
Invasive Everywhere? Phylogeographic Analysis of the Globally Distributed Tree Pathogen Lasiodiplodia theobromae


Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolate Collections and DNA Extractions
2.2. PCR Amplifications, DNA Sequencing, and Confirmation of Species Identity
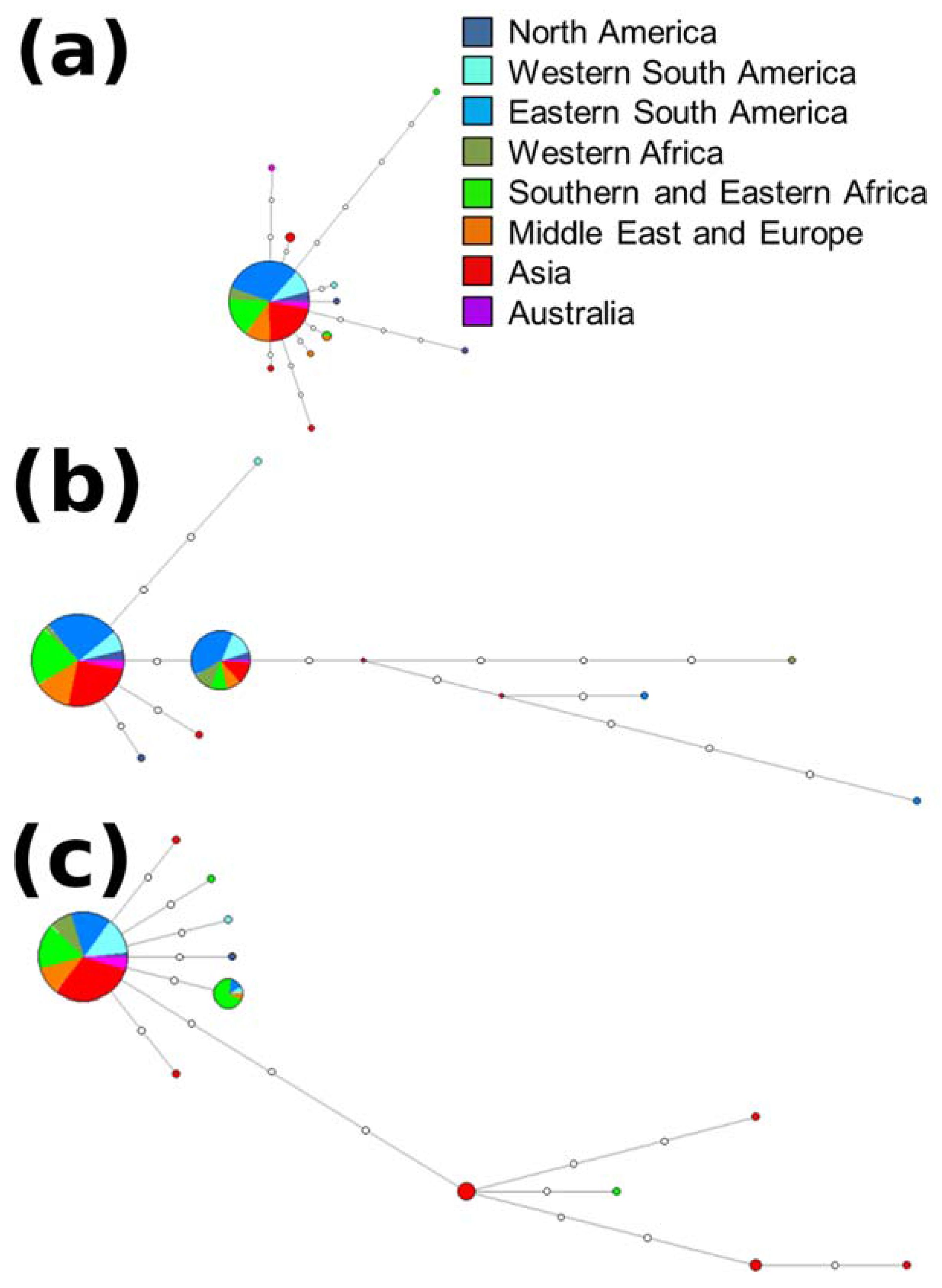
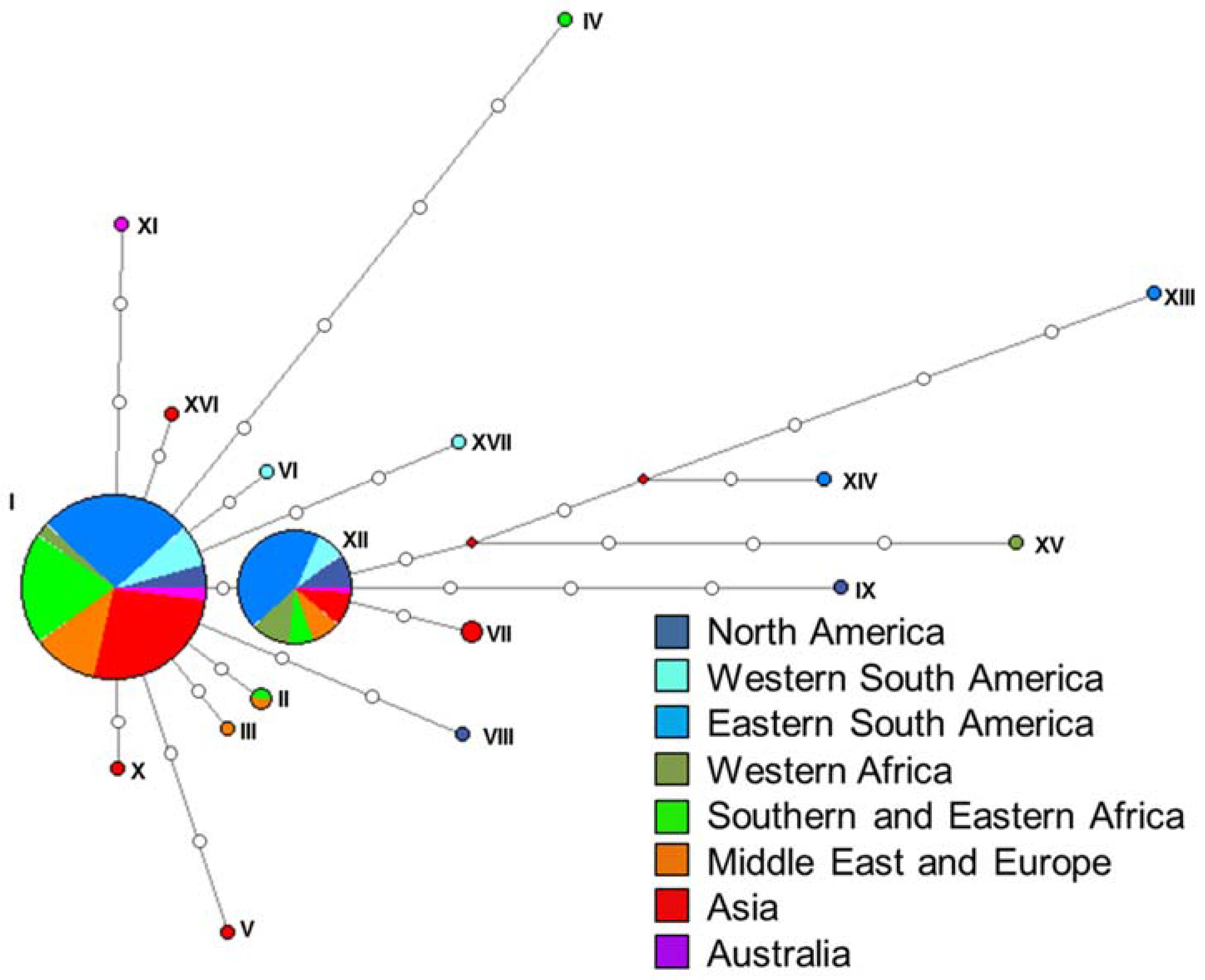
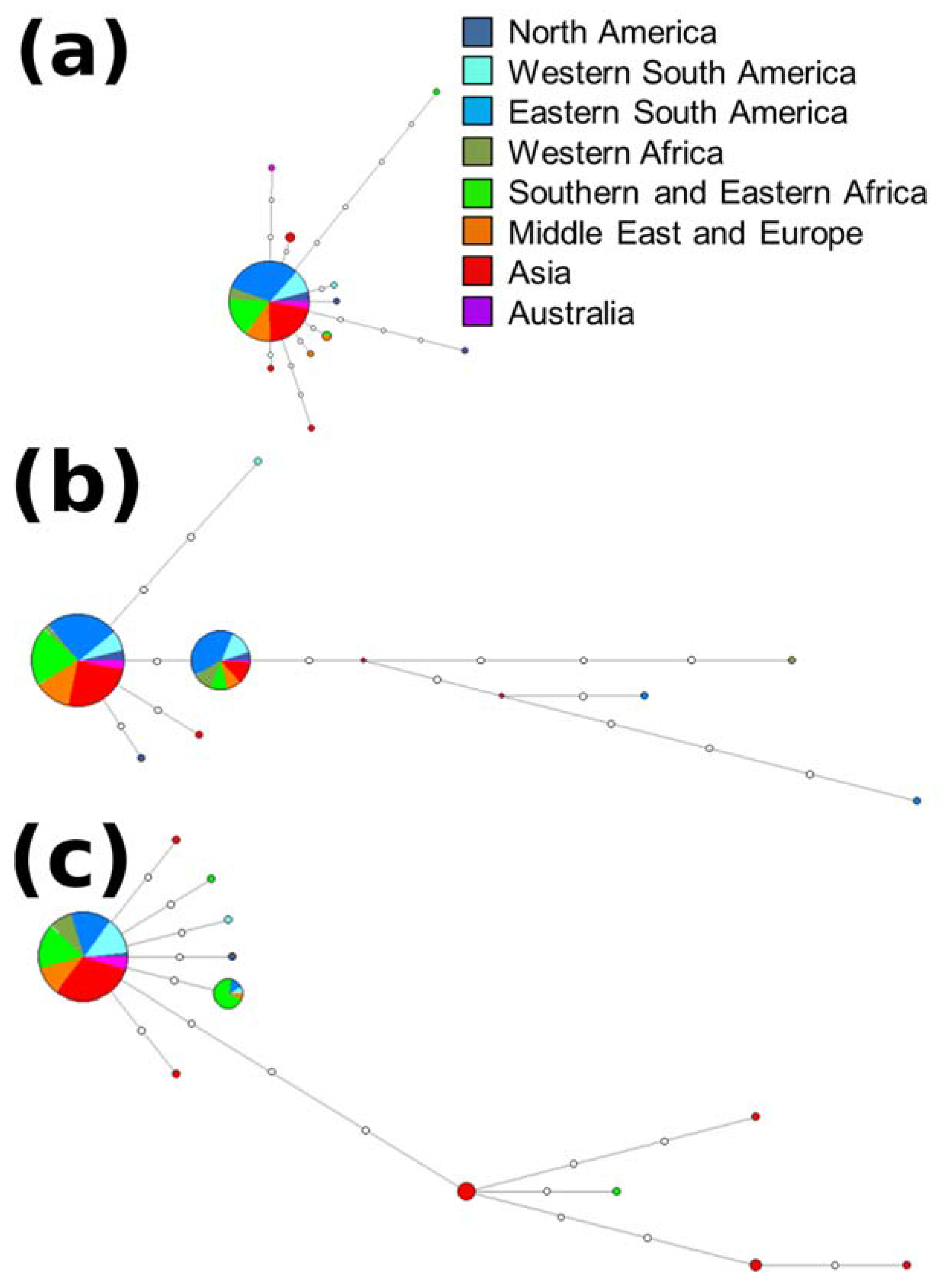
2.3. Haplotype Assignment and Networks
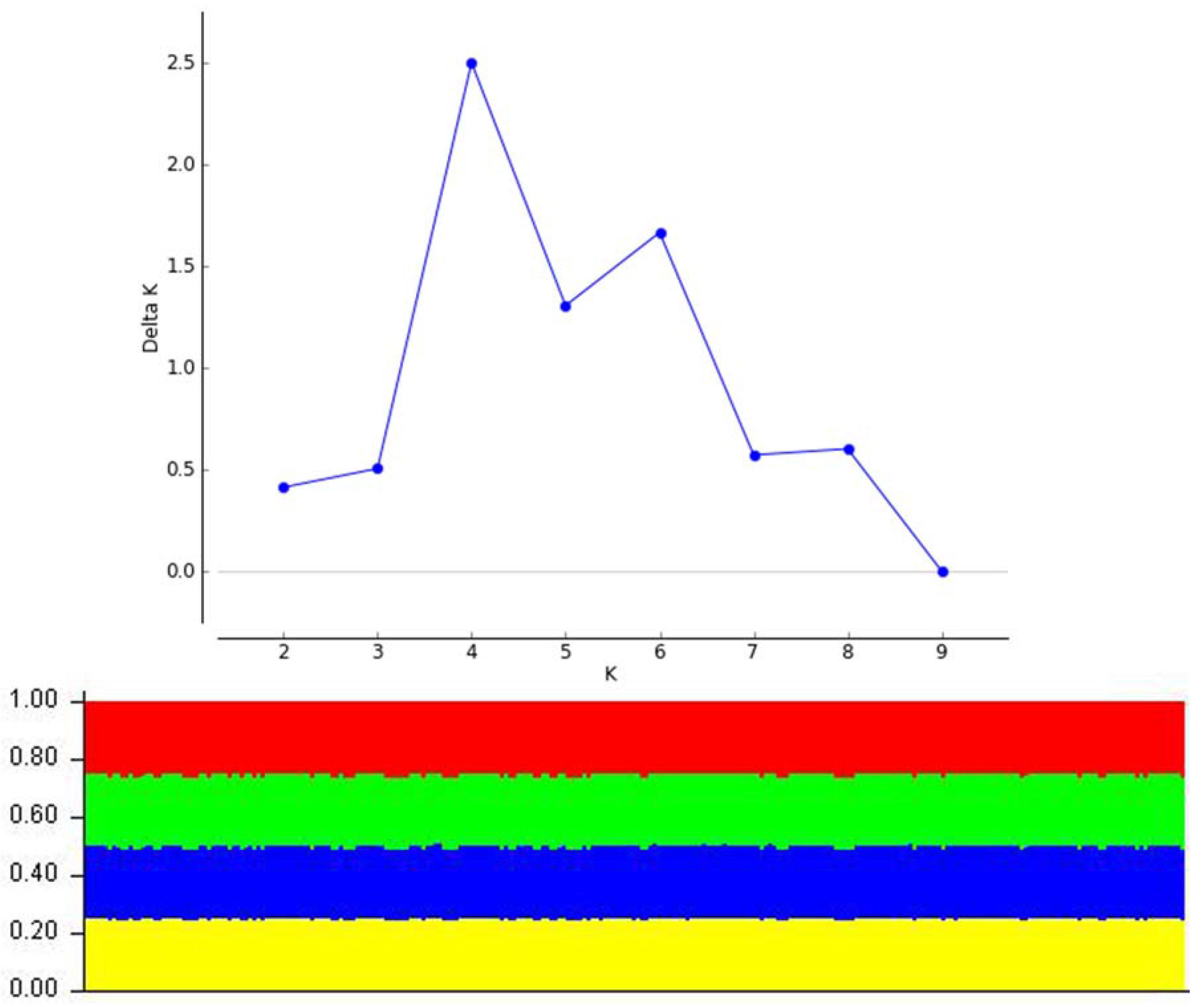
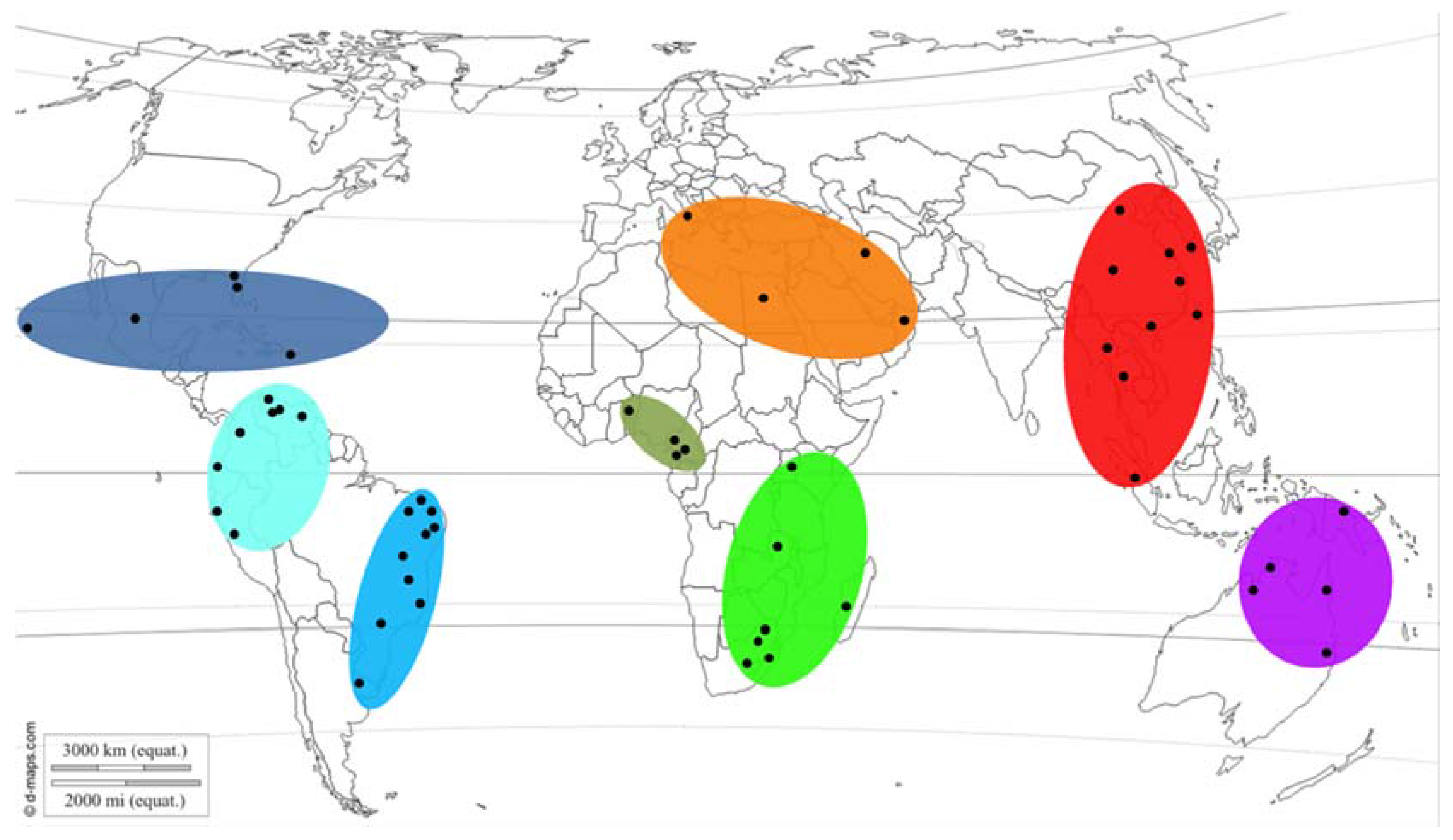
2.4. Population and Regional Structure and Diversity
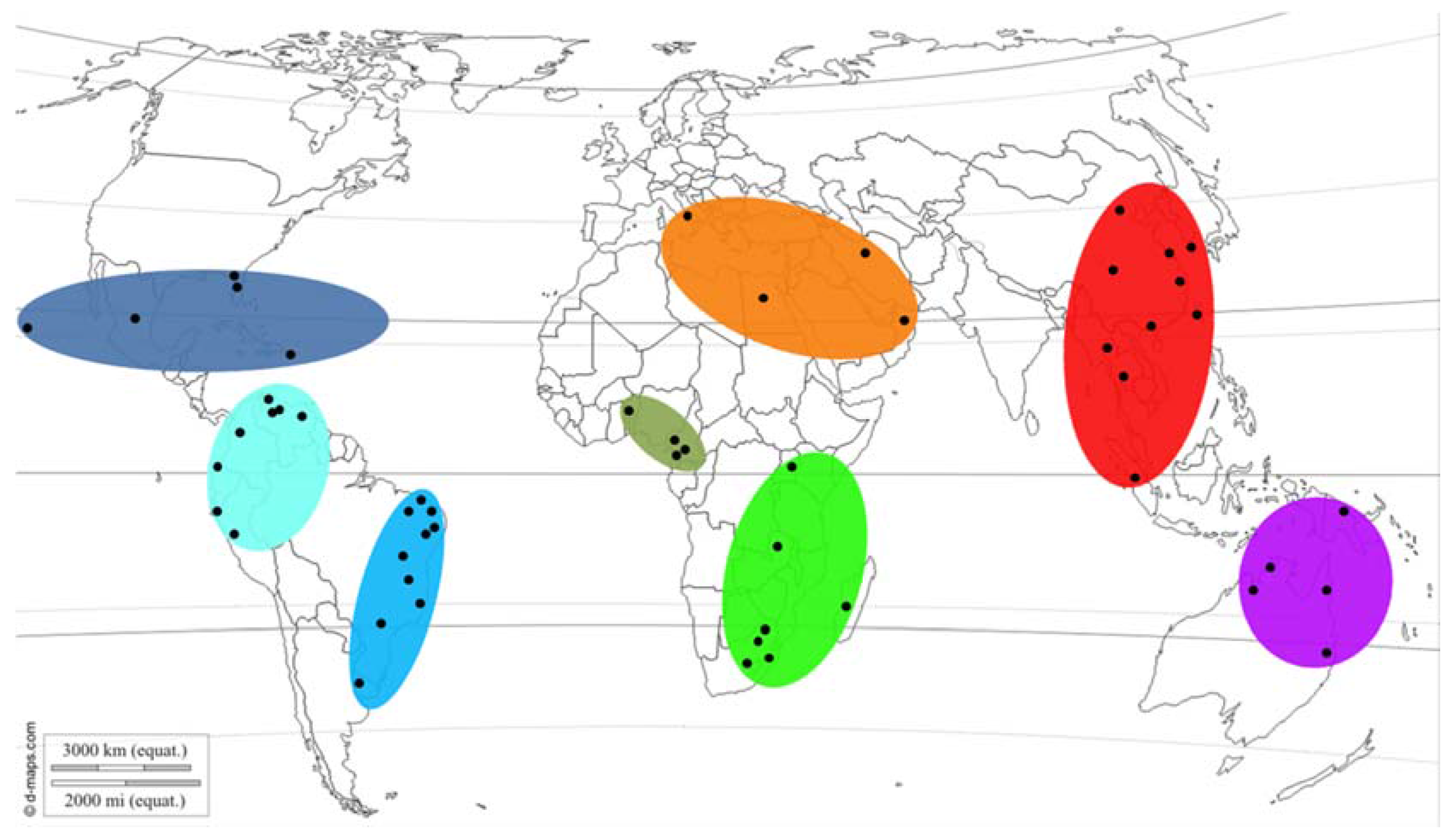
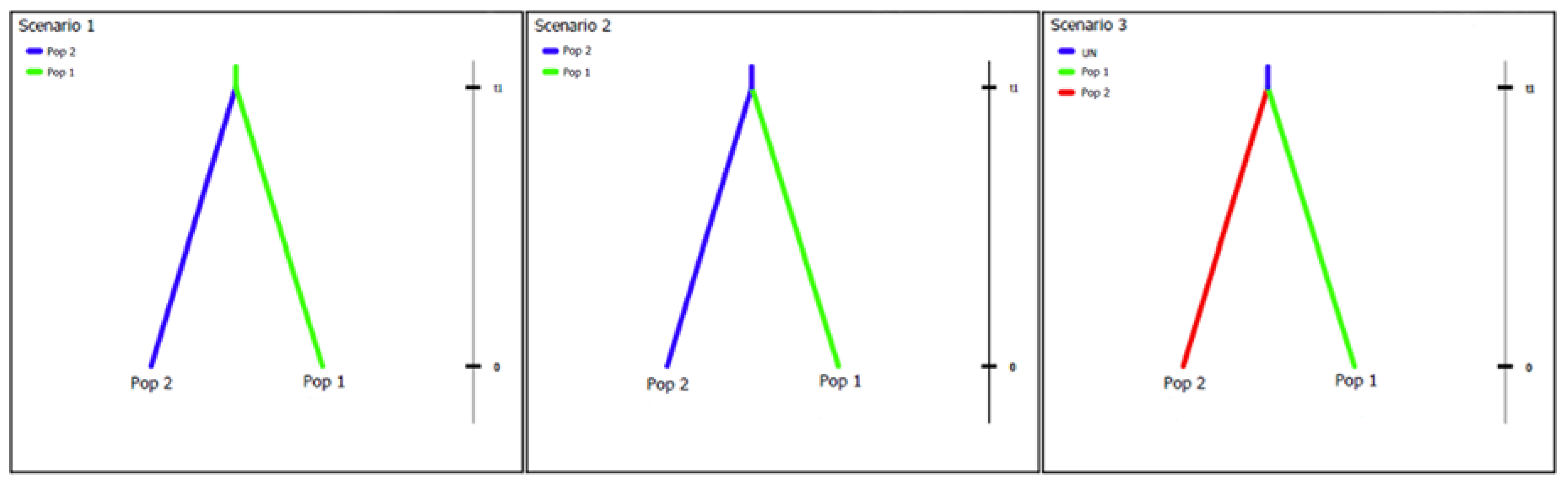
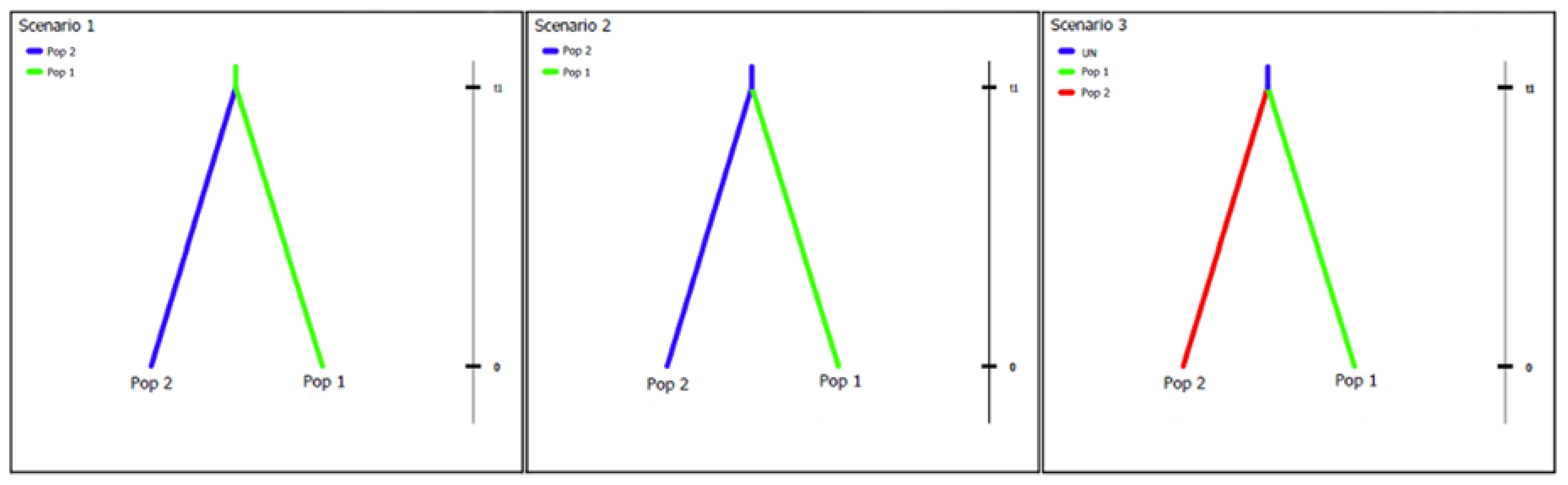
2.5. Putative Geographic Origin of Lasiodiplodia theobromae
3. Results
3.1. Isolate Collections and Confirmation of Species Identity
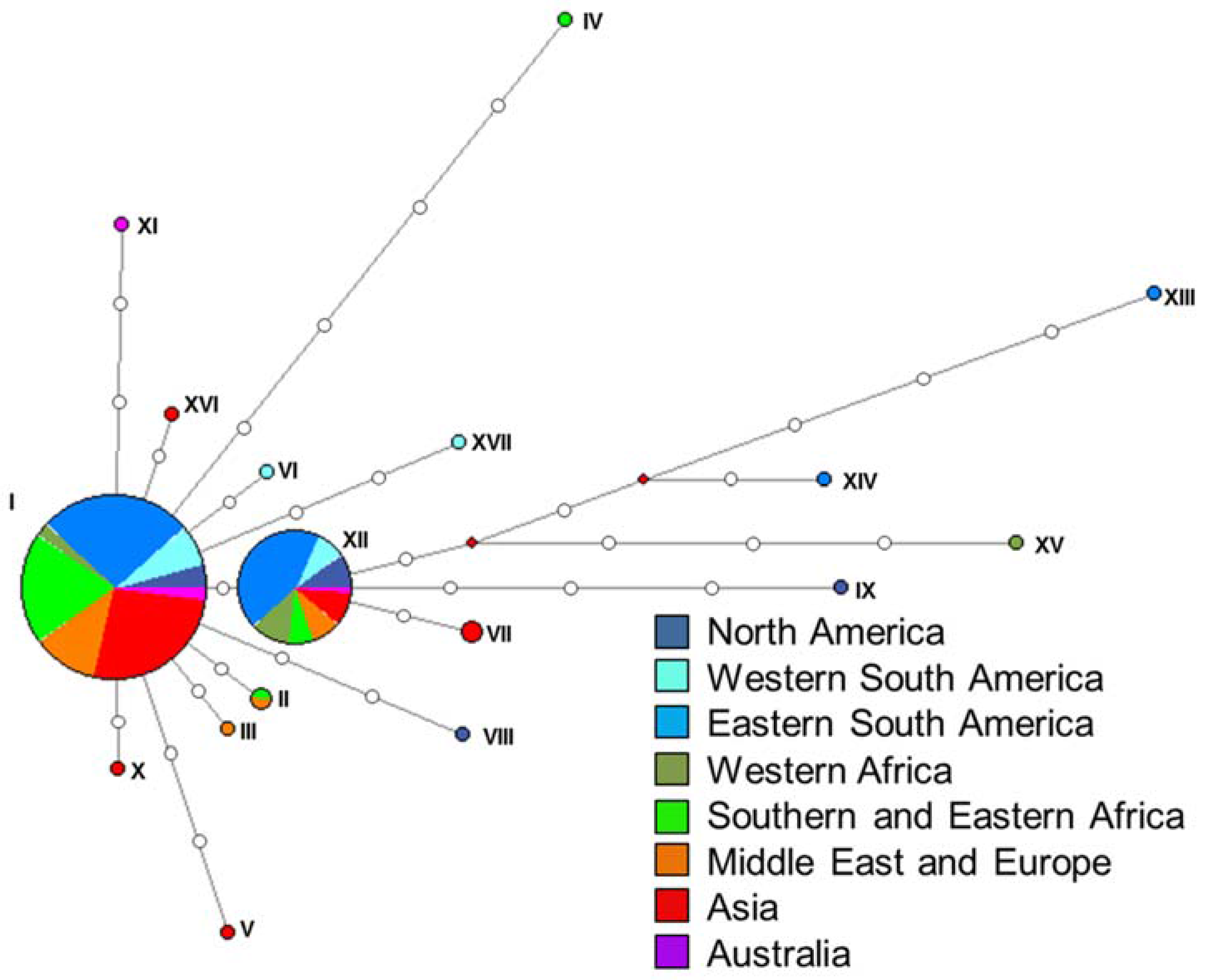
3.2. Haplotype Assignment and Networks
3.3. Population and Regional Structure and Diversity
3.4. Putative Geographic Origin of Lasiodiplodia theobromae
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wingfield, M.J.; Brockerhoff, E.G.; Wingfield, B.D.; Slippers, B. Planted forest health: The need for a global strategy. Science 2015, 349, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Ghelardini, L.; Pepori, A.L.; Luchi, N.; Capretti, P.; Santini, A. Drivers of emerging fungal diseases of forest trees. For. Ecol. Manag. 2016, 381, 235–246. [Google Scholar] [CrossRef]
- Burgess, T.I.; Crous, C.J.; Slippers, B.; Hantula, J.; Wingfield, M.J. Tree invasions and biosecurity: Eco-evolutionary dynamics of hitchhiking fungi. AoB Plants 2016, 8, plw076. [Google Scholar] [CrossRef] [PubMed]
- Slippers, B.; Wingfield, M.J. Botryosphaeriaceae as endophytes and latent pathogens of woody plants: Diversity, ecology and impact. Fungal Biol. Rev. 2007, 21, 90–106. [Google Scholar] [CrossRef]
- Slippers, B.; Crous, P.W.; Jami, F.; Groenewald, J.Z.; Wingfield, M.J. Diversity in the Botryosphaeriales: Looking back, looking forward. Fungal Biol. 2017, 121, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Mehl, J.W.M.; Slippers, B.; Roux, J.; Wingfield, M.J. Cankers and other diseases caused by the Botryosphaeriaceae. In Infectious Forest Diseases; Gonthier, P., Nicolotti, G., Eds.; CAB International: Boston, MN, USA, 2013; pp. 298–317. [Google Scholar]
- Taylor, K.; Barber, P.A.; Hardy, G.E.S.J.; Burgess, T.I. Botryosphaeriaceae from tuart (Eucalyptus gomphocephala) woodland, including descriptions of four new species. Mycol. Res. 2009, 113, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.A.; Wingfield, M.J.; Slippers, B.; Altier, N.A.; Blanchette, R.A. Endophytic and canker-associated Botryosphaeriaceae occurring on non-native Eucalyptus and native Myrtaceae trees in Uruguay. Fungal Divers. 2010, 41, 53–69. [Google Scholar] [CrossRef]
- Bihon, W.; Burgess, T.I.; Slippers, B.; Wingfield, M.J.; Wingfield, B.D. Distribution of Diplodia pinea and its genotypic diversity within asymptomatic Pinus patula trees. Australas. Plant Pathol. 2011, 40, 540–548. [Google Scholar] [CrossRef]
- Sakalidis, M.L.; Hardy, G.E.S.J.; Burgess, T.I. Class III endophytes, clandestine movement amongst hosts and habitats and their potential for disease; a focus on Neofusicoccum australe. Australas. Plant Pathol. 2011, 40, 510–521. [Google Scholar] [CrossRef]
- Jami, F.; Slippers, B.; Wingfield, M.J.; Gryzenhout, M. Botryosphaeriaceae species overlap on four unrelated, native South African hosts. Fungal Biol. 2014, 118, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.J.L.; Alves, A.; Abdollahzadeh, J.; Slippers, B.; Wingfield, M.J.; Groenewald, J.Z.; Crous, P.W. The Botryosphaeriaceae: Genera and species known from culture. Stud. Mycol. 2013, 76, 51–167. [Google Scholar] [CrossRef] [PubMed]
- Slippers, B.; Roux, J.; Wingfield, M.J.; Van der Walt, F.J.J.; Jami, F.; Mehl, J.W.M.; Marais, G.J. Confronting the constraints of morphological taxonomy in the Botryosphaeriales. Persoonia 2014, 33, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Jami, F.; Slippers, B.; Wingfield, M.J.; Loots, M.T.; Gryzenhout, M. Temporal and spatial variation of Botryosphaeriaceae associated with Acacia karroo in South Africa. Fungal Ecol. 2015, 15, 51–62. [Google Scholar] [CrossRef]
- Pavlic-Zupanc, D.; Wingfield, M.J.; Boissin, E.; Slippers, B. The distribution of genetic diversity in the Neofusicoccum parvum/N. ribis complex suggests structure correlated with level of disturbance. Fungal Ecol. 2015, 13, 93–102. [Google Scholar] [CrossRef]
- Úrbez-Torres, J.R.; Battany, M.; Bettiga, L.J.; Gispert, C.; McGourty, G.; Roncoroni, J.; Smith, R.J.; Verdegaal, P.; Gubler, W.D. Botryosphaeriaceae species spore-trapping studies in California vineyards. Plant Dis. 2010, 94, 717–724. [Google Scholar] [CrossRef]
- Punithalingam, E. Botryodiplodia theobromae. C.M.I. Descript. Fungi Bact. 1976, 519, 1–2. [Google Scholar]
- Sakalidis, M.L.; Slippers, B.; Wingfield, B.D.; Hardy, G.E.S.J.; Burgess, T.I. The challenge of understanding the origin, pathways and extent of fungal invasions: Global populations of the Neofusicoccum parvum-N. ribis species complex. Divers. Distrib. 2013, 19, 873–883. [Google Scholar] [CrossRef]
- Marsberg, A.; Kemler, M.; Jami, F.; Nagel, J.H.; Postma-Smidt, A.; Naidoo, S.; Wingfield, M.J.; Crous, P.W.; Spatafora, J.W.; Hesse, C.N.; et al. Botryosphaeria dothidea: A latent pathogen of global importance to woody plant health. Mol. Plant Pathol. 2017, in press. [Google Scholar]
- Dissanayake, A.J.; Phillips, A.J.L.; Li, X.H.; Hyde, K.D. Botryosphaeriaceae: Current status of genera and species. Mycosphere 2016, 7, 1001–1073. [Google Scholar]
- Burgess, T.; Wingfield, M.J. Quarantine is important in restricting the spread of exotic seed-borne tree pathogens in the southern hemisphere. Int. For. Rev. 2002, 4, 56–65. [Google Scholar]
- Burgess, T.I.; Wingfield, M.J.; Wingfield, B.D. Global distribution of Diplodia pinea genotypes revealed using simple sequence repeat (SSR) markers. Australas. Plant Pathol. 2004, 33, 513–519. [Google Scholar] [CrossRef]
- Bihon, W.; Burgess, T.; Slippers, B.; Wingfield, M.J.; Wingfield, B.D. High levels of genetic diversity and cryptic recombination is widespread in introduced Diplodia pinea populations. Australas. Plant Pathol. 2012, 41, 41–46. [Google Scholar] [CrossRef]
- Sarr, M.P.; Ndiaye, M.; Groenewald, J.Z.; Crous, P.W. Genetic diversity in Macrophomina phaseolina, the causal agent of charcoal rot. Phytopathol. Mediterr. 2014, 53, 250–268. [Google Scholar]
- Farr, D.F.; Rossman, A.Y. Fungal Databases. Systematic Mycology and Microbiology Laboratory, ARS, USDA. Available online: http://nt.ars-grin.gov/fungaldatabases/ (accessed on 28 February 2017).
- Coutinho, I.B.L.; Freire, F.C.O.; Lima, C.S.; Lima, J.S.; Gonçalves, F.J.T.; Machado, A.R.; Silva, A.M.S.; Cardoso, J.E. Diversity of genus Lasiodiplodia associated with perennial tropical fruit plants in northeastern Brazil. Plant Pathol. 2017, in press. [Google Scholar] [CrossRef]
- Cruywagen, E.M.; Slippers, B.; Roux, J.; Wingfield, M.J. Phylogenetic Species Recognition and hybridisation in Lasiodiplodia: A case study on species from baobabs. Fungal Biol. 2017, 121, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Netto, M.S.B.; Lima, W.G.; Correia, K.C.; da Silva, C.F.B.; Thon, M.; Martins, R.B.; Miller, R.N.G.; Michereff, S.J.; Câmara, M.P.S. Analysis of phylogeny, distribution and pathogenicity of Botryosphaeriaceae species associated with gummosis of Anacardium in Brazil, with a new species of Lasiodiplodia. Fungal Biol. 2017, 121, 437–451. [Google Scholar] [CrossRef] [PubMed]
- World Europe and Africa Centered. Available online: http://www.d-maps.com/carte.php?num_car=126805&lang=en (accessed on 28 February 2017).
- Mehl, J.W.M.; Slippers, B.; Roux, J.; Wingfield, M.J. Botryosphaeriaceae associated with Pterocarpus angolensis (kiaat) in South Africa. Mycologia 2011, 103, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.P.; Davis, A.J.; Wingfield, B.D.; Crous, P.W.; Brenneman, T.; Wingfield, M.J. Population structure of Cylindrocladium parasiticum infecting peanuts (Arachis hypogaea) in Georgia, USA. Eur. J. Plant Pathol. 2010, 127, 199–206. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Jacobs, K.; Bergdahl, D.R.; Wingfield, M.J.; Halik, S.; Seifert, K.A.; Bright, D.E.; Wingfield, B.D. Leptographium wingfieldii introduced into North America and found associated with exotic Tomicus piniperda and native bark beetles. Mycol. Res. 2004, 108, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.; Crous, P.W.; Correia, A.; Phillips, A.J.L. Morphological and molecular data reveal cryptic speciation in Lasiodiplodia theobromae. Fungal Divers. 2008, 28, 1–13. [Google Scholar]
- Glass, N.L.; Donaldson, G.C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microb. 1995, 61, 1323–1330. [Google Scholar]
- Mehl, J.W.M.; Slippers, B.; Roux, J.; Wingfield, M.J. Overlap of latent pathogens in the Botryosphaeriaceae on a native and agricultural host. Fungal Biol. 2017, 121, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Mehl, J.W.M.; Slippers, B.; Roux, J.; Wingfield, M.J. Botryosphaeriaceae associated with die-back of Schizolobium parahyba trees in South Africa and Ecuador. For. Pathol. 2014, 44, 396–408. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JMODELTEST 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PHYML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Monacell, J.T.; Carbone, I. Mobyle SNAP Workbench: A web-based analysis portal for population genetics and evolutionary genomics. Bioinformatics 2014, 30, 1488–1490. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Fluxus Technology Ltd. NETWORK Version 4.6.1.3. Available online: http://www.fluxus-engineering.com/sharenet.htm (accessed on 28 February 2017).
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E. ARLEQUIN suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.M.; Pudlo, P.; Veyssier, J.; Dehne-Garcia, A.; Gautier, M.; Leblois, R.; Marin, J.M.; Estoup, A. DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 2014, 30, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Evolution and the Genetics of Populations: A Treatise in Four Volumes: Vol. 4: Variability within and among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1978. [Google Scholar]
- O’Donnell, K.; Kistler, H.; Tacke, B.; Casper, H. Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc. Natl. Acad. Sci. USA 2000, 97, 7905–7910. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, T.; White, T.J.; Koenig, G.; Mcewen, J.; Restrepo, A.; Castaneda, E.; Da Silva Lacaz, C.; Heins-Vaccari, E.M.; De Freitas, R.S.; Zancopé-Oliveira, R.M.; et al. Phylogeography of the fungal pathogen Histoplasma capsulatum. Mol. Ecol. 2003, 12, 3383–3401. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Turner, E.; Townsend, J.P.; Dettman, J.R.; Jacobson, D. Eukaryotic microbes, species recognition and the geographic limits of species: Examples from the kingdom Fungi. Philos. Trans. R. Soc. B 2006, 361, 1947–1963. [Google Scholar] [CrossRef] [PubMed]
- Gladieux, P.; Feurtey, A.; Hood, M.E.; Snirc, A.; Clavel, T.J.; Dutech, C.; Roy, M.; Giraud, T. The population biology of fungal invasions. Mol. Ecol. 2015, 24, 1969–1986. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.; Ward, T.J.; Geiser, D.M.; Kistler, H.C.; Aoki, T. Genealogical concordance between the mating type locus and seven other nuclear genes supports formal recognition of nine phylogenetically distinct species within the Fusarium graminearum clade. Fungal Genet. Biol. 2004, 41, 600–623. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.D.M.; Patané, J.S.; Taylor, M.L.; Gómez, B.L.; Theodoro, R.C.; de Hoog, S.; Engelthaler, D.M.; Zancopé-Oliveira, R.M.; Felipe, M.S.; Barker, B.M. Worldwide phylogenetic distributions and population dynamics of the genus Histoplasma. PLoS Negl. Trop. Dis. 2016, 10, e0004732. [Google Scholar] [CrossRef] [PubMed]
- Pringle, A.; Baker, D.M.; Platt, J.L.; Wares, J.P.; Latge, J.P.; Taylor, J.W. Cryptic speciation in the cosmopolitan and clonal human pathogenic fungus Aspergillus fumigatus. Evolution 2005, 59, 1886–1899. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Camejo, L.A.; Zuluaga-Montero, A.; Lázaro-Escudero, M.; Hernández-Kendall, V.; Bayman, P. Phylogeography of the cosmopolitan fungus Aspergillus flavus: Is everything everywhere? Fungal Biol. 2012, 116, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Swart, W.J.; Wingfield, M.J.; Knox-Davies, P.S. Conidial dispersal of Sphaeropsis sapinea in three climatic regions of South Africa. Plant Dis. 1987, 71, 1038–1040. [Google Scholar] [CrossRef]
- Pusey, P.L. Availability and dispersal of ascospores and conidia of Botryosphaeria in peach orchards. Phytopathology 1989, 79, 635–639. [Google Scholar] [CrossRef]
- Amponsah, N.T.; Jones, E.E.; Ridgway, H.J.; Jaspers, M.V. Rainwater dispersal of Botryosphaeria conidia from infected grapevines. N. Z. Plant Prot. 2009, 62, 228–233. [Google Scholar]
- Santini, A.; Ghelardini, L.; Pace, C.D.; Desprez-Loustau, M.L.; Capretti, P.; Chandelier, A.; Cech, T.; Chira, D.; Diamandis, S.; Gaitniekis, T.; et al. Biogeographical patterns and determinants of invasion by forest pathogens in Europe. New Phytol. 2013, 197, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Slippers, B.; Smit, W.A.; Crous, P.W.; Coutinho, T.A.; Wingfield, B.D.; Wingfield, M.J. Taxonomy, phylogeny and identification of Botryosphaeriaceae associated with pome and stone fruit trees in South Africa and other regions of the world. Plant Pathol. 2007, 56, 128–139. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z.; Slippers, B.; Wingfield, M.J. Global food and fibre security threatened by current inefficiencies in fungal identification. Philos. Trans. R. Soc. B 2016, 371. [Google Scholar] [CrossRef] [PubMed]
- Mohali, S.; Burgess, T.I.; Wingfield, M.J. Diversity and host association of the tropical tree endophyte Lasiodiplodia theobromae revealed using simple sequence repeat markers. For. Pathol. 2005, 35, 385–396. [Google Scholar] [CrossRef]
- Shah, M.D.; Verma, K.S.; Singh, K.; Kaur, R. Morphological, pathological and molecular variability in Botryodiplodia theobromae (Botryosphaeriaceae) isolates associated with die-back and bark canker of pear trees in Punjab, India. Genet. Mol. Res. 2010, 9, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Begoude, A.D.B.; Slippers, B.; Perez, G.; Wingfield, M.J.; Roux, J. High gene flow and outcrossing within populations of two cryptic fungal pathogens on a native and non-native host in Cameroon. Fungal Biol. 2012, 116, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Kolbe, J.J.; Glor, R.E.; Schettino, L.R.; Lara, A.C.; Larson, A.; Losos, J.B. Genetic variation increases during biological invasion by a Cuban lizard. Nature 2004, 431, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, R.N.; Frankel, S.J.; Brown, A.V.; Hennon, P.E.; Kliejunas, J.T.; Lewis, K.J.; Worrall, J.J.; Woods, A.J. Climate change and forest diseases. Plant Pathol. 2011, 60, 133–149. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Country, Locality | Isolate | Host | Plant Family | ITS | tef1α | tub2 | rpb2 |
|---|---|---|---|---|---|---|---|---|
| North America | Hawaii | CBS111530 | Leucospermum sp. | Proteaceae | FJ150695 | EF622054 | KU887531 | KU696382 |
| Mexico | BOM230 | Carica papaya | Caricaceae | KR001856 | KT075154 | |||
| Mexico | BOS104 | Car. papaya | Caricaceae | KR001857 | KT075158 | |||
| Mexico | BOT112 | Car. papaya | Caricaceae | KT075139 | KT075155 | |||
| Mexico | BOT359 | Car. papaya | Caricaceae | KR001859 | KT075159 | |||
| Mexico | LAM118 | Car. papaya | Caricaceae | KT075141 | KT075156 | |||
| Puerto Rico | K286 | Mangifera indica | Anacardiaceae | KC631660 | KC631656 | KC631652 | ||
| Puerto Rico | K8 | Man. indica | Anacardiaceae | KC631659 | KC631655 | KC631651 | ||
| Puerto Rico | PHLO10 | Dimocarpus longan | Sapindaceae | KC964547 | KC964554 | KC964550 | ||
| Puerto Rico | PHLO9 | Dim. longan | Sapindaceae | KC964546 | KC964553 | KC964549 | ||
| USA | CBS124.13 | Unknown | DQ458890 | DQ458875 | DQ458858 | KY472887 | ||
| USA, Florida | CMW34107 | Eucalyptus amplifolia | Myrtaceae | KY473070 | KY473018 | |||
| USA, Florida | SEFL3 | Vaccinium sp. | Ericaceae | JN607091 | JN607114 | JN607138 | ||
| USA, Florida, Apopka | UF05161 | Vacc. corymbosum | Ericaceae | GQ845096 | GQ850468 | |||
| USA, Florida, Alaucha Country | WFF92 | Vacc. corymbosum | Ericaceae | GQ845095 | GQ850467 | |||
| Western South America | Colombia, Andes | CMW34303 | Unknown | KY473031 | KY472979 | |||
| Ecuador | CMW4694 | Schizolobium parahyba | Fabaceae | KY473033 | KY472981 | KY472913 | KY472842 | |
| Ecuador | CMW4695 | Sch. parahyba | Fabaceae | KF886707 | KF886730 | KY472914 | KY472843 | |
| Ecuador | CMW4696 | Sch. parahyba | Fabaceae | KY473034 | KY472982 | KY472915 | ||
| Ecuador | CMW9273 | Sch. parahyba | Fabaceae | KY473035 | KY472983 | KY472916 | KY472844 | |
| Ecuador, Esmeraldas | CMW22924 | Sch. parahyba | Fabaceae | KF886709 | KF886732 | KY472911 | KY472840 | |
| Ecuador, Esmeraldas | CMW22926 | Sch. parahyba | Fabaceae | KY473032 | KY472980 | KY472912 | KY472841 | |
| Peru | CMW31861 | Theobroma cacao | Malvaceae | KY473048 | KY472996 | KY472935 | ||
| Peru | CMW31867 | Th. cacao | Malvaceae | KY473049 | KY472997 | KY472936 | KY472862 | |
| Peru | CMW31899 | Th. cacao | Malvaceae | KY473050 | KY472998 | KY472937 | KY472863 | |
| Peru, Cienneguillo Norte, Piura | LA-SJ1 | Vitis vinifera | Vitaceae | KM401976 | KM401973 | |||
| Peru, Sol-Sol, Piura | LA-SOL1 | Vts. vinifera | Vitaceae | KM401974 | KM401971 | |||
| Peru, San Vicente, Piura | LA-SV1 | Vts. vinifera | Vitaceae | KM401975 | KM401972 | |||
| Venezuela, Guayana | A10 | Acacia mangium | Fabaceae | JX545093 | JX545113 | JX545133 | ||
| Venezuela, Guayana | A13 | Ac. mangium | Fabaceae | JX545094 | JX545114 | JX545134 | ||
| Venezuela, Acarigua | CMW13490 | Euc. urophylla | Myrtaceae | KY473071 | KY473019 | KY472962 | KY472888 | |
| Venezuela, Cojedes | CMW13501 | Ac. mangium | Fabaceae | KY473072 | KY473020 | KY472963 | KY472889 | |
| Venezuela, Falcon State | CMW13519 | Pinus caribaea var. hondurensis | Pinaceae | KY473073 | KY473021 | KY472964 | KY472890 | |
| Venezuela, Falcon State | CMW13527 | Pin. caribaea var. hondurensis | Pinaceae | KY473074 | KY473022 | KY472965 | KY472891 | |
| Eastern South America | Brazil | ARM122 | Jatropha curcas | Euphorbiaceae | KF553895 | KF553896 | ||
| Brazil, Vicosa, MG | CDA 425 | Cocos nucifera | Arecaceae | KP244697 | KP308475 | KP308531 | ||
| Brazil, Vicosa, MG | CDA 444 | Coc. nucifera | Arecaceae | KP244699 | KP308477 | KP308532 | ||
| Brazil, Vicosa, MG | CDA 450 | Coc. nucifera | Arecaceae | KP244688 | KP308478 | KP308533 | ||
| Brazil, Vicosa, MG | CDA 455 | Coc. nucifera | Arecaceae | KP244689 | KP308463 | KP308534 | ||
| Brazil, Juazeiro, BA | CDA 465 | Coc. nucifera | Arecaceae | KP244701 | KP308465 | KP308535 | ||
| Brazil, Juazeiro, BA | CDA 467 | Coc. nucifera | Arecaceae | KP244702 | KP308473 | KP308536 | ||
| Brazil, Juazeiro, BA | CDA 469 | Coc. nucifera | Arecaceae | KP244691 | KP308466 | KP308537 | ||
| Brazil, Juazeiro, BA | CDA 472 | Coc. nucifera | Arecaceae | KP244692 | KP308467 | KP308538 | ||
| Brazil, Sao Francisco Valley | CMM 0307 | Vts. vinifera | Vitaceae | KJ450879 | KJ417879 | |||
| Brazil, Sao Francisco Valley | CMM 0310 | Vts. vinifera | Vitaceae | KJ450880 | KJ417880 | |||
| Brazil, Sao Francisco Valley | CMM 0384 | Vts. vinifera | Vitaceae | KJ450876 | KJ417876 | |||
| Brazil, Sao Francisco Valley | CMM 0455 | Vts. vinifera | Vitaceae | KJ450878 | KJ417878 | |||
| Brazil, Sao Francisco Valley | CMM 0820 | Vts. vinifera | Vitaceae | KJ450877 | KJ417877 | |||
| Brazil | CMM1476 | Man. indica | Anacardiaceae | JX464083 | JX464057 | |||
| Brazil | CMM1481 | Man. indica | Anacardiaceae | JX464095 | JX464021 | |||
| Brazil | CMM1517 | Man. indica | Anacardiaceae | JX464060 | JX464054 | |||
| Brazil | CMM2168 | Car. papaya | Caricaceae | KC484817 | KC481572 | |||
| Brazil | CMM2179 | Car. papaya | Caricaceae | KC484787 | KC481569 | |||
| Brazil | CMM2183 | Car. papaya | Caricaceae | KC484824 | KC481573 | |||
| Brazil | CMM2190 | Car. papaya | Caricaceae | KC484780 | KC481518 | |||
| Brazil | CMM2193 | Car. papaya | Caricaceae | KC484826 | KC481550 | |||
| Brazil | CMM2208 | Car. papaya | Caricaceae | KC484776 | KC481575 | |||
| Brazil | CMM2209 | Car. papaya | Caricaceae | KC484784 | KC481578 | |||
| Brazil | CMM2210 | Car. papaya | Caricaceae | KC484783 | KC481577 | |||
| Brazil | CMM2231 | Car. papaya | Caricaceae | KC484775 | KC481515 | |||
| Brazil | CMM2232 | Car. papaya | Caricaceae | KC484785 | KC481521 | |||
| Brazil | CMM2235 | Car. papaya | Caricaceae | KC484779 | KC481517 | |||
| Brazil | CMM2237 | Car. papaya | Caricaceae | KC484819 | KC481547 | |||
| Brazil | CMM2238 | Car. papaya | Caricaceae | KC484771 | KC481512 | |||
| Brazil | CMM2239 | Car. papaya | Caricaceae | KC484786 | KC481522 | |||
| Brazil | CMM2241 | Car. papaya | Caricaceae | KC484790 | KC481571 | |||
| Brazil | CMM2261 | Car. papaya | Caricaceae | KC484789 | KC481579 | |||
| Brazil | CMM2262 | Car. papaya | Caricaceae | KC484822 | KC481581 | |||
| Brazil | CMM2265 | Car. papaya | Caricaceae | KC484772 | KC481574 | |||
| Brazil | CMM2267 | Car. papaya | Caricaceae | KC484777 | KC481576 | |||
| Brazil | CMM2268 | Car. papaya | Caricaceae | KC484818 | KC481580 | |||
| Brazil | CMM2269 | Car. papaya | Caricaceae | KC484821 | KC481585 | |||
| Brazil | CMM2276 | Car. papaya | Caricaceae | KC484820 | KC481548 | |||
| Brazil | CMM2278 | Car. papaya | Caricaceae | KC484781 | KC481519 | |||
| Brazil | CMM2280 | Car. papaya | Caricaceae | KC484773 | KC481513 | |||
| Brazil | CMM2282 | Car. papaya | Caricaceae | KC484827 | KC481551 | |||
| Brazil | CMM2294 | Car. papaya | Caricaceae | KC484828 | KC481552 | |||
| Brazil | CMM2295 | Car. papaya | Caricaceae | KC484774 | KC481514 | |||
| Brazil | CMM2297 | Car. papaya | Caricaceae | KC484823 | KC481582 | |||
| Brazil | CMM2303 | Car. papaya | Caricaceae | KC484816 | KC481546 | |||
| Brazil | CMM2306 | Car. papaya | Caricaceae | KC484788 | KC481570 | |||
| Brazil | CMM2310 | Car. papaya | Caricaceae | KC484782 | KC481520 | |||
| Brazil | CMM2327 | Car. papaya | Caricaceae | KC484778 | KC481516 | |||
| Brazil | CMM2328 | Car. papaya | Caricaceae | KC484825 | KC481549 | |||
| Brazil | CMM3612 | Jat. curcas | Euphorbiaceae | KF234546 | KF226692 | KF254929 | ||
| Brazil | CMM3647 | Jat. curcas | Euphorbiaceae | KF234548 | KF226704 | KF254932 | ||
| Brazil | CMM3654 | Jat. curcas | Euphorbiaceae | KF234555 | KF226716 | KF254939 | ||
| Brazil | CMM3831 | Jat. curcas | Euphorbiaceae | KF234556 | KF226717 | KF254940 | ||
| Brazil | CMM4019 | Mangifera indica | Anacardiaceae | JX464096 | JX464026 | |||
| Brazil | CMM4021 | Man. indica | Anacardiaceae | JX464064 | JX464047 | |||
| Brazil | CMM4033 | Man. indica | Anacardiaceae | JX464081 | JX464032 | |||
| Brazil | CMM4039 | Man. indica | Anacardiaceae | JX464065 | JX464041 | |||
| Brazil | CMM4041 | Man. indica | Anacardiaceae | KC184891 | JX464042 | |||
| Brazil | CMM4042 | Man. indica | Anacardiaceae | JX464070 | JX464017 | |||
| Brazil | CMM4043 | Man. indica | Anacardiaceae | JX464087 | JX464056 | |||
| Brazil | CMM4046 | Man. indica | Anacardiaceae | JX464091 | JX464027 | |||
| Brazil | CMM4047 | Man. indica | Anacardiaceae | JX464082 | JX464025 | |||
| Brazil | CMM4048 | Man. indica | Anacardiaceae | JX464093 | JX464048 | |||
| Brazil | CMM4050 | Man. indica | Anacardiaceae | JX464062 | JX464024 | |||
| Brazil | CMM4499 | Anacardium occidentale | Anacardiaceae | KT325578 | KT325587 | |||
| Brazil | CMM4508 | Ana. occidentale | Anacardiaceae | KT325576 | KT325588 | |||
| Brazil | CMM4513 | Ana. occidentale | Anacardiaceae | KT325577 | KT325589 | |||
| Brazil | CMW32099 | Unknown | KY473028 | KY472971 | KY472897 | |||
| Brazil, Vicosa, MG | COAD 1788 | Coc. nucifera | Arecaceae | KP244698 | KP308476 | KP308528 | ||
| Brazil, Vicosa, MG | COAD 1789 | Coc. nucifera | Arecaceae | KP244700 | KP308474 | KP308529 | ||
| Brazil, Juazeiro, BA | COAD 1790 | Coc. nucifera | Arecaceae | KP244703 | KP308468 | KP308530 | ||
| Brazil, Catuana, Ceará | IBL340 | Spondias purpurea | Anacardiaceae | KT247466 | KT247472 | KT247475 | ||
| Brazil, Itapipoca, Ceara | IBL375 | Talisia esculenta | Sapindaceae | KT247467 | KT247473 | KT247474 | ||
| Brazil, Buique, Piauí | IBL404 | Ana. occidentale | Anacardiaceae | KT247468 | KT247470 | KT247476 | ||
| Brazil, Buique, Piauí | IBL405 | Ana. occidentale | Anacardiaceae | KT247469 | KT247471 | KT247477 | ||
| Uruguay, Paysandú | Fi2359 | Malus domestica | Rosaceae | KR071127 | KT191041 | |||
| Western Africa | Benin | CMW33290 | Adansonia digitata | Bombacaceae | KY473027 | KY472970 | KY472896 | KY472828 |
| Cameroon, Mbalmayo-Bilink | CMW28311 | Terminalia ivorensis | Combretaceae | GQ469932 | GQ469898 | KY472898 | KY472829 | |
| Cameroon, Kribi | CMW28317 | Ter. catappa | Combretaceae | FJ900602 | FJ900648 | KY472899 | KY472830 | |
| Cameroon, Kribi | CMW28319 | Ter. catappa | Combretaceae | FJ900603 | FJ900650 | |||
| Cameroon, Kribi | CMW28547 | Ter. mentaly | Combretaceae | GQ469919 | KY472972 | KY472900 | KY472831 | |
| Cameroon, Kribi | CMW28548 | Ter. mentaly | Combretaceae | GQ469920 | KY472973 | KY472901 | KY472832 | |
| Cameroon, Kribi | CMW28550 | Ter. mentaly | Combretaceae | GQ469921 | KY472974 | KY472902 | KY472833 | |
| Cameroon, Mbalmayo-Ebogo | CMW28570 | Ter. ivorensis | Combretaceae | GQ469923 | GQ469896 | KY472903 | KY472834 | |
| Cameroon, Mbalmayo-Ebogo | CMW28571 | Ter. ivorensis | Combretaceae | GQ469924 | GQ469897 | KY472904 | KY472835 | |
| Cameroon, Mbalmayo-Ebogo | CMW28573 | Ter. ivorensis | Combretaceae | GQ469925 | KY472975 | KY472905 | KY472836 | |
| Cameroon, Mbalmayo-Ekombitie | CMW28625 | Ter. ivorensis | Combretaceae | GQ469933 | KY472976 | KY472906 | KY472837 | |
| Cameroon, Lombel | CMW36127 | Ad. digitata | Bombacaceae | KY473029 | KY472977 | KY472907 | ||
| Southern and Eastern Africa | Madagascar, Madamo | CMW27810 | Ter. catappa | Combretaceae | FJ900605 | FJ900651 | KY472923 | KY472851 |
| South Africa, Mpumalanga | CMW18422 | Pin. patula | Pinaceae | DQ103544 | DQ103562 | |||
| South Africa, Mpumalanga | CMW18423 | Pin. patula | Pinaceae | DQ103545 | DQ103563 | |||
| South Africa, Mpumalanga | CMW18425 | Pin. patula | Pinaceae | DQ103546 | DQ103561 | KY472864 | ||
| South Africa, Mpumalanga | CMW22663 | Pterocarpus angolensis | Fabaceae | FJ888468 | FJ888450 | KY472865 | ||
| South Africa, Mpumalanga | CMW22664 | Pt. angolensis | Fabaceae | FJ888469 | FJ888451 | |||
| South Africa, Kwazulu-Natal | CMW24125 | Sclerocarya birrea | Anacardiaceae | KU997372 | KU997111 | KY472866 | ||
| South Africa, Mpumalanga | CMW25212 | Man. indica | Anacardiaceae | KU997392 | KU997128 | KU997566 | ||
| South Africa, Limpopo | CMW26616 | Euphorbia ingens | Euphorbiaceae | KY473051 | KY472999 | KY472941 | KY472867 | |
| South Africa, Limpopo | CMW26630 | Euph. ingens | Euphorbiaceae | KY473052 | KY473000 | KY472942 | KY472868 | |
| South Africa, Kwazulu-Natal | CMW26715 | Ter. catappa | Combretaceae | FJ900604 | FJ900649 | KY472943 | KY472869 | |
| South Africa, Kwazulu-Natal | CMW32018 | Pin. elliottii | Pinaceae | KY473053 | KY473001 | KY472944 | KY472870 | |
| South Africa, Mpumalanga | CMW32498 | Pin. patula | Pinaceae | KY473054 | KY473002 | KY472945 | KY472871 | |
| South Africa, Mpumalanga | CMW32536 | Pin. elliottii | Pinaceae | KY473055 | KY473003 | KY472946 | KY472872 | |
| South Africa, Kwazulu-Natal | CMW32544 | Pin. elliottii | Pinaceae | KY473056 | KY473004 | KY472947 | KY472873 | |
| South Africa, Kwazulu-Natal | CMW32549 | Pin. elliottii | Pinaceae | KY473057 | KY473005 | KY472948 | KY472874 | |
| South Africa, Kwazulu-Natal | CMW32571 | Pin. elliottii | Pinaceae | KY473058 | KY473006 | KY472949 | KY472875 | |
| South Africa, Kwazulu-Natal | CMW32603 | Pin. elliottii | Pinaceae | KY473059 | KY473007 | KY472950 | KY472876 | |
| South Africa, Kwazulu-Natal | CMW32604 | Pin. elliottii | Pinaceae | KY473060 | KY473008 | KY472951 | KY472877 | |
| South Africa, Kwazulu-Natal | CMW32606 | Pin. elliottii | Pinaceae | KY473061 | KY473009 | KY472952 | KY472878 | |
| South Africa, Kwazulu-Natal | CMW32651 | Pin. elliottii | Pinaceae | KY473062 | KY473010 | KY472953 | KY472879 | |
| South Africa, Kwazulu-Natal | CMW32666 | Pin. elliottii | Pinaceae | KY473063 | KY473011 | KY472954 | ||
| South Africa, Kwazulu-Natal | CMW32669 | Pin. elliottii | Pinaceae | KY473064 | KY473012 | KY472955 | KY472880 | |
| South Africa, Mpumalanga | CMW33658 | Man. indica | Anacardiaceae | KY473065 | KY473013 | KY472956 | ||
| South Africa, Gauteng | CMW38120 | Vachellia karroo | Fabaceae | KC769935 | KC769843 | KC769887 | ||
| South Africa, Gauteng | CMW38121 | Vac. karroo | Fabaceae | KC769936 | KC769844 | KC769888 | ||
| South Africa, Gauteng | CMW38122 | Vac. karroo | Fabaceae | KC769937 | KC769845 | KC769889 | ||
| South Africa, Gauteng | CMW39290 | Vac. karroo | Fabaceae | KF270061 | KF270021 | |||
| South Africa, Gauteng | CMW39291 | Vac. karroo | Fabaceae | KF270062 | KF270022 | |||
| South Africa, Kwazulu-Natal | CMW41214 | Barringtonia racemosa | Lecythidaceae | KP860842 | KU666547 | KP860765 | KU587889 | |
| South Africa, Kwazulu-Natal | CMW41222 | Bar. racemosa | Lecythidaceae | KP860836 | KU666549 | KP860759 | KU587881 | |
| South Africa, Kwazulu-Natal | CMW41223 | Bar. racemosa | Lecythidaceae | KP860837 | KU666548 | KP860760 | KU587882 | |
| South Africa, Kwazulu-Natal | CMW41360 | Bar. racemosa | Lecythidaceae | KP860841 | KP860686 | KP860764 | KU587888 | |
| South Africa, Kwazulu-Natal | CMW42341 | Bar. racemosa | Lecythidaceae | KP860843 | KU587945 | KU587866 | ||
| South Africa, Kwazulu-Natal | MTU53 | Sygygium cordatum | Myrtaceae | KY052943 | KY024622 | KY000125 | ||
| Uganda | CMW10130 | Vitex donniana | Lamiaceae | AY236951 | AY236900 | AY236929 | KY472883 | |
| Uganda, Mbale | CMW18420 | Casuarina cunninghamii | Casuarinaceae | DQ103534 | DQ103564 | KY472959 | KY472884 | |
| Uganda, Mbale | CMW32245 | Cas. cunninghamii | Casuarinaceae | KY473068 | KY473016 | KY472960 | KY472885 | |
| Uganda, Mbale | CMW32246 | Cas. cunninghamii | Casuarinaceae | KY473069 | KY473017 | KY472961 | KY472886 | |
| Zambia, Samfya | CMW30103 | Syz. cordatum | Myrtaceae | FJ747640 | FJ871114 | |||
| Zambia, Samfya | CMW30104 | Syz. cordatum | Myrtaceae | FJ747641 | FJ871115 | |||
| Zambia, Samfya | CMW30105 | Syz. cordatum | Myrtaceae | FJ747642 | FJ871116 | |||
| Middle East and Europe | Egypt | BOT23 | Man. indica | Anacardiaceae | JN814400 | JN814427 | ||
| Egypt | BOT4 | Man. indica | Anacardiaceae | JN814395 | JN814422 | |||
| Egypt | BOT5 | Man. indica | Anacardiaceae | JN814376 | JN814403 | |||
| Egypt | BOT6 | Man. indica | Anacardiaceae | JN814399 | JN814426 | |||
| Egypt | BOT7 | Man. indica | Anacardiaceae | JN814396 | JN814423 | |||
| Egypt | BOT9 | Man. indica | Anacardiaceae | JN814392 | JN814419 | |||
| Iran | CJA198 | Unknown | GU973871 | GU973863 | ||||
| Iran | CJA199 | Unknown | GU973872 | GU973864 | ||||
| Iran | IRAN1233C | Unknown | GU973868 | GU973860 | ||||
| Iran | IRAN1496C | Man. indica | Anacardiaceae | GU973869 | GU973861 | |||
| Iran | IRAN1499C | Man. indica | Anacardiaceae | GU973870 | GU973862 | |||
| Italy, Foggia | B159 | Vts. vinifera | Vitaceae | KM675760 | KM822731 | |||
| Italy, Cerignola | B202 | Vts. vinifera | Vitaceae | KM675761 | KM822732 | |||
| Italy, Cerignola | B215 | Vts. vinifera | Vitaceae | KM675762 | KM822733 | |||
| Italy, Cerignola | B342 | Vts. vinifera | Vitaceae | KM675763 | KM822734 | |||
| Italy, Cerignola | B85 | Vts. vinifera | Vitaceae | KM675759 | KM822730 | |||
| Oman, Barka | CMW20506 | Man. indica | Anacardiaceae | KY473037 | KY472985 | KY472924 | KY472852 | |
| Oman, Barka | CMW20508 | Man. indica | Anacardiaceae | KY473038 | KY472986 | KY472925 | KY472853 | |
| Oman, Barka | CMW20511 | Man. indica | Anacardiaceae | KY473039 | KY472987 | KY472926 | KY472854 | |
| Oman, Barka | CMW20512 | Man. indica | Anacardiaceae | KY473040 | KY472988 | KY472927 | KY472855 | |
| Oman | CMW20537 | Unknown | KY473041 | KY472989 | KY472928 | KY472856 | ||
| Oman | CMW20542 | Unknown | KY473042 | KY472990 | KY472929 | |||
| Oman | CMW20543 | Unknown | KY473043 | KY472991 | KY472930 | KY472857 | ||
| Oman | CMW20546 | Unknown | KY473044 | KY472992 | KY472931 | KY472858 | ||
| Oman | CMW20560 | Unknown | KY473045 | KY472993 | KY472932 | KY472859 | ||
| Oman | CMW20573 | Unknown | KY473046 | KY472994 | KY472933 | KY472860 | ||
| Oman | CMW20579 | Unknown | KY473047 | KY472995 | KY472934 | KY472861 | ||
| Asia | China, Fangshan, Pingtung | B838 | Man. indica | Anacardiaceae | GQ502456 | GQ980001 | GU056852 | |
| China, Guantian, Tainan | B852 | Man. indica | Anacardiaceae | GQ502457 | GQ980002 | GU056851 | ||
| China, Chiayi | B886 | Man. indica | Anacardiaceae | GQ502452 | GQ980005 | GU056847 | ||
| China, Guantian, Tainan | B902 | Man. indica | Anacardiaceae | GQ502459 | GQ980004 | GU056849 | ||
| China, Guantian, Tainan | B918 | Man. indica | Anacardiaceae | GQ502458 | GQ980003 | GU056850 | ||
| China, Guantian, Tainan | B961 | Man. indica | Anacardiaceae | GQ502453 | GQ979999 | GU056845 | ||
| China, Guantian, Tainan | B965 | Man. indica | Anacardiaceae | GQ502454 | GQ980000 | GU056854 | ||
| China | BL1331 | Albizia falcataria | Fabaceae | KU712499 | KU712500 | KU712501 | ||
| China | CBS122127 | Homo sapiens | EF622017 | EF622018 | ||||
| China, GuangDong Province | CERC1983 | Polyscias balfouriana | Araliaceae | KP822979 | KP822997 | KP823012 | ||
| China, GuangDong Province | CERC1985 | Pol. balfouriana | Araliaceae | KP822980 | KP822998 | KP823013 | ||
| China, GuangDong Province | CERC1988 | Pol. balfouriana | Araliaceae | KP822981 | KP822999 | KP823014 | ||
| China, GuangDong Province | CERC1989 | Euc. GU hybrid | Myrtaceae | KP822982 | KP823000 | KP823015 | ||
| China, GuangDong Province | CERC1991 | Euc. GU hybrid | Myrtaceae | KP822983 | KP823001 | KP823016 | ||
| China, GuangDong Province | CERC1996 | Euc. GU hybrid | Myrtaceae | KP822984 | KP823002 | KP823017 | ||
| China, GuangDong Province | CERC2049 | Bougainvillea spectabilis | Nyctaginaceae | KP822985 | KP823003 | KP823018 | ||
| China, GuangDong Province | CERC3820 | Rosa rugosa | Rosaceae | KR816831 | KR816837 | KR816843 | ||
| China, GuangDong Province | CERC3821 | R. rugosa | Rosaceae | KR816832 | KR816838 | KR816844 | ||
| China, GuangDong Province | CERC3822 | R. rugosa | Rosaceae | KR816833 | KR816839 | KR816845 | ||
| China, GuangDong Province | CERC3823 | R. rugosa | Rosaceae | KR816834 | KR816840 | KR816846 | ||
| China, GuangDong Province | CERC3824 | R. rugosa | Rosaceae | KR816835 | KR816841 | KR816847 | ||
| China, GuangDong Province | CERC3825 | R. rugosa | Rosaceae | KR816836 | KR816842 | KR816848 | ||
| China, Dong Men Forest Farm | CMW24701 | Euc. GU hybrid | Myrtaceae | HQ332193 | HQ332209 | KY472908 | KY472838 | |
| China, Dong Men Forest Farm | CMW24702 | Euc. GU hybrid | Myrtaceae | HQ332194 | HQ332210 | KY472909 | KY472839 | |
| China | CMW33957 | Eucalyptus sp. | Myrtaceae | KY473030 | KY472978 | KY472910 | ||
| China | FXPZ | Vts. vinifera | Vitaceae | KR232666 | KR232660 | KR232674 | ||
| China | HD1332 | Alb. falcataria | Fabaceae | KU712502 | KU712503 | KU712504 | ||
| China | HN74 | Hevea brasiliensis | Euphorbiaceae | KT947466 | KU925617 | KU925616 | ||
| China, Guangxi Province | L1 | Man. indica | Anacardiaceae | KR260791 | KR260808 | KR260820 | ||
| China, Guangxi Province | L2 | Man. indica | Anacardiaceae | KR260792 | KR260809 | KR260821 | ||
| China, Guangxi Province | L3 | Man. indica | Anacardiaceae | KR260793 | KR260810 | KR260822 | ||
| China, Guangxi Province | L4 | Man. indica | Anacardiaceae | KR260794 | KR260811 | KR260823 | ||
| China, Guangxi Province | L5 | Man. indica | Anacardiaceae | KR260795 | KR260812 | KR260824 | ||
| China, Guangxi Province | L6 | Man. indica | Anacardiaceae | KR260796 | KR260813 | KR260825 | ||
| China, Guangxi Province | L7 | Man. indica | Anacardiaceae | KR260797 | KR260814 | KR260826 | ||
| China, Guangxi Province | L8 | Man. indica | Anacardiaceae | KR260798 | KR260815 | KR260827 | ||
| China, Guangxi Province | L9 | Man. indica | Anacardiaceae | KR260799 | KR260816 | KR260828 | ||
| China, Guangxi Province | L10 | Man. indica | Anacardiaceae | KR260800 | KR260817 | KR260829 | ||
| China, Guangxi Province | L11 | Man. indica | Anacardiaceae | KR260801 | KR260818 | KR260830 | ||
| China, Guangxi Province | L15 | Man. indica | Anacardiaceae | KR260802 | KR260819 | KR260831 | ||
| China, Sichuan | Mht-5 | Actinidia deliciosa | Actinidiaceae | JQ658976 | JQ658977 | JQ658978 | ||
| China, Shanghai | SHYAG | Vitis vinifera | Vitaceae | JX275794 | JX462302 | JX462276 | ||
| China, Zhejiang | ZHn411 | Pyrus pyrifolia | Rosaceae | KC960899 | KC961038 | KC960992 | ||
| Indonesia, Sumatra | CMW22881 | Euc. grandis | Myrtaceae | KY473036 | KY472984 | KY472917 | KY472845 | |
| Indonesia, Logas | CMW23003 | Ac. mangium | Fabaceae | EU588629 | EU588609 | KY472918 | KY472846 | |
| Indonesia, Logas | CMW23008 | Ac. mangium | Fabaceae | EU588630 | EU588610 | KY472919 | KY472847 | |
| Indonesia, Logas | CMW23018 | Ac. mangium | Fabaceae | EU588633 | EU588613 | KY472920 | KY472848 | |
| Indonesia, Teso | CMW23031 | Ac. mangium | Fabaceae | EU588631 | EU588611 | KY472921 | KY472849 | |
| Indonesia, Logas | CMW23073 | Ac. mangium | Fabaceae | EU588632 | EU588612 | KY472922 | KY472850 | |
| Korea | ML1001 | Man. indica | Anacardiaceae | JN542561 | JN542563 | |||
| Korea | ML1005 | Man. indica | Anacardiaceae | JN542562 | JN542564 | |||
| Thailand, Prajinburi | CMW15680 | Euc. camaldulensis | Myrtaceae | KY473066 | KY473014 | KY472957 | KY472881 | |
| Thailand, Prajinburi | CMW15682 | Euc. camaldulensis | Myrtaceae | KY473067 | KY473015 | KY472958 | KY472882 | |
| Thailand, Chiang Mai | CPC 22766 | Pin. kesiya | Pinaceae | KM006436 | KM006467 | |||
| Thailand, Chiang Mai | CPC 22780 | Manilkara zapota | Sapotaceae | KM006442 | KM006473 | |||
| Thailand, Chiang Mai | CPC 22798 | Syz. samarangense | Myrtaceae | KM006454 | KM006485 | |||
| Thailand, Chiang Mai | MFLUCC12 0293 | Tectona grandis | Lamiaceae | KM396896 | KM409634 | KM510354 | ||
| Australasia | Australia | CMW40630 | Syzygium sp. | Myrtaceae | KY473023 | KY472966 | KY472892 | KY472825 |
| Australia | CMW40635 | Syz. novosum | Myrtaceae | KY473024 | KY472967 | KY472893 | ||
| Australia | CMW40636 | Syz. novosum | Myrtaceae | KY473025 | KY472968 | KY472894 | KY472826 | |
| Australia | CMW40637 | Syz. novosum | Myrtaceae | KY473026 | KY472969 | KY472895 | KY472827 | |
| Darwin, Australia | MUCC737 | Ad. gregorii | Bombacaceae | GU199387 | GU199407 | |||
| Papua New Guinea, Madang | CBS164.96 | Fruit along coral reef coast | AY640255 | AY640258 | KU887532 | KU696383 |
| Region | Country | N | ITS | tef1α | ITS + tef1α | tub2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H | HE | π (×10−3) | H | HE | π (×10−3) | H | HE | π (×10−3) | N | H | HE | π (×10−3) | |||
| North America | Hawaii | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 |
| Mexico | 5 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Puerto Rico | 4 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 4 | 1 | 0 | 0 | |
| USA | 5 | 3 | 0.356 | 4.271 | 2 | 0.356 | 1.646 | 4 | 0.385 | 4.210 | 2 | 2 | 0.667 | 2.157 | |
| Total | 15 | 3 | 0.129 | 1.546 | 3 | 0.405 | 3.746 | 4 | 0.193 | 2.110 | 7 | 2 | 0.264 | 0.854 | |
| Western South America | Colombia | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ecuador | 6 | 1 | 0 | 0 | 3 | 0.384 | 5.331 | 3 | 0.384 | 2.097 | 6 | 1 | 0 | 0 | |
| Peru | 6 | 1 | 0 | 0 | 2 | 0.303 | 1.403 | 2 | 0.303 | 0.552 | 3 | 2 | 0.533 | 1.726 | |
| Venezuela | 6 | 2 | 0.303 | 0.910 | 2 | 0.303 | 1.403 | 3 | 0.303 | 1.104 | 6 | 2 | 0.303 | 0.981 | |
| Total | 19 | 2 | 0.102 | 0.308 | 3 | 0.201 | 2.792 | 4 | 0.176 | 1.285 | 15 | 3 | 0.129 | 0.833 | |
| Eastern South America | Brazil | 76 | 1 | 0 | 0 | 4 | 0.097 | 3.131 | 4 | 0.097 | 1.232 | 19 | 2 | 0.185 | 0.597 |
| Uruguay | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Total | 77 | 1 | 0 | 0 | 4 | 0.096 | 3.106 | 4 | 0.096 | 1.222 | 19 | 2 | 0.185 | 0.597 | |
| Western Africa | Benin | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 |
| Cameroon | 11 | 1 | 0 | 0 | 3 | 0.222 | 5.131 | 3 | 0.222 | 2.019 | 10 | 1 | 0 | 0 | |
| Total | 12 | 1 | 0 | 0 | 3 | 0.220 | 5.099 | 3 | 0.220 | 2.006 | 11 | 1 | 0 | 0 | |
| Southern and Eastern Africa | Madagascar | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 |
| South Africa | 32 | 3 | 0.064 | 0.953 | 2 | 0.112 | 0.520 | 3 | 0.062 | 0.560 | 29 | 4 | 0.119 | 2.302 | |
| Uganda | 4 | 1 | 0 | 0 | 2 | 0.429 | 1.984 | 2 | 0.429 | 0.781 | 4 | 2 | 0.429 | 1.387 | |
| Zambia | 3 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Total | 40 | 3 | 0.051 | 0.760 | 2 | 0.248 | 1.147 | 4 | 0.072 | 0.782 | 34 | 4 | 0.119 | 2.301 | |
| Middle East and Europe | Egypt | 6 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Iran | 5 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Italy | 5 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Oman | 11 | 3 | 0.173 | 1.040 | 1 | 0 | 0 | 3 | 0.173 | 0.631 | 11 | 2 | 0.173 | 0.560 | |
| Total | 27 | 3 | 0.073 | 0.436 | 2 | 0.308 | 1.424 | 4 | 0.151 | 0.825 | 11 | 2 | 0.173 | 0.560 | |
| Asia | China | 43 | 3 | 0.606 | 0.546 | 3 | 0.108 | 1.003 | 5 | 0.080 | 0.726 | 42 | 7 | 0.153 | 4.939 |
| Indonesia | 6 | 1 | 0 | 0 | 2 | 0.485 | 2.245 | 2 | 0.485 | 0.883 | 6 | 1 | 0 | 0 | |
| Korea | 2 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Thailand | 6 | 2 | 0.303 | 0.910 | 2 | 0.485 | 2.245 | 3 | 0.394 | 1.435 | 3 | 1 | 0 | 0 | |
| Total | 57 | 4 | 0.043 | 0.518 | 3 | 0.139 | 1.288 | 6 | 0.075 | 0.821 | 51 | 7 | 0.130 | 4.202 | |
| Australasia | Australia | 5 | 2 | 0.356 | 2.135 | 1 | 0 | 0 | 2 | 0.356 | 1.295 | 4 | 1 | 0 | 0 |
| Papua New Guinea | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | |
| Total | 6 | 2 | 0.303 | 1.820 | 2 | 0.303 | 1.403 | 3 | 0.303 | 1.656 | 5 | 1 | 0 | 0 | |
| All | 255 | 11 | 0.001 | 8 | 0.003 | 17 | 0.001 | 153 | 12 | 0.002 | |||||
| Region | N | North America | Western South America | Eastern South America | Western Africa | Southern and Eastern Africa | Middle East and Europe | Asia | Australasia |
|---|---|---|---|---|---|---|---|---|---|
| North America | 15 | ||||||||
| Western South America | 19 | 0.047 | |||||||
| Eastern South America | 77 | 0.026 | 0.014 | ||||||
| Western Africa | 12 | 0.040 | 0.165 | 0.121 | - | ||||
| Southern and Eastern Africa | 40 | 0.189 | 0.051 | 0.105 | 0.367 | ||||
| Middle East and Europe | 27 | 0.109 | 0.008 | 0.045 | 0.272 | 0.01 | |||
| Asia | 57 | 0.166 | 0.032 | 0.080 | 0.343 | 0.006 | 0.002 | ||
| Australasia | 6 | 0.087 | 0.041 | 0.087 | 0.205 | 0.075 | 0.056 | 0.068 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehl, J.; Wingfield, M.J.; Roux, J.; Slippers, B. Invasive Everywhere? Phylogeographic Analysis of the Globally Distributed Tree Pathogen Lasiodiplodia theobromae. Forests 2017, 8, 145. https://doi.org/10.3390/f8050145
Mehl J, Wingfield MJ, Roux J, Slippers B. Invasive Everywhere? Phylogeographic Analysis of the Globally Distributed Tree Pathogen Lasiodiplodia theobromae. Forests. 2017; 8(5):145. https://doi.org/10.3390/f8050145
Chicago/Turabian StyleMehl, James, Michael J. Wingfield, Jolanda Roux, and Bernard Slippers. 2017. "Invasive Everywhere? Phylogeographic Analysis of the Globally Distributed Tree Pathogen Lasiodiplodia theobromae" Forests 8, no. 5: 145. https://doi.org/10.3390/f8050145
APA StyleMehl, J., Wingfield, M. J., Roux, J., & Slippers, B. (2017). Invasive Everywhere? Phylogeographic Analysis of the Globally Distributed Tree Pathogen Lasiodiplodia theobromae. Forests, 8(5), 145. https://doi.org/10.3390/f8050145