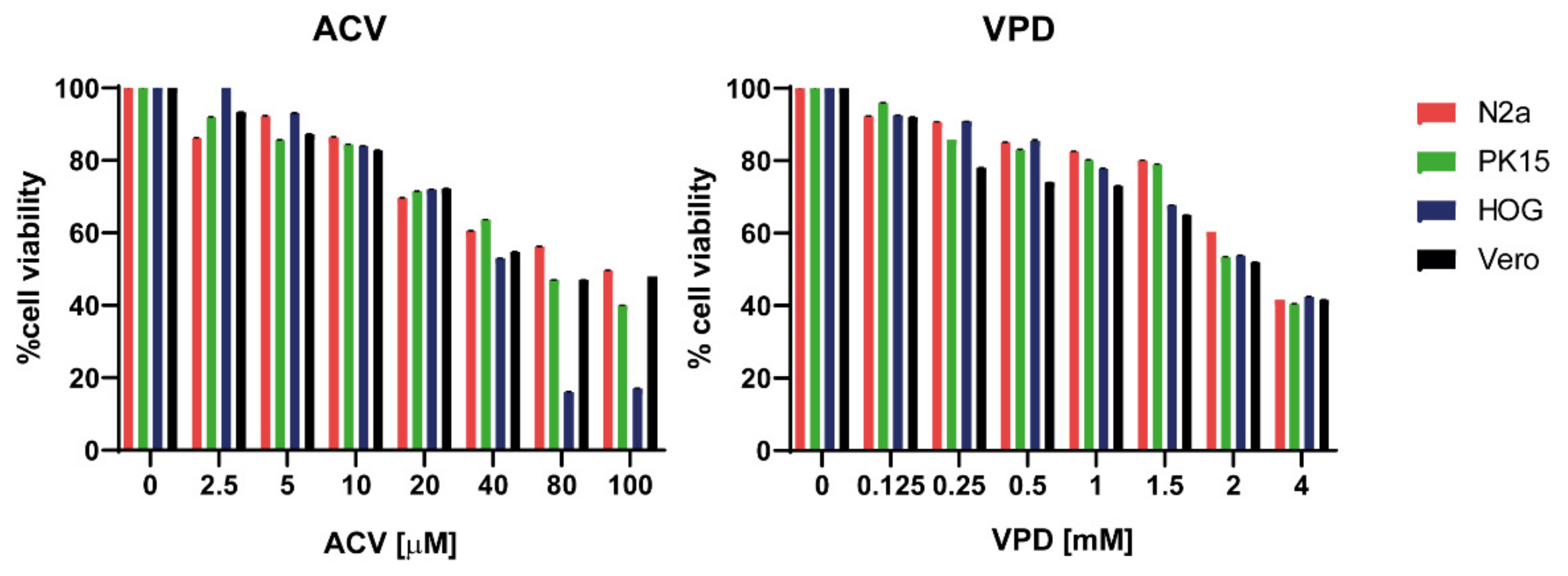
3.1. VPD and ACV Are Non-Toxic in Neither N2a nor PK15 Cells at Clinically Relevant Concentrations
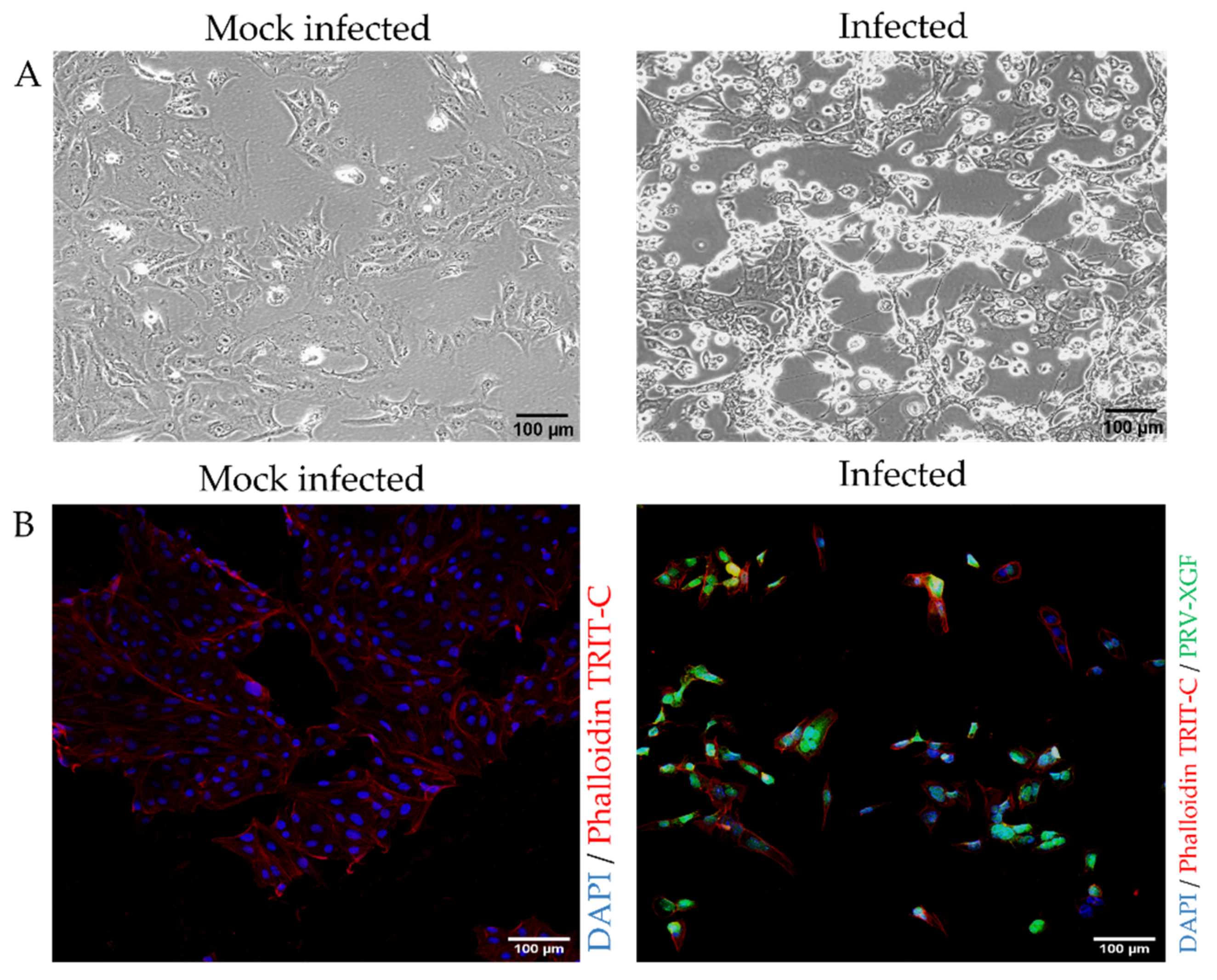
3.2. Infection of PK15 Cells with PRV
3.3. VPD Shows No Virucidal Effect against PRV
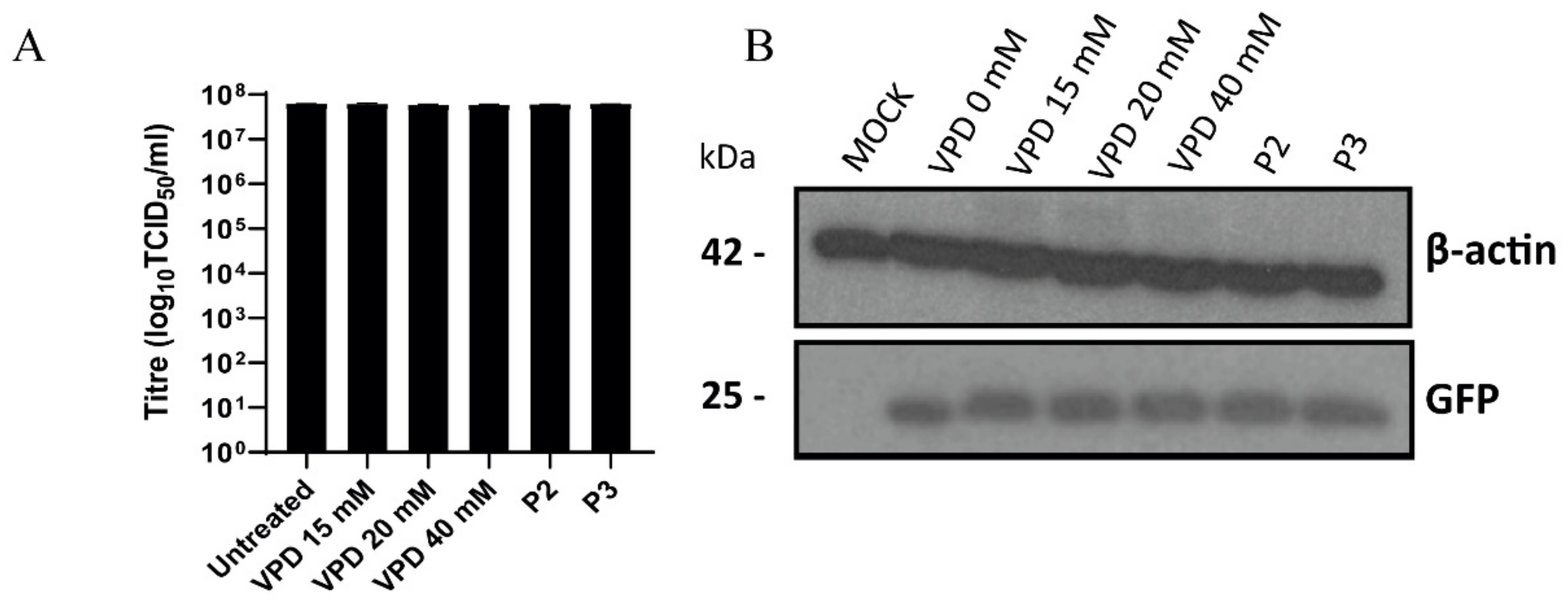
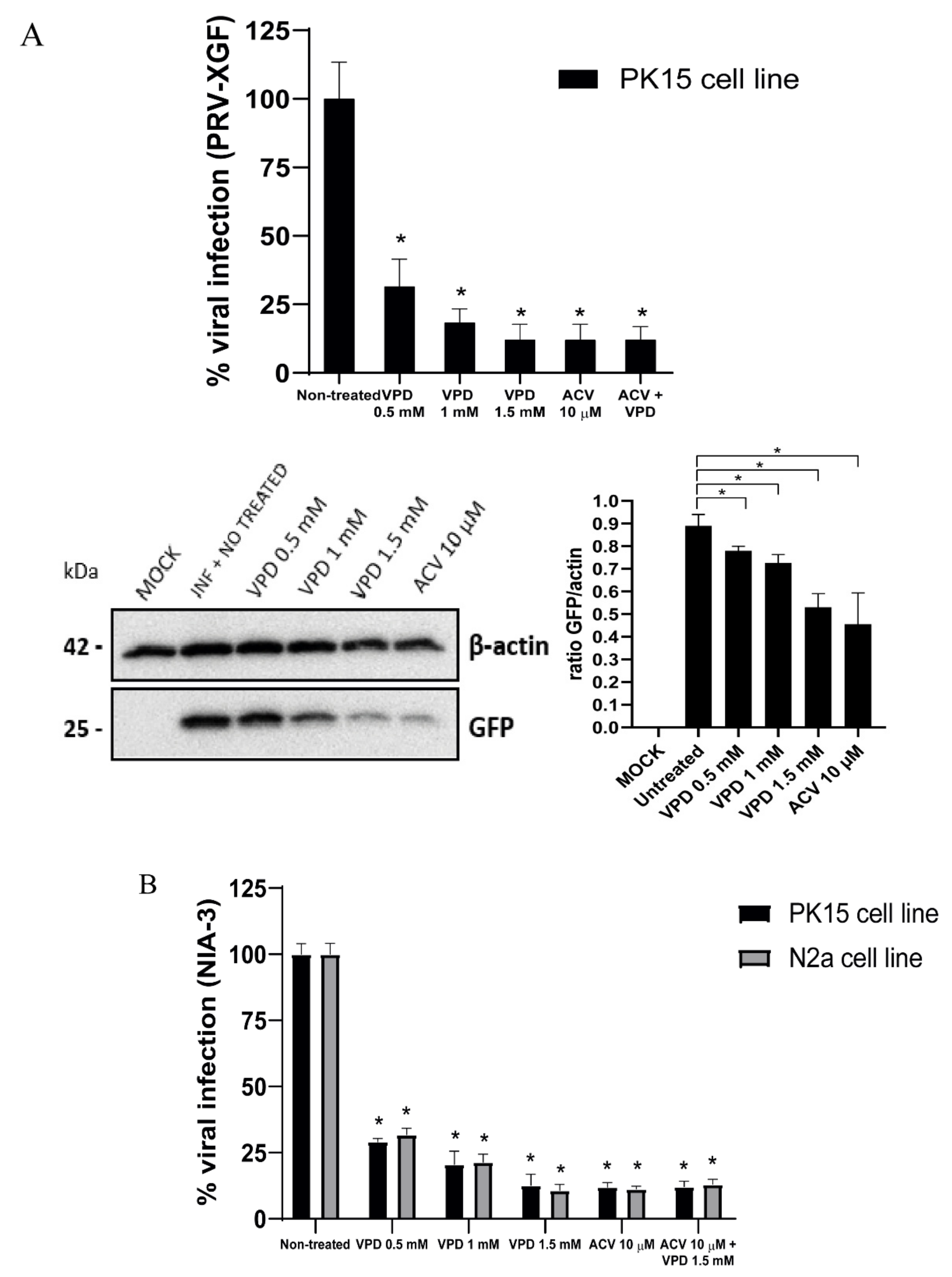
3.4. Antiviral Effect of VPD and ACV on PRV Infection of PK15 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular Biology of Pseudorabies Virus: Impact on Neurovirology and Veterinary Medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettenleiter, T.C. Aujeszky’s disease (pseudorabies) virus: The virus and molecular pathogenesis - State of the art, June 1999. Vet. Res. 2000, 31, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Laval, K.; Enquist, L.W. The Neuropathic Itch Caused by Pseudorabies Virus. Pathogens 2020, 9, 254. [Google Scholar] [CrossRef] [Green Version]
- Nauwynck, H.; Glorieux, S.; Favoreel, H.; Pensaert, M. Cell biological and molecular characteristics of pseudorabies virus infections in cell cultures and in pigs with emphasis on the respiratory tract. Vet. Res. 2007, 38, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabo, A.; Rajcani, J. Latent pseudorabies virus infection in pigs. Acta Virol. 1976, 20, 208–214. [Google Scholar]
- Sun, Y.; Luo, Y.; Wang, C.H.; Yuan, J.; Li, N.; Song, K.; Qiu, H.J. Control of swine pseudorabies in China: Opportunities and limitations. Vet. Microbiol. 2016, 183, 119–124. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Auclert, L.; Zhai, X.; Wong, G.; Zhang, C.; Zhu, H.; Xing, G.; Wang, S.; He, W.; Li, K.; et al. Interspecies transmission, genetic diversity, and evolutionary dynamics of pseudorabies virus. J. Infect. Dis. 2019, 219, 1705–1715. [Google Scholar] [CrossRef]
- Thiry, E.; Addie, D.; Belák, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Hosie, M.J.; Lloret, A.; et al. Aujeszky’s Disease/Pseudorabies in Cats: ABCD guidelines on prevention and management. J. Feline Med. Surg. 2013, 15, 555–556. [Google Scholar] [CrossRef]
- Tu, L.; Lian, J.; Pang, Y.; Liu, C.; Cui, S.; Lin, W. Retrospective detection and phylogenetic analysis of pseudorabies virus in dogs in China. Arch. Virol. 2021, 166, 91–100. [Google Scholar] [CrossRef]
- Cheng, Z.; Kong, Z.; Liu, P.; Fu, Z.; Zhang, J.; Liu, M.; Shang, Y. Natural infection of a variant pseudorabies virus leads to bovine death in China. Transbound. Emerg. Dis. 2020, 67, 518–522. [Google Scholar] [CrossRef]
- Müller, T.; Hahn, E.C.; Tottewitz, F.; Kramer, M.; Klupp, B.G.; Mettenleiter, T.C.; Freuling, C. Pseudorabies virus in wild swine: A global perspective. Arch. Virol. 2011, 156, 1691–1705. [Google Scholar] [CrossRef]
- Yang, X.; Guan, H.; Li, C.; Li, Y.; Wang, S.; Zhao, X.; Zhao, Y.; Liu, Y. Characteristics of human encephalitis caused by pseudorabies virus: A case series study. Int. J. Infect. Dis. 2019, 87, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Ai, J.W.; Weng, S.S.; Cheng, Q.; Cui, P.; Li, Y.J.; Wu, H.L.; Zhu, Y.M.; Xu, B.; Zhang, W.H. Human endophthalmitis caused by pseudorabies virus infection, China, 2017. Emerg. Infect. Dis. 2018, 24, 1087–1090. [Google Scholar] [CrossRef] [Green Version]
- Li, X.D.; Fu, S.H.; Chen, L.Y.; Li, F.; Deng, J.H.; Lu, X.C.; Wang, H.Y.; Tian, K.G. Detection of Pseudorabies Virus Antibodies in Human Encephalitis Cases. Biomed. Environ. Sci. 2020, 33, 444–447. [Google Scholar] [CrossRef]
- Fan, S.; Yuan, H.; Liu, L.; Li, H.; Wang, S.; Zhao, W.; Wu, Y.; Wang, P.; Hu, Y.; Han, J.; et al. Pseudorabies virus encephalitis in humans: A case series study. J. Neurovirol. 2020, 26, 556–564. [Google Scholar] [CrossRef]
- Q, L.; X, W.; C, X.; S, D.; H, Y.; S, G.; J, L.; L, Q.; F, B.; D, W.; et al. A novel human acute encephalitis caused by pseudorabies virus variant strain. Clin. Infect. Dis. 2020, 73(11), e3690–e3700. [Google Scholar] [CrossRef]
- Freuling, C.M.; Müller, T.F.; Mettenleiter, T.C. Vaccines against pseudorabies virus (PrV). Vet. Microbiol. 2017, 206, 3–9. [Google Scholar] [CrossRef]
- Zhou, J.; Li, S.; Wang, X.; Zou, M.; Gao, S. Bartha-k61 vaccine protects growing pigs against challenge with an emerging variant pseudorabies virus. Vaccine 2017, 35, 1161–1166. [Google Scholar] [CrossRef]
- Delva, J.L.; Nauwynck, H.J.; Mettenleiter, T.C.; Favoreel, H.W. The attenuated pseudorabies virus vaccine strain bartha K61: A brief review on the knowledge gathered during 60 years of research. Pathogens 2020, 9, 897. [Google Scholar] [CrossRef]
- Yu, X.; Zhou, Z.; Hu, D.; Zhang, Q.; Han, T.; Li, X.; Gu, X.; Yuan, L.; Zhang, S.; Wang, B.; et al. Pathogenic Pseudorabies Virus, China, 2012. Emerg. Infect. Dis. 2012, 20, 102. [Google Scholar] [CrossRef] [Green Version]
- An, T.Q.; Peng, J.M.; Tian, Z.J.; Zhao, H.Y.; Li, N.; Liu, Y.M.; Chen, J.Z.; Leng, C.L.; Sun, Y.; Chang, D.; et al. Pseudorabies virus variant in Bartha-K61-vaccinated pigs, China, 2012. Emerg. Infect. Dis. 2013, 19, 1749–1755. [Google Scholar] [CrossRef]
- Wang, G.-s.; Du, Y.; Wu, J.-q.; Tian, F.-l.; Yu, X.-j.; Wang, J.-b. Vaccine resistant pseudorabies virus causes mink infection in China. BMC Vet. Res. 2018, 14, 1–8. [Google Scholar] [CrossRef]
- Shiraki, K. Antiviral drugs against alphaherpesvirus. In Advances in Experimental Medicine and Biology; Springer New York LLC: New York, NY, USA, 2018; Volume 1045, pp. 103–122. [Google Scholar]
- Kukhanova, M.K.; Korovina, A.N.; Kochetkov, S.N. Human herpes simplex virus: Life cycle and development of inhibitors. Biochemistry 2014, 79, 1635–1652. [Google Scholar] [CrossRef]
- Ahrens, K.A.; Anderka, M.T.; Feldkamp, M.L.; Canfield, M.A.; Mitchell, A.A.; Werler, M.M. Antiherpetic medication use and the risk of gastroschisis: Findings from the national birth defects prevention study, 1997-2007. Paediatr. Perinat. Epidemiol. 2013, 27, 340–345. [Google Scholar] [CrossRef] [Green Version]
- Andreu, S.; Ripa, I.; Bello-Morales, R.; López-Guerrero, J.A. Valproic Acid and Its Amidic Derivatives as New Antivirals against Alphaherpesviruses. Viruses 2020, 12, 1356. [Google Scholar] [CrossRef]
- Ghodke-Puranik, Y.; Thorn, C.F.; Lamba, J.K.; Leeder, J.S.; Song, W.; Birnbaum, A.K.; Altman, R.B.; Klein, T.E. Valproic acid pathway: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genom. 2013, 23, 236–241. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Calvo, A.; Saiz, J.-C.; Sobrino, F.; Martin-Acebes, M.A. Inhibition of Enveloped Virus Infection of Cultured Cells by Valproic Acid. J. Virol. 2011, 85, 1267–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespillo, A.J.; Praena, B.; Bello-Morales, R.; Lerma, L.; Vázquez-Calvo, A.; Martín-Acebes, M.A.; Tabarés, E.; Sobrino, F.; López-Guerrero, J.A. Inhibition of herpes virus infection in oligodendrocyte cultured cells by valproic acid. Virus Res. 2016, 214, 71–79. [Google Scholar] [CrossRef]
- Gil, M.; González-González, R.; Vázquez-Calvo, A.; Álvarez-Gutiérrez, A.; Martín-Acebes, M.A.; Praena, B.; Bello-Morales, R.; Saiz, J.-C.; López-Guerrero, J.A.; Tabarés, E. Clinical Infections by Herpesviruses in Patients Treated with Valproic Acid: A Nested Case-Control Study in the Spanish Primary Care Database, BIFAP. J. Clin. Med. 2019, 8, 1442. [Google Scholar] [CrossRef] [Green Version]
- Unal, G.; Turan, B.; Balcioglu, Y.H. Immunopharmacological management of COVID-19: Potential therapeutic role of valproic acid. Med. Hypotheses 2020, 143, 109891. [Google Scholar] [CrossRef]
- De León, P.; Bustos, M.J.; Torres, E.; Cañas-Arranz, R.; Sobrino, F.; Carrascosa, A.L. Inhibition of porcine viruses by different cell-targeted antiviral drugs. Front. Microbiol. 2019, 10, 1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello-Morales, R.; Fedetz, M.; Alcina, A.; Tabarés, E.; López-Guerrero, J.A. High susceptibility of a human oligodendroglial cell line to herpes simplex type 1 infection. J. Neurovirol. 2005, 11, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Mawasi, H.; Eisenkraft, A.; Elger, C.E.; Bialer, M.; Kunz, W.S. Mitochondrial liver toxicity of valproic acid and its acid derivatives is related to inhibition of α-lipoamide dehydrogenase. Int. J. Mol. Sci. 2017, 18, 1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialer, M.; Haj-Yehia, A.; Barzaghi, N.; Pisani, F.; Perucca, E. Pharmacokinetics of a valpromide isomer, valnoctamide, in healthy subjects. Eur. J. Clin. Pharmacol. 1990, 38, 289–291. [Google Scholar] [CrossRef]
- Radatz, M.; Ehlers, K.; Yagen, B.; Bialer, M.; Nau, H. Valnoctamide, valpromide and valnoctic acid are much less teratogenic in mice than valproic acid. Epilepsy Res. 1998, 30, 41–48. [Google Scholar] [CrossRef]
- Okada, A.; Kurihara, H.; Aoki, Y.; Bialer, M.; Fujiwara, M. Amidic Modification of Valproic Acid Reduces Skeletal Teratogenicity in Mice. Birth Defects Res. Part B - Dev. Reprod. Toxicol. 2004, 71, 47–53. [Google Scholar] [CrossRef]
- Praena, B.; Bello-Morales, R.; de Castro, F.; López-Guerrero, J.A. Amidic derivatives of valproic acid, valpromide and valnoctamide, inhibit HSV-1 infection in oligodendrocytes. Antiviral Res. 2019, 168, 91–99. [Google Scholar] [CrossRef]
- Isoherranen, N.; Yagen, B.; Bialer, M. New CNS-active drugs which are second-generation valproic acid: Can they lead to the development of a magic bullet? Curr. Opin. Neurol. 2003, 16, 203–211. [Google Scholar] [CrossRef]
- Bialer, M.; Haj-Yehia, A.; Badir, K.; Hadad, S. Can we develop improved derivatives of valproic acid? Pharm. World Sci. 1994, 16, 2–6. [Google Scholar] [CrossRef]
- Bialer, M.; Yagen, B. Valproic Acid: Second Generation. Neurotherapeutics 2007, 4, 130–137. [Google Scholar] [CrossRef]
- Lin, Y.L.; Bialer, M.; Cabrera, R.M.; Finnell, R.H.; Wlodarczyk, B.J. Teratogenicity of valproic acid and its constitutional isomer, amide derivative valnoctamide in mice. Birth Defects Res. 2019, 111, 1013–1023. [Google Scholar] [CrossRef]
- Todaro, G.J.; Benveniste, R.E.; Lieber, M.M.; Sherr, C.J. Characterization of a type C virus released from the porcine cell line PK(15). Virology 1974, 58, 65–74. [Google Scholar] [CrossRef]
- Post, G.R.; Dawson, G. Characterization of a cell line derived from a human oligodendroglioma. Mol. Chem. Neuropathol. 1992, 16, 303–317. [Google Scholar] [CrossRef]
- Viejo-Borbolla, A.; Muñoz, A.; Tabarés, E.; Alcamí, A. Glycoprotein G from pseudorabies virus binds to chemokines with high affinity and inhibits their function. J. Gen. Virol. 2010, 91, 23–31. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Jiang, Y.C.; Feng, H.; Lin, Y.C.; Guo, X.R. New strategies against drug resistance to herpes simplex virus. Int. J. Oral Sci. 2016, 8, 1–6. [Google Scholar] [CrossRef]
- Levin, M.J.; Bacon, T.H.; Leary, J.J. Resistance of herpes simplex virus infections to nucleoside analogues in HIV-infected patients. Clin. Infect. Dis. 2004, 39, S248–S257. [Google Scholar] [CrossRef]
- Gorres, K.L.; Daigle, D.; Mohanram, S.; McInerney, G.E.; Lyons, D.E.; Miller, G. Valpromide inhibits Lytic cycle reactivation of epstein-Barr virus. MBio 2016, 7, e00113–e00116. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.G.; Gaffy, C.B.; Weseli, J.R.; Gorres, K.L. Inhibition of epstein-barr virus lytic reactivation by the atypical antipsychotic drug clozapine. Viruses 2019, 11, 450. [Google Scholar] [CrossRef] [Green Version]
- Ornaghi, S.; Davis, J.N.; Gorres, K.L.; Miller, G.; Paidas, M.J.; van den Pol, A.N. Mood stabilizers inhibit cytomegalovirus infection. Virology 2016, 499, 121–135. [Google Scholar] [CrossRef]
- Davison, A.J. Comparative analysis of the genomes. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007; pp. 10–26. ISBN 9780511545313. [Google Scholar]
- King, D.H. History, pharmacokinetics, and pharmacology of acyclovir. J. Am. Acad. Dermatol. 1988, 18, 176–179. [Google Scholar] [CrossRef]
- Wagstaff, A.J.; Faulds, D.; Goa, K.L. Aciclovir. A reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic efficacy. Drugs 1994, 47, 153–205. [Google Scholar] [CrossRef]
- Diederich, M.; Chateauvieux, S.; Morceau, F.; Dicato, M. Molecular and therapeutic potential and toxicity of valproic acid. J. Biomed. Biotechnol. 2010, 2010, 479364. [Google Scholar]
- Farber, N.B.; Jiang, X.P.; Heinkel, C.; Nemmers, B. Antiepileptic drugs and agents that inhibit voltage-gated sodium channels prevent NMDA antagonist neurotoxicity. Mol. Psychiatry 2002, 7, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Calvo, Á.; Martín-Acebes, M.A.; Sáiz, J.C.; Ngo, N.; Sobrino, F.; de la Torre, J.C. Inhibition of multiplication of the prototypic arenavirus LCMV by valproic acid. Antiviral Res. 2013, 99, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Bialer, M. Pharmacokinetic considerations in the design of better and safer new antiepileptic drugs. J. Control. Release 1999, 62, 187–192. [Google Scholar] [CrossRef]
- Bialer, M.; Rubinstein, A. Pharmacokinetics of valpromide in dogs after various modes of administration. Biopharm. Drug Dispos. 1984, 5, 177–183. [Google Scholar] [CrossRef]
- Martínez, R.; Vaquero, J.; De La Morena, L.V.; Tendillo, F.; Aragonés, P. Toxicology and kinetics of long-term intraventricular infusion of phenytoin and valproic acid in pigs: Experimental study. Acta Neurochir. Suppl. 1991, 52, 3–4. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreu, S.; Ripa, I.; Praena, B.; López-Guerrero, J.A.; Bello-Morales, R. The Valproic Acid Derivative Valpromide Inhibits Pseudorabies Virus Infection in Swine Epithelial and Mouse Neuroblastoma Cell Lines. Viruses 2021, 13, 2522. https://doi.org/10.3390/v13122522
Andreu S, Ripa I, Praena B, López-Guerrero JA, Bello-Morales R. The Valproic Acid Derivative Valpromide Inhibits Pseudorabies Virus Infection in Swine Epithelial and Mouse Neuroblastoma Cell Lines. Viruses. 2021; 13(12):2522. https://doi.org/10.3390/v13122522
Chicago/Turabian StyleAndreu, Sabina, Inés Ripa, Beatriz Praena, José Antonio López-Guerrero, and Raquel Bello-Morales. 2021. "The Valproic Acid Derivative Valpromide Inhibits Pseudorabies Virus Infection in Swine Epithelial and Mouse Neuroblastoma Cell Lines" Viruses 13, no. 12: 2522. https://doi.org/10.3390/v13122522
APA StyleAndreu, S., Ripa, I., Praena, B., López-Guerrero, J. A., & Bello-Morales, R. (2021). The Valproic Acid Derivative Valpromide Inhibits Pseudorabies Virus Infection in Swine Epithelial and Mouse Neuroblastoma Cell Lines. Viruses, 13(12), 2522. https://doi.org/10.3390/v13122522