Transcriptome Analysis of Rice Reveals the lncRNA–mRNA Regulatory Network in Response to Rice Black-Streaked Dwarf Virus Infection

 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Growth, Virus Inoculation, and RNA Isolation
2.2. Preparation of Transcriptome Libraries and Deep Sequencing
2.3. Assembly and Annotation of Transcriptome Data
2.4. Identification of lncRNAs
2.5. Differential Expression Analysis of mRNAs and lncRNAs
2.6. Analyses of GO and KEGG Enrichment
2.7. Prediction of DElncRNA-Targeted mRNAs and Construction of lncRNA–mRNA Regulatory Network
2.8. Validation of DEmRNAs and DElncRNAs Using Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
2.9. Experimental Validation of the lncRNA–mRNA Regulatory Relationship
2.10. Data Availability
3. Results
3.1. RNA Sequencing Results
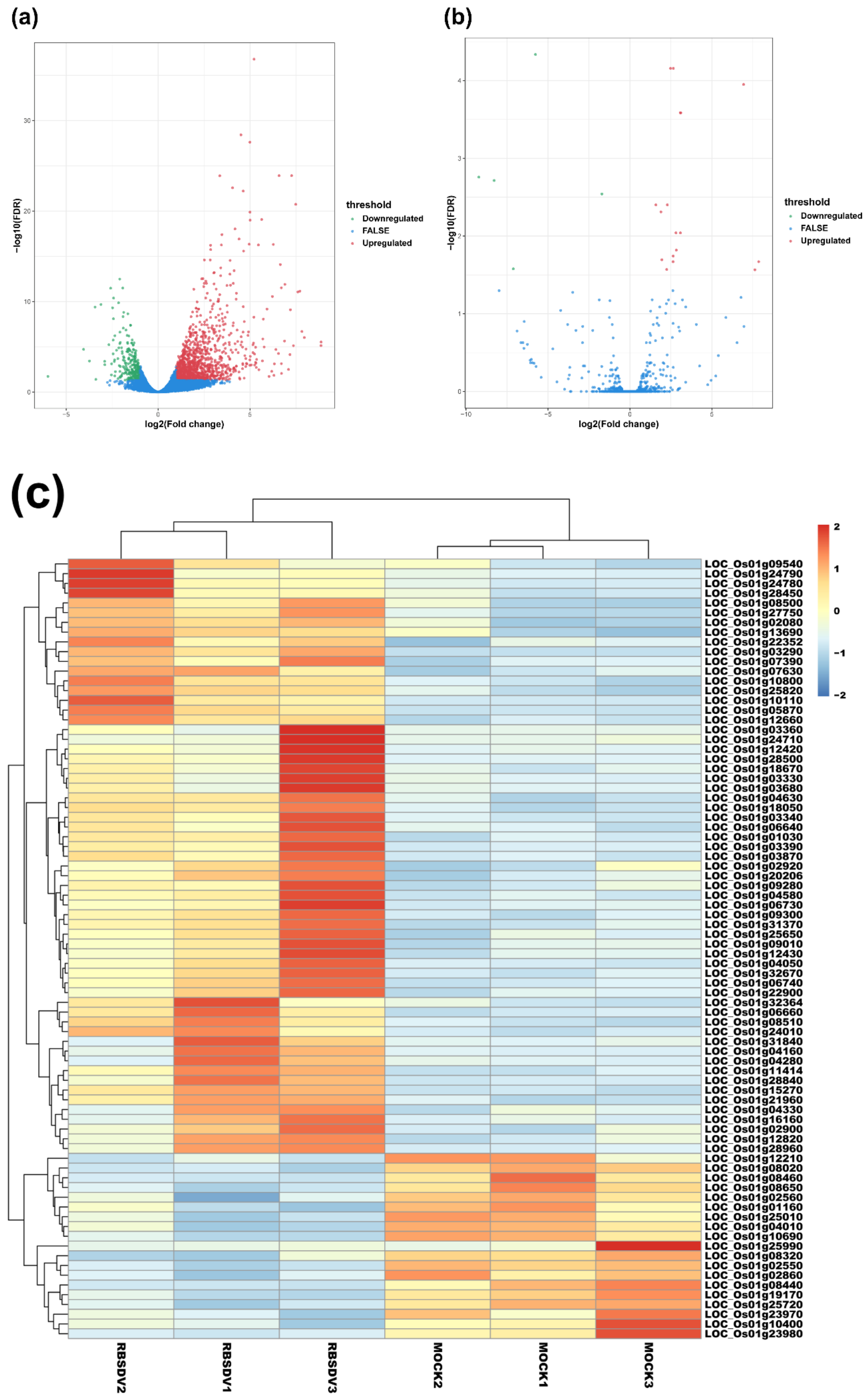
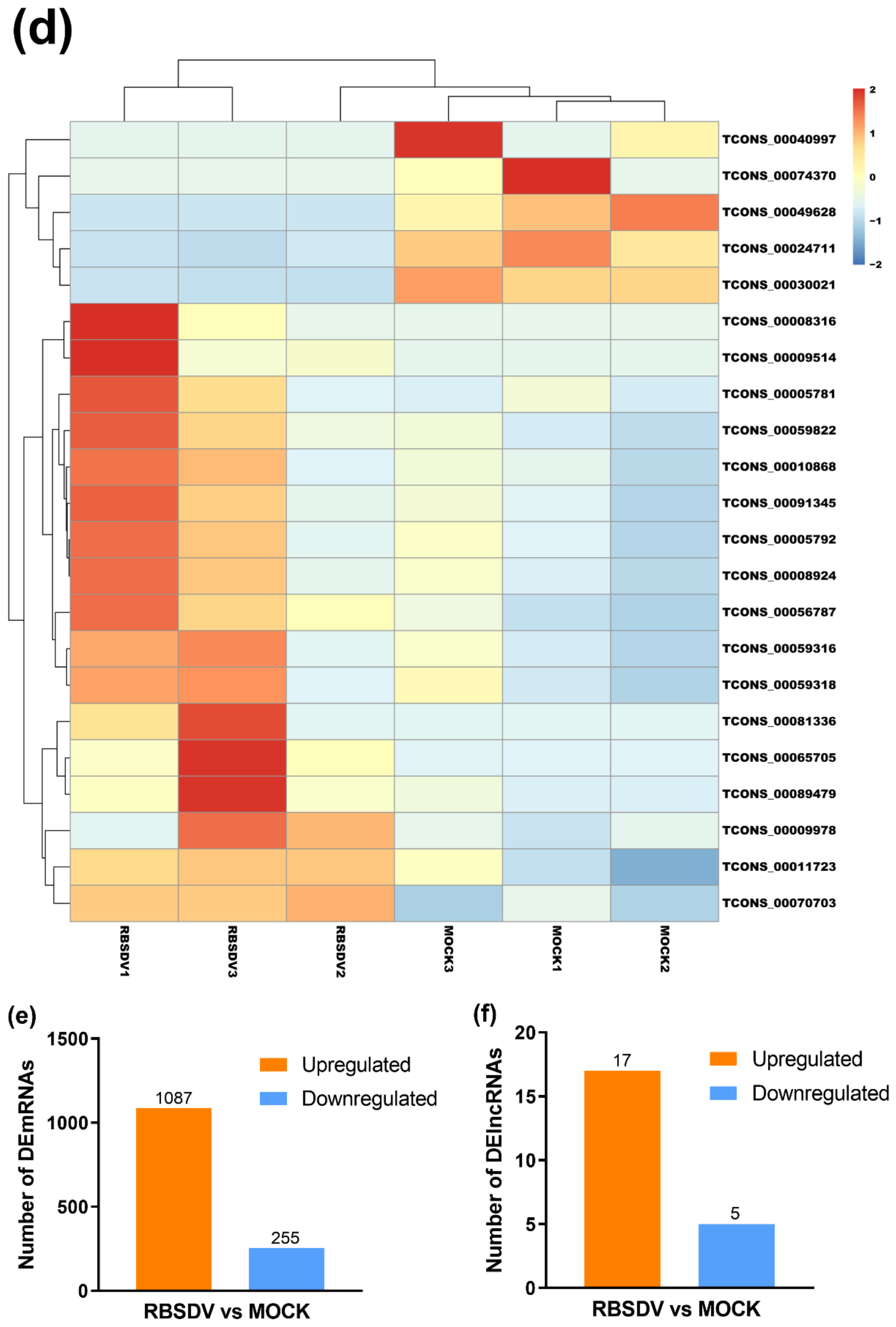
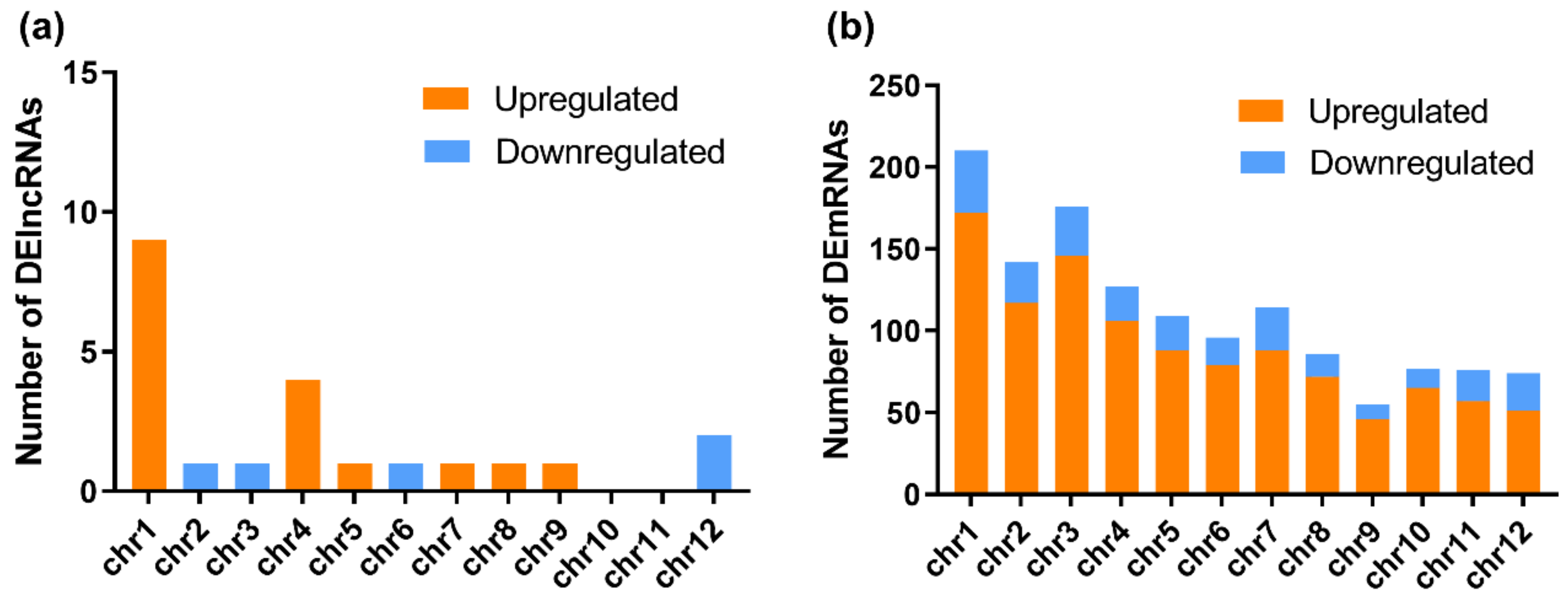
3.2. RBSDV Infection-Induced Differential Expression Patterns of mRNAs and lncRNAs
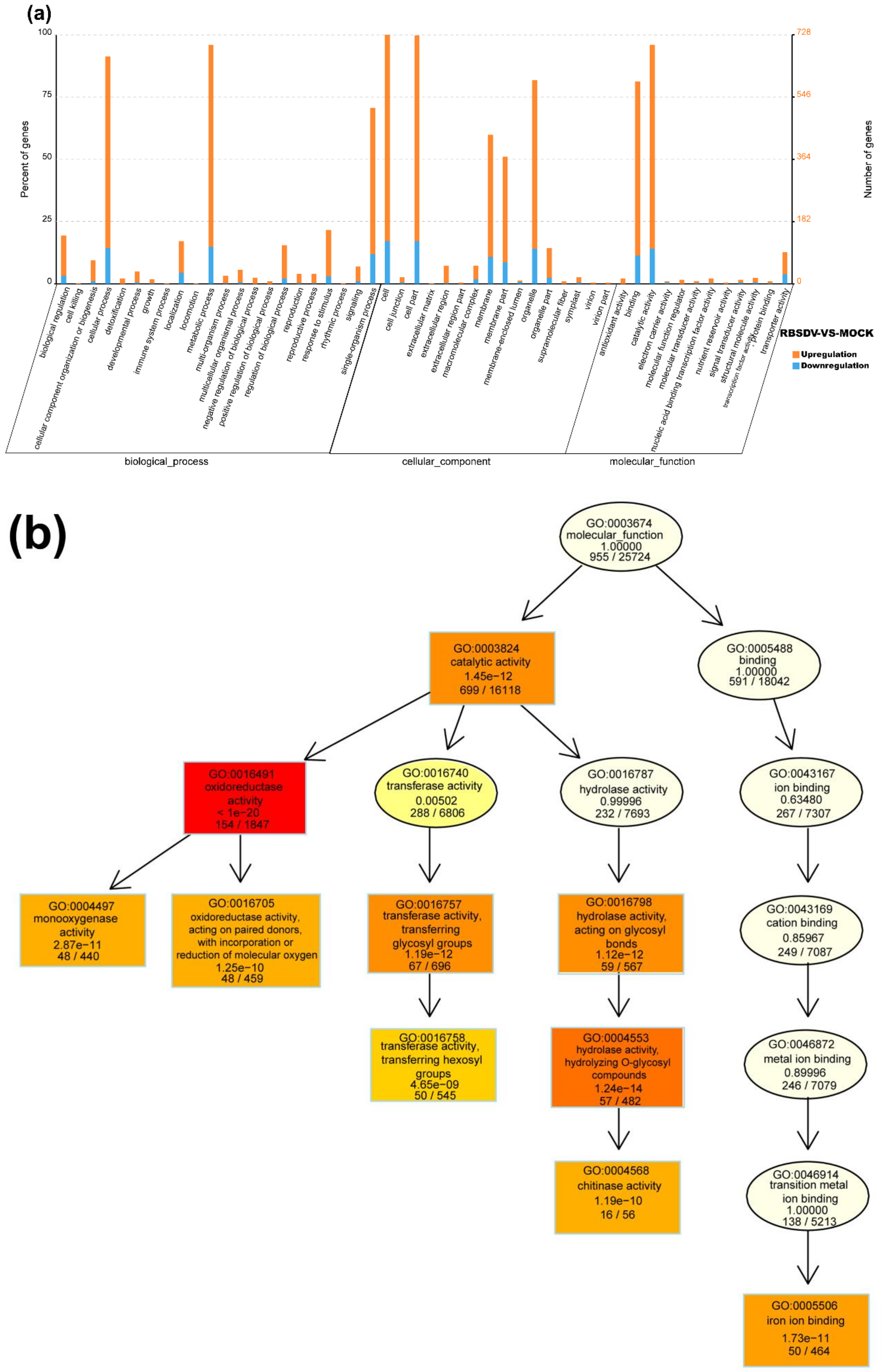
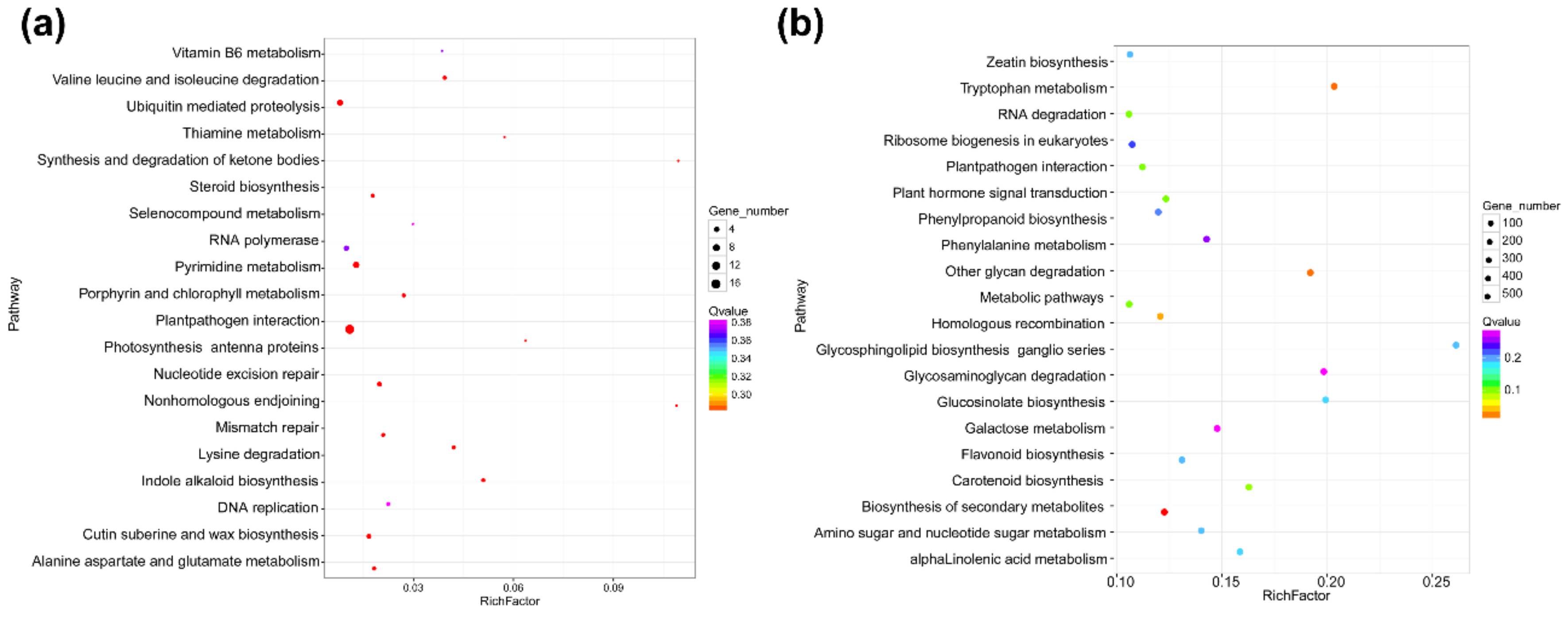
3.3. Functional Characteristics of DEmRNAs
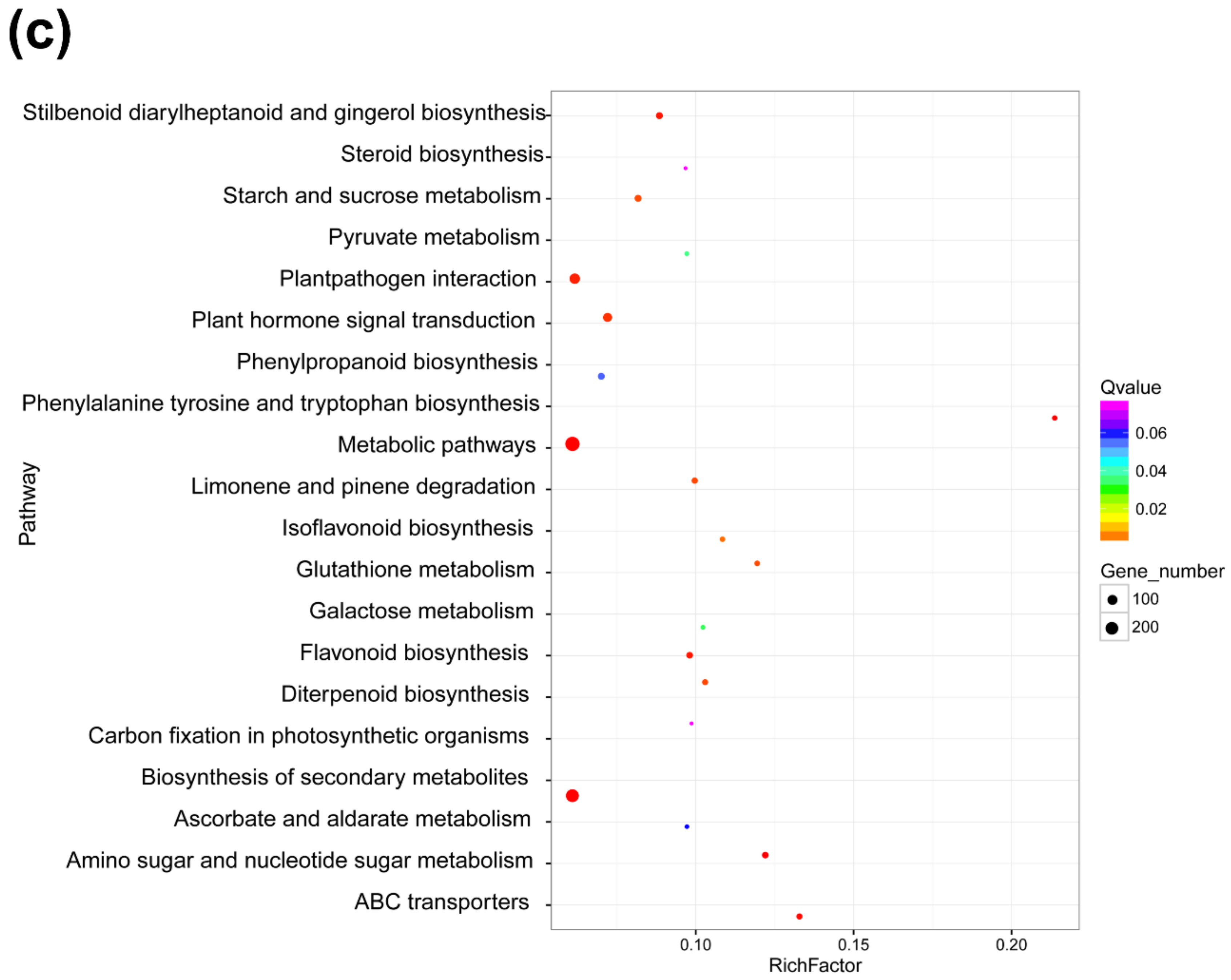
3.4. Functional Analyses of DElncRNA-Targeted Genes
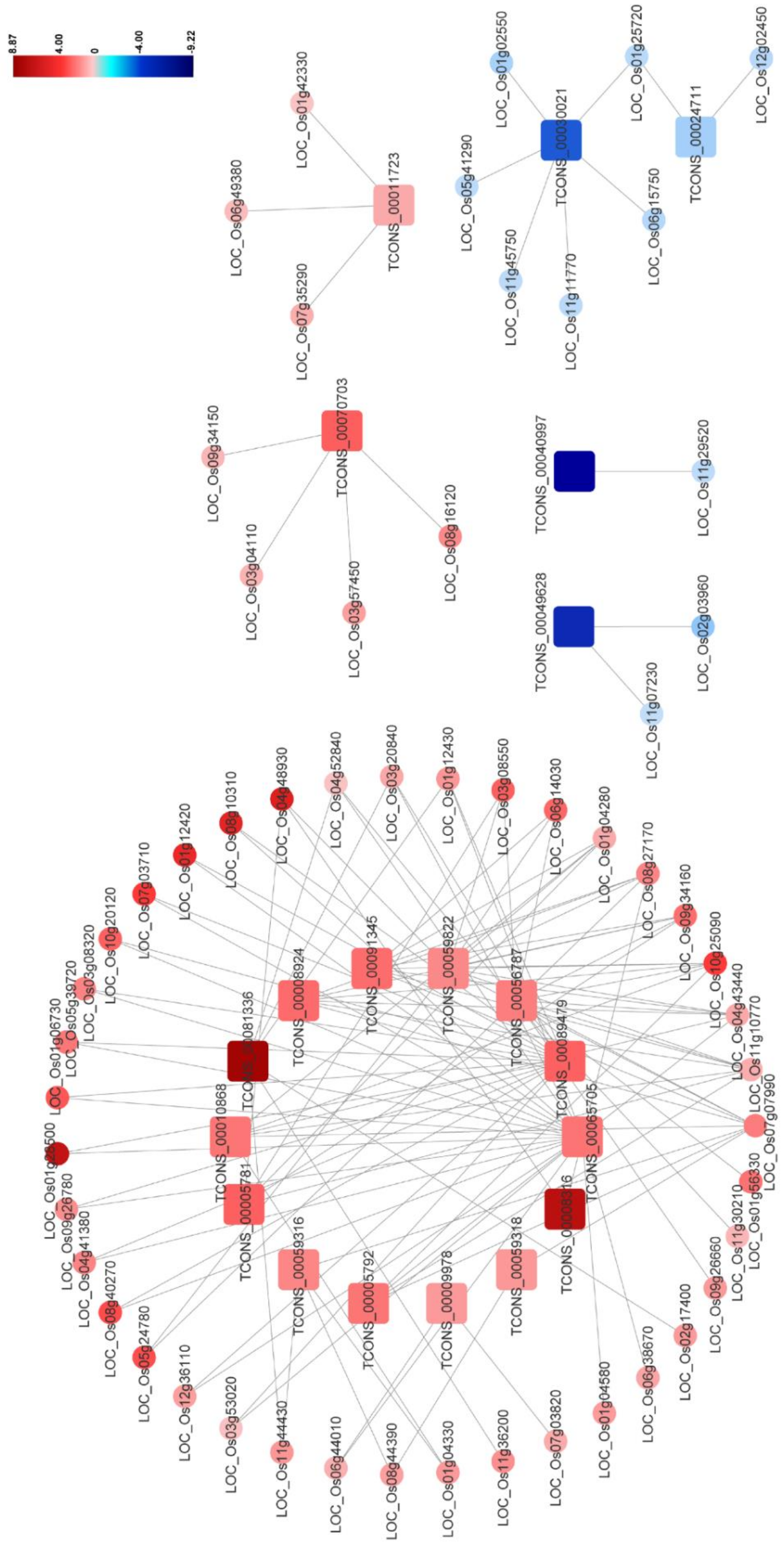
3.5. DElncRNA–DEmRNA Co-Expression Network
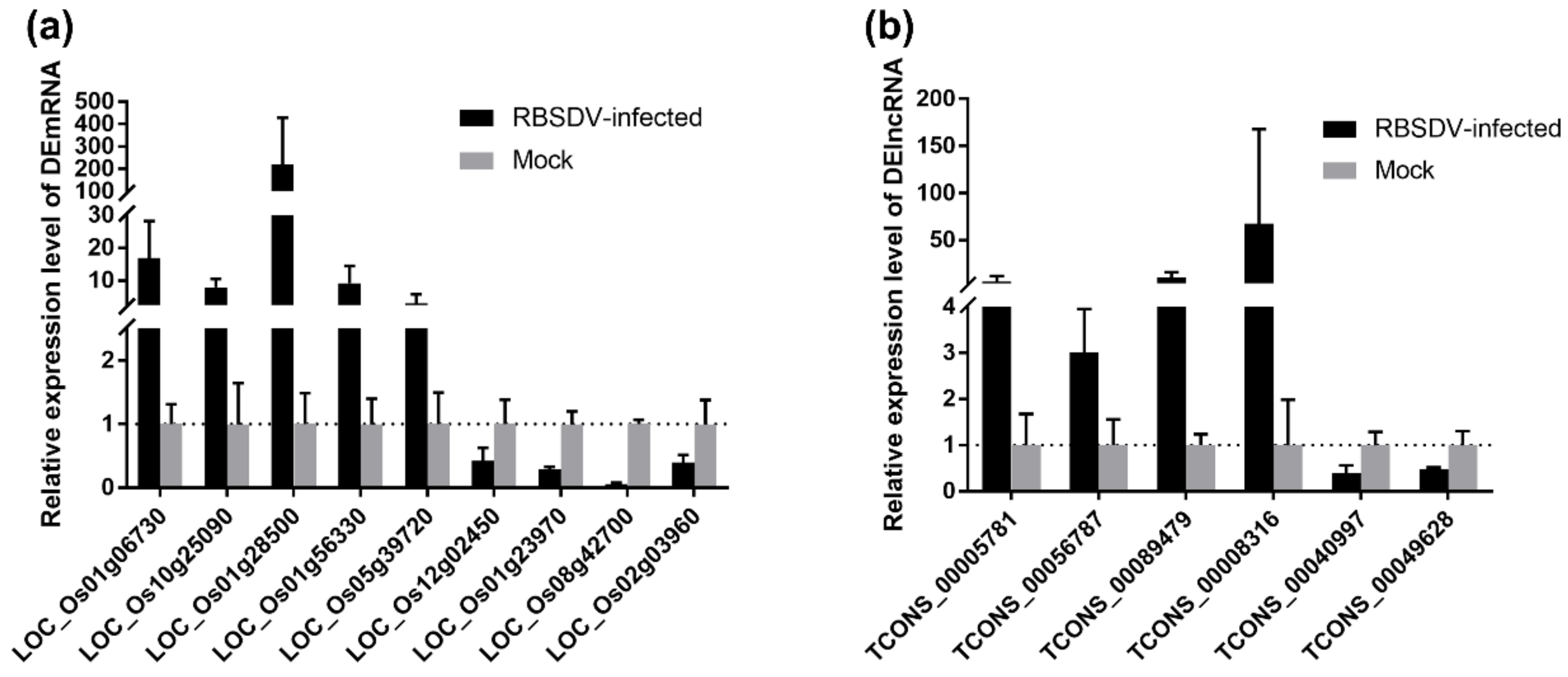
3.6. Validation of DEmRNA and DElncRNA in Response to Viral Infection
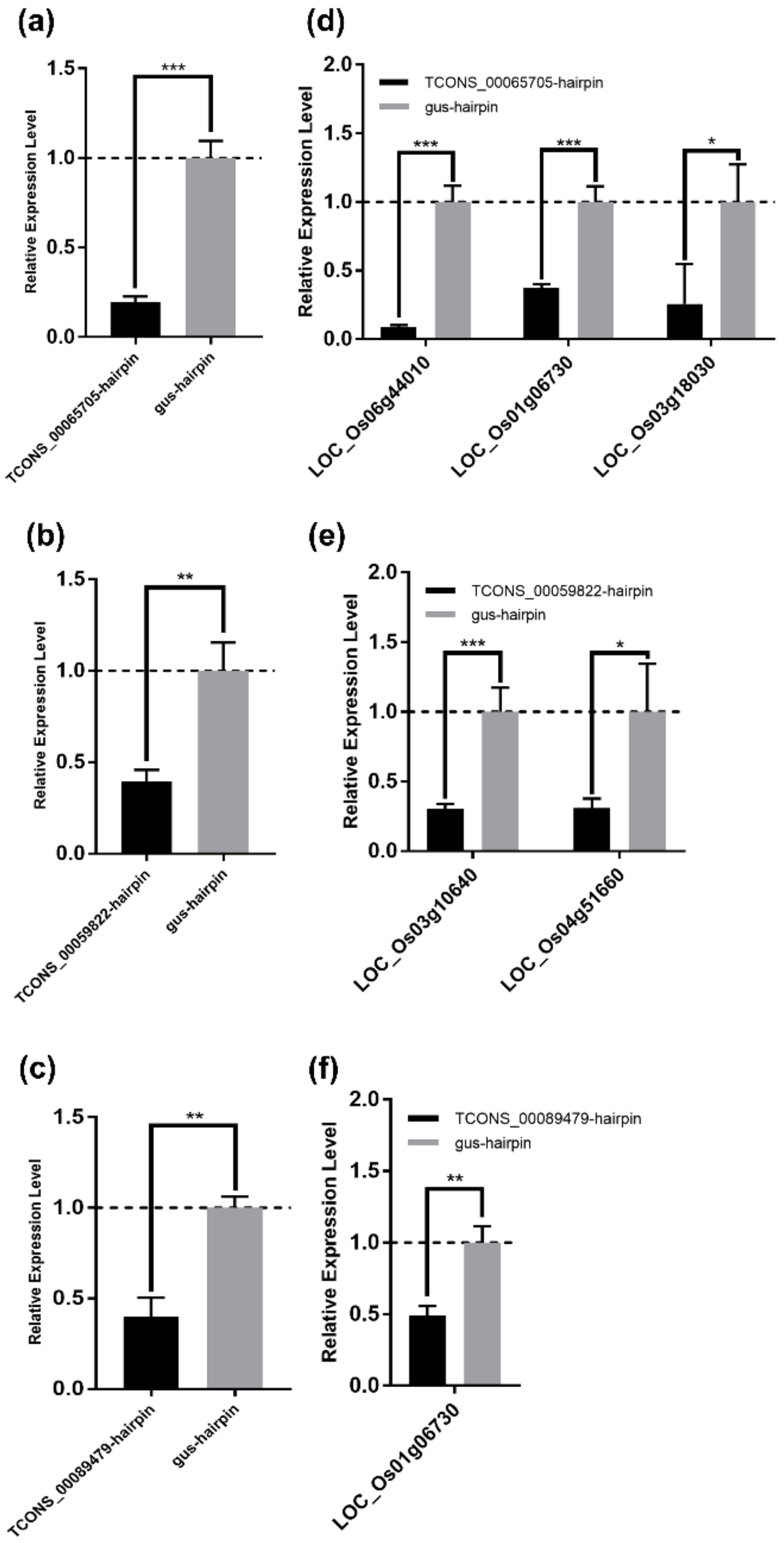
3.7. Experimental Validation of the lncRNA–mRNA Regulatory Relationship in the DElncRNA–DEmRNA Co-Expression Network
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RBSDV | Rice black-streaked dwarf virus |
| SBPH | small brown planthopper |
| FPKM | fragments per kilo-base per million reads |
| FDR | false discovery rate |
| qRT-PCR | quantitative reverse transcription-polymerase chain reaction |
| DEmRNA | differentially expressed mRNA |
| DElncRNA | differentially expressed lncRNA |
References
- Zhang, C.; Liu, Y.; Liu, L.; Lou, Z.; Zhang, H.; Miao, H.; Hu, X.; Pang, Y.; Qiu, B. Rice black streaked dwarf virus P9-1, an α-helical protein, self-interacts and forms viroplasms in vivo. J. Gen. Virol. 2008, 89, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fang, S.; Zhang, Z.; Han, C.; Li, D.; Yu, J. Development of an ID-ELISA for the detection of Rice black-streaked dwarf virus in plants. J. Virol. Methods 2006, 134, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Shikata, E.; Kitagawa, Y. Rice black-streaked dwarf virus: Its properties, morphology and intracellular localization. Virology 1977, 77, 826–842. [Google Scholar] [CrossRef]
- Bai, F.W.; Qu, Z.C.; Yan, J.; Zhang, H.W.; Xu, J.; Ye, M.M.; Wu, H.L.; Liao, X.G.; Shen, D.L. Identification of rice black streaked dwarf virus in different cereal crops with dwarfing symptoms in China. Acta Virol. 2001, 45, 335–339. [Google Scholar]
- Fang, S.; Yu, J.; Feng, J.; Han, C.; Li, D.; Liu, Y. Identification of rice black-streaked dwarf fijivirus in maize with rough dwarf disease in China. Arch. Virol. 2001, 146, 167–170. [Google Scholar] [CrossRef]
- Luan, J.; Wang, F.; Li, Y.; Zhang, B.; Zhang, J. Mapping quantitative trait loci conferring resistance to rice black-streaked virus in maize (Zea mays L.). Theor. Appl. Genet. 2012, 125, 781–791. [Google Scholar] [CrossRef]
- Wu, J.; Ni, Y.; Liu, H.; Rao, L.; Zhou, Y.; Zhou, X. Development and use of three monoclonal antibodies for the detection of rice black-streaked dwarf virus in field plants and planthopper vectors. Virol. J. 2013, 10, 114. [Google Scholar] [CrossRef]
- Zhang, H.M.; Chen, J.P.; Adams, M.J. Molecular characterisation of segments 1 to 6 of Rice black-streaked dwarf virus from China provides the complete genome. Arch. Virol. 2001, 146, 2331–2339. [Google Scholar] [CrossRef]
- Hibino, H. Biology and Epidemiology. Annu. Rev. Phytopathol. 1996, 34, 249–274. [Google Scholar] [CrossRef]
- Conti, M.; Lovisolo, O. Tubular Structures Associated with Maize Rough Dwarf Virus Particles in Crude Extracts: Electron Microscopy Study. J. Gen. Virol. 1971, 13, 173–176. [Google Scholar] [CrossRef]
- Zhang, H.; Tan, X.; Li, L.; He, Y.; Hong, G.; Li, J.; Lin, L.; Cheng, Y.; Yan, F.; Chen, J.; et al. Suppression of auxin signalling promotes rice susceptibility to Rice black streaked dwarf virus infection. Mol. Plant Pathol. 2019, 20, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Li, L.; Zhang, H.; Wang, R.; Tan, X.; He, Y.; Hong, G.; Li, J.; Ming, F.; Yao, X.; et al. Abscisic acid negatively modulates plant defence against rice black-streaked dwarf virus infection by suppressing the jasmonate pathway and regulating reactive oxygen species levels in rice. Plant. Cell Environ. 2018, 41, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, Z.; Duan, C.; Chen, Y.; Meng, Q.; Wu, J.; Hao, Z.; Wang, Z.; Li, M.; Yong, H.; et al. Dual transcriptome analysis reveals insights into the response to Rice black-streaked dwarf virus in maize. J. Exp. Bot. 2016, 67, 4593–4609. [Google Scholar] [CrossRef] [PubMed]
- Chaloner, T.; van Kan, J.A.L.; Grant-Downton, R.T. RNA ‘Information Warfare’ in Pathogenic and Mutualistic Interactions. Trends Plant Sci. 2016, 21, 738–748. [Google Scholar] [CrossRef]
- Xu, L.; Hu, Y.; Cao, Y.; Li, J.; Ma, L.; Li, Y.; Qi, Y. An expression atlas of miRNAs in Arabidopsis thaliana. Sci. China Life Sci. 2018, 61, 178–189. [Google Scholar] [CrossRef]
- Deng, P.; Liu, S.; Nie, X.; Weining, S.; Wu, L. Conservation analysis of long non-coding RNAs in plants. Sci. China Life Sci. 2018, 61, 190–198. [Google Scholar] [CrossRef]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601–614. [Google Scholar] [CrossRef]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, X.; Sun, F.; Hu, J.; Zha, X.; Su, W.; Yang, J. Overexpressing lncRNA LAIR increases grain yield and regulates neighbouring gene cluster expression in rice. Nat. Commun. 2018, 9, 3516. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Chua, N.-H. Long noncoding RNA transcriptome of plants. Plant Biotechnol. J. 2015, 13, 319–328. [Google Scholar] [CrossRef]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.-H. Genome-Wide Analysis Uncovers Regulation of Long Intergenic Noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.-H.; Stephen, S.; Taylor, J.; Helliwell, C.A.; Wang, M.-B. Long noncoding RNAs responsive to Fusarium oxysporum infection in Arabidopsis thaliana. New Phytol. 2014, 201, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.S.; Sun, H.-X.; Park, B.S.; Huang, C.-H.; Yeh, S.-D.; Jung, C.; Chua, N.-H. ELF18-INDUCED LONG-NONCODING RNA Associates with Mediator to Enhance Expression of Innate Immune Response Genes in Arabidopsis. Plant Cell 2017, 29, 1024–1038. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Liu, P.; Irwanto, N.; Loh, D.R.; Wong, S.-M. Upregulation of LINC-AP2 is negatively correlated with AP2 gene expression with Turnip crinkle virus infection in Arabidopsis thaliana. Plant Cell Rep. 2016, 35, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, T.; Shen, D.; Wang, J.; Ling, X.; Hu, Z.; Chen, T.; Hu, J.; Huang, J.; Yu, W.; et al. Tomato yellow leaf curl virus intergenic siRNAs target a host long noncoding RNA to modulate disease symptoms. PLoS Pathog. 2019, 15, 1–22. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Mistry, J.; Bateman, A.; Finn, R.D. Predicting active site residue annotations in the Pfam database. BMC Bioinform. 2007, 8, 298. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Warnes, G.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Mächler, M.; Magnusson, A.; Möller, S. gplots: Various R programming tools for plotting data. Available online: https://cran.r-project.org/package=gplots (accessed on 25 February 2018).
- Fang, P.; Lu, R.; Sun, F.; Lan, Y.; Shen, W.; Du, L.; Zhou, Y.; Zhou, T. Assessment of reference gene stability in Rice stripe virus and Rice black streaked dwarf virus infection rice by quantitative Real-time PCR. Virol. J. 2015, 12, 175. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hiei, Y.; Komari, T. Agrobacterium-mediated transformation of rice using immature embryos or calli induced from mature seed. Nat. Protoc. 2008, 3, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Quinodoz, S.; Guttman, M. Long noncoding RNAs: An emerging link between gene regulation and nuclear organization. Trends Cell Biol. 2014, 24, 651–663. [Google Scholar] [CrossRef]
- Kumar, M.; Brar, A.; Yadav, M.; Chawade, A.; Vivekanand, V.; Pareek, N. Chitinases—Potential Candidates for Enhanced Plant Resistance towards Fungal Pathogens. Agriculture 2018, 8, 88. [Google Scholar] [CrossRef]
- He, Y.; Zhang, H.; Sun, Z.; Li, J.; Hong, G.; Zhu, Q.; Zhou, X.; MacFarlane, S.; Yan, F.; Chen, J. Jasmonic acid-mediated defense suppresses brassinosteroid-mediated susceptibility to Rice black streaked dwarf virus infection in rice. New Phytol. 2017, 214, 388–399. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- He, L.; Chen, X.; Yang, J.; Zhang, T.; Li, J.; Zhang, S.; Zhong, K.; Zhang, H.; Chen, J.; Yang, J. Rice black-streaked dwarf virus-encoded P5-1 regulates the ubiquitination activity of SCF E3 ligases and inhibits jasmonate signaling to benefit its infection in rice. New Phytol. 2020, 225, 896–912. [Google Scholar] [CrossRef]
- Mierziak, J.; Kostyn, K.; Kulma, A. Flavonoids as Important Molecules of Plant Interactions with the Environment. Molecules 2014, 19, 16240–16265. [Google Scholar] [CrossRef]
- Aldon, D.; Mbengue, M.; Mazars, C.; Galaud, J.P. Calcium Signalling in Plant Biotic Interactions. Int. J. Mol. Sci. 2018, 19, 665. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.P.; Somssich, I.E. The Role of WRKY Transcription Factors in Plant Immunity: Figure 1. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Niu, Q.; Wu, H.; Liu, J.; Ye, J.; Yu, N.; Chua, N. Analysis of non-coding transcriptome in rice and maize uncovers roles of conserved lncRNAs associated with agriculture traits. Plant J. 2015, 84, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, Evolution, and Mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef]
- Zhang, Y.-C.; Liao, J.-Y.; Li, Z.-Y.; Yu, Y.; Zhang, J.-P.; Li, Q.-F.; Qu, L.-H.; Shu, W.-S.; Chen, Y.-Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.-T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E.; et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef]
- Wang, T.-Z.; Liu, M.; Zhao, M.-G.; Chen, R.; Zhang, W.-H. Identification and characterization of long non-coding RNAs involved in osmotic and salt stress in Medicago truncatula using genome-wide high-throughput sequencing. BMC Plant Biol. 2015, 15, 131. [Google Scholar] [CrossRef]
- Ahmed, M.M.S.; Ji, W.; Wang, M.; Bian, S.; Xu, M.; Wang, W.; Zhang, J.; Xu, Z.; Yu, M.; Liu, Q.; et al. Transcriptional changes of rice in response to rice black-streaked dwarf virus. Gene 2017, 628, 38–47. [Google Scholar] [CrossRef]
- Lee, H.-A.; Yeom, S.-I. Plant NB-LRR proteins: Tightly regulated sensors in a complex manner. Brief. Funct. Genomics 2015, 14, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Du, L.; Poovaiah, B.W. Calcium signaling and biotic defense responses in plants. Plant Signal. Behav. 2014, 9, e973818. [Google Scholar] [CrossRef] [PubMed]
- Campos, L.; López-Gresa, M.P.; Fuertes, D.; Bellés, J.M.; Rodrigo, I.; Lisón, P. Tomato glycosyltransferase Twi1 plays a role in flavonoid glycosylation and defence against virus. BMC Plant Biol. 2019, 19, 450. [Google Scholar] [CrossRef] [PubMed]
- French, C.J.; Towers, G.H.N. Inhibition of infectivity of potato virus X by flavonoids. Phytochemistry 1992, 31, 3017–3020. [Google Scholar] [CrossRef]
- Dai, Z.; Tan, J.; Zhou, C.; Yang, X.; Yang, F.; Zhang, S.; Sun, S.; Miao, X.; Shi, Z. The OsmiR396-OsGRF8-OsF3H-flavonoid pathway mediates resistance to the brown planthopper in rice (Oryza sativa). Plant Biotechnol. J. 2019, 17, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, S.; Ashikari, M.; Fujioka, S.; Takatsuto, S.; Yoshida, S.; Yano, M.; Yoshimura, A.; Kitano, H.; Matsuoka, M.; Fujisawa, Y.; et al. A Novel Cytochrome P450 Is Implicated in Brassinosteroid Biosynthesis via the Characterization of a Rice Dwarf Mutant, dwarf11, with Reduced Seed Length. Plant Cell 2005, 17, 776–790. [Google Scholar] [CrossRef]
- He, Y.; Hong, G.; Zhang, H.; Tan, X.; Li, L.; Kong, Y.; Sang, T.; Xie, K.; Wei, J.; Li, J.; et al. The OsGSK2 Kinase Integrates Brassinosteroid and Jasmonic Acid Signaling by Interacting with OsJAZ4. Plant Cell 2020, 3. [Google Scholar] [CrossRef]
- Chujo, T.; Miyamoto, K.; Shimogawa, T.; Shimizu, T.; Otake, Y.; Yokotani, N.; Nishizawa, Y.; Shibuya, N.; Nojiri, H.; Yamane, H.; et al. OsWRKY28, a PAMP-responsive transrepressor, negatively regulates innate immune responses in rice against rice blast fungus. Plant Mol. Biol. 2013, 82, 23–37. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mock-Inoculated | RBSDV-Inoculated | |||||
|---|---|---|---|---|---|---|
| MOCK1 | MOCK2 | MOCK3 | RBSDV1 | RBSDV2 | RBSDV3 | |
| Total Reads | 52,107,508 | 50,199,476 | 50,199,546 | 48,580,152 | 50,198,952 | 48,580,180 |
| Clean Reads | 47,629,488 | 46,285,766 | 46,451,138 | 45,328,332 | 46,432,674 | 45,452,636 |
| Reads-rRNAs | 46,936,588 | 45,549,054 | 45,510,752 | 44,740,026 | 45,793,276 | 44,324,792 |
| Reads Mapped to Rice Genome | 42,336,802 (90.20%) | 37,350,224 (82.00%) | 36,545,133 (80.30%) | 36,060,460 (80.60%) | 33,291,711 (72.70%) | 29,520,311 (66.60%) |
| Q20 (%) | 97.37 | 96.28 | 96.25 | 96.48 | 96.15 | 96.42 |
| Q30 (%) | 92.65 | 90.14 | 90.10 | 90.62 | 89.85 | 90.49 |
| Gene ID | Length | log2Ratio (RBSDV/MOCK) | Up-Down-Regulation (RBSDV/MOCK) | p-Value | FDR | Chromosome Location |
|---|---|---|---|---|---|---|
| TCONS_00081336 | 4448 | 7.86 | up | 3.21 × 10−4 | 2.14 × 10−2 | chr7 |
| TCONS_00009514 | 714 | 7.62 | up | 4.88 × 10−4 | 2.71 × 10−2 | chr1 |
| TCONS_00008316 | 1887 | 6.93 | up | 3.67 × 10−7 | 1.12 × 10−4 | chr1 |
| TCONS_00070703 | 1608 | 3.10 | up | 1.27 × 10−6 | 2.60 × 10−4 | chr8 |
| TCONS_00005781 | 11439 | 3.08 | up | 1.04 × 10−4 | 9.10 × 10−3 | chr1 |
| TCONS_00089479 | 1652 | 3.07 | up | 1.27× 10−6 | 2.60 × 10−4 | chr1 |
| TCONS_00008924 | 1722 | 2.83 | up | 1.86 × 10−4 | 1.52 × 10−2 | chr1 |
| TCONS_00091345 | 4501 | 2.81 | up | 9.69 × 10−5 | 9.10 × 10−3 | chr9 |
| TCONS_00065705 | 430 | 2.65 | up | 1.71 × 10−7 | 6.96 × 10−5 | chr5 |
| TCONS_00005792 | 12346 | 2.64 | up | 2.36 × 10−4 | 1.81 × 10−2 | chr1 |
| TCONS_00010868 | 1028 | 2.63 | up | 3.32 × 10−4 | 2.14 × 10−2 | chr1 |
| TCONS_00056787 | 4596 | 2.48 | up | 1.30 × 10−7 | 6.96 × 10−5 | chr4 |
| TCONS_00059316 | 5727 | 2.28 | up | 3.32 × 10−5 | 3.97 × 10−3 | chr4 |
| TCONS_00059822 | 2929 | 2.24 | up | 4.63 × 10−4 | 2.69 × 10−2 | chr4 |
| TCONS_00059318 | 11526 | 1.95 | up | 2.80 × 10−4 | 2.02 × 10−2 | chr4 |
| TCONS_00009978 | 1177 | 1.89 | up | 4.79 × 10−5 | 4.89 × 10−3 | chr1 |
| TCONS_00011723 | 2453 | 1.58 | up | 3.57 × 10−5 | 3.97 × 10−3 | chr1 |
| TCONS_00024711 | 4310 | −1.71 | down | 2.11 × 10−5 | 2.87 × 10−3 | chr12 |
| TCONS_00030021 | 3026 | −5.76 | down | 3.77 × 10−8 | 4.61 × 10−5 | chr12 |
| TCONS_00049628 | 1917 | −7.12 | down | 4.32 × 10−4 | 2.64 × 10−2 | chr3 |
| TCONS_00040997 | 10059 | −8.28 | down | 1.26 × 10−5 | 1.93 × 10−3 | chr2 |
| TCONS_00074370 | 6771 | −9.22 | down | 9.99 × 10−6 | 1.75 × 10−3 | chr6 |
| DElncRNA | Co-Located DEmRNA | |||||||
|---|---|---|---|---|---|---|---|---|
| lncRNA ID | lncRNA Exon Position | Up or Down-Regulation (RBSDV/MOCK) | log2Ratio (RBSDV/MOCK) | mRNA_ID | mRNA_Position | Up or Down-Regulation (RBSDV/MOCK) | log2Ratio (RBSDV/MOCK) | Gene Product Name |
| TCONS_00089479 | 17556112–17556165, 17558595–17560192 | up | 3.07 | LOC_Os08g28710 | 17558062–17560192 | up | 3.06 | Receptor protein kinase CRINKLY4 precursor |
| TCONS_00065705 | 8902479–8902875, 8949120–8949152 | up | 2.65 | LOC_Os05g15770 | 8902479–8903793 | up | 2.31 | Glycosyl hydrolase |
| TCONS_00010868 | 36308049–36308470, 36310641–36311246 | up | 2.63 | LOC_Os01g62630 | 36268063–36269737 | up | 2.97 | Aspartic proteinase nepenthesin precursor |
| TCONS_00056787 | 6938662–6942391, 6942487–6943054, 6943635–6943932 | up | 2.48 | LOC_Os04g12540 | 6939046–6942716 | up | 2.43 | Receptor-like protein kinase |
| TCONS_00059316 | 27164826–27165925, 27166037–27166095, 27166309–27166512, 27171990–27175910, 27177026–27177468 | up | 2.28 | LOC_Os04g45890 | 27172115–27173657 | up | 1.73 | Retrotransposon protein |
| TCONS_00059822 | 30607493–30609094, 30609233–30610559 | up | 2.24 | LOC_Os04g51660 | 30608765–30610406 | up | 1.79 | Transferase family protein |
| TCONS_00059822 | 30607493–30609094, 30609233–30610559 | up | 2.24 | LOC_Os04g51796 | 30697527–30701645 | up | 6.33 | DNA repair ATPase-related |
| TCONS_00009978 | 30512276–30512751, 30512877–30513308, 30513415–30513683 | up | 1.89 | LOC_Os01g53090 | 30512276–30521565 | up | 1.59 | Pathogen-related protein |
| TCONS_00011723 | 41949333–41951662, 41951830–41951887, 41952060–41952124 | up | 1.58 | LOC_Os01g72270 | 41900002–41901651 | up | 3.89 | Cytochrome P450-dependent fatty acid hydroxylase |
| TCONS_00024711 | 819049–819195, 819546–819756, 820494–824445 | down | −1.71 | LOC_Os12g02330 | 746155–747300 | up | 1.47 | LTPL13—Protease inhibitor |
| TCONS_00030021 | 19637837–19638788, 19638882–19640733, 19641766–19641987 | down | −5.76 | LOC_Os12g32610 | 19675353–19676314 | up | 2.57 | - |
| TCONS_00049628 | 22799469–22800028, 22800496–22801659, 22803529–22803721 | down | −7.12 | LOC_Os03g40930 | 22744328–22746916 | up | 1.49 | - |
| DElncRNA | DEmRNA | Verified? | Gene Product Name |
|---|---|---|---|
| TCONS_00065705 | LOC_Os06g44010 | Yes | OsWRKY28 |
| LOC_Os01g06730 | Yes | Receptor-like protein | |
| LOC_Os03g18030 | Yes | Leucocyanidin oxygenase | |
| LOC_Os01g28500 | No | PR1 protein | |
| LOC_Os03g03034 | No | Flavonol synthase/flavanone 3-hydroxylase | |
| TCONS_00059822 | LOC_Os03g10640 | Yes | Calcium-transporting ATPase |
| LOC_Os04g51660 | Yes | Malonyl transferase | |
| LOC_Os02g47470 | No | Cytochrome P450 | |
| TCONS_00089479 | LOC_Os01g06730 | Yes | Receptor-like protein |
| LOC_Os01g28500 | No | PR1 protein |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Liang, Q.; Li, C.; Fu, S.; Kundu, J.K.; Zhou, X.; Wu, J. Transcriptome Analysis of Rice Reveals the lncRNA–mRNA Regulatory Network in Response to Rice Black-Streaked Dwarf Virus Infection. Viruses 2020, 12, 951. https://doi.org/10.3390/v12090951
Zhang T, Liang Q, Li C, Fu S, Kundu JK, Zhou X, Wu J. Transcriptome Analysis of Rice Reveals the lncRNA–mRNA Regulatory Network in Response to Rice Black-Streaked Dwarf Virus Infection. Viruses. 2020; 12(9):951. https://doi.org/10.3390/v12090951
Chicago/Turabian StyleZhang, Tianze, Qian Liang, Chenyang Li, Shuai Fu, Jiban Kumar Kundu, Xueping Zhou, and Jianxiang Wu. 2020. "Transcriptome Analysis of Rice Reveals the lncRNA–mRNA Regulatory Network in Response to Rice Black-Streaked Dwarf Virus Infection" Viruses 12, no. 9: 951. https://doi.org/10.3390/v12090951
APA StyleZhang, T., Liang, Q., Li, C., Fu, S., Kundu, J. K., Zhou, X., & Wu, J. (2020). Transcriptome Analysis of Rice Reveals the lncRNA–mRNA Regulatory Network in Response to Rice Black-Streaked Dwarf Virus Infection. Viruses, 12(9), 951. https://doi.org/10.3390/v12090951