3.1. The MC Conformational Analysis
The stability, stereometric configuration and packing of the MC molecules are influenced by various interactions, including the electrostatic, hydrogen bonding, aromatic π-stacking, and hydrophobic and hydrophilic interactions with the neighboring MC and solvent molecules. The C10NH6 groups in both the fixed and rocking segments of the MC molecules minimize their contact with the OH- groups of the solvent molecules, significantly affecting the packing of the MC molecules. Additionally, the flexible wagging and rocking fragments can adjust their orientations to enhance their interactions with adjacent MC and solvent molecules.
The time averaged distribution of angles between the normal vectors to the average planes defined by the fixed and rocking fragments of MC molecules reveals key insights into their conformational behavior across different environments (
Figure S2, Supplementary Materials). In the acetonitrile-solvated system (MCA), the angular distribution is broad, with a moderate preference centered around 90–120°, indicating substantial orientational flexibility of the rocking fragment relative to the fixed one. Upon desolvation (MC(A)), this distribution remains broad, but develops more distinct peaks, suggesting the emergence of multiple preferred conformations, due to the increased relevance of inter-MC interactions in the absence of solvent screening. Acetonitrile, with its low hydrogen-bonding capacity, imposes minimal conformational constraints on the MC molecules, allowing both solvated and desolvated systems to sample a wide range of rocking orientations.
The ethanol-solvated system (MCE) shows a slightly sharper distribution, with a peak near 130°, indicating a modest stabilization of the rocking fragment compared to MCA. After desolvation (MC(E)), the distribution broadens slightly while retaining its general shape, implying that ethanol imparts a degree of angular ordering, albeit without fully constraining flexibility.
In contrast, methanol (MCM) exhibits a more defined angular distribution, mostly spanning 60–130°, suggesting that methanol promotes more structured configurations than ethanol or acetonitrile. In the desolvated counterpart (MC(M)), the distribution broadens again, indicating that while methanol promotes tighter packing through its stronger polarity and smaller size, this effect is partially lost upon solvent removal.
Water has the most pronounced influence. In the solvated system (MCW), the rocking–fixed fragment angle distribution is the narrowest and right-shifted, with sharp peaks around 130–150°. This reflects the strong conformational restriction imposed by the extensive hydrogen-bonding network of water, which significantly structures the MC conformation. Even in the desolvated state (MC(W)), while the angular distribution becomes broader and less sharply peaked, it still resembles the solvated profile, indicating a lasting structural imprint from prior water interactions.
Overall, the ability of the solvent to form hydrogen bonds—ranked as water > methanol > ethanol > acetonitrile—correlates directly with its capacity to modulate the conformational landscape of the MC rocking fragment, progressively stabilizing distinct angular domains as hydrogen-bonding strength and polarity increase.
The average distributions of angles formed between the normal vector to the average plane of the fixed fragment and the orientation vector of the wagging fragment reveal that the wagging segment exhibits substantial conformational flexibility (see
Figure S3 in Supplementary Materials). In all solvated systems, the angular distributions are broad, with the highest probability density centered around perpendicular orientations (~90–130°) relative to the fixed fragment. This indicates that the wagging fragment frequently adopts diverse conformations but retains the freedom to have a wide range of orientations.
In the acetonitrile-solvated system (MCA), the distribution remains broad, reflecting minimal steric or hydrogen bonding constraints. Due to acetonitrile poor donor and moderate acceptor character, its weak hydrogen-bonding capabilities result in limited interaction with the sulfonic acid or the polar linker atoms (O and N), thereby allowing greater wagging flexibility. Upon desolvation (MC(A)), the angular distribution shifts, exhibiting increased population at larger angles, suggesting that inter-MC interactions help stabilize more compact conformations of the wagging fragment in the absence of solvent.
The ethanol-solvated system (MCE) presents a more defined angular distribution, with a prominent peak near 110–140°, indicative of a preferred extended conformation. This suggests that ethanol can stabilize specific wagging orientations, likely through moderate hydrogen bonding with the sulfonic acid group. In the desolvated system (MC(E)), the distribution becomes broader and diffuse, with peaks spanning ~80–130°, reflecting an increase in conformational flexibility once ethanol is removed.
In methanol (MCM), the angular distribution is very broad (30–150°) and slightly bimodal, implying that methanol allows a diverse set of orientations due to its strong hydrogen-bonding capacity and small molecular size, which facilitates dynamic reorientation. After desolvation (MC(M)), the distribution sharpens considerably around 130°, indicating that the removal of methanol restricts the wagging motion and stabilizes a preferred orientation, likely through enhanced inter-MC packing and reduced steric competition.
The water-solvated system (MCW) displays a broad bimodal distribution with a notable peak between 110 and 130°, suggesting some degree of conformational preference imposed by water’s extensive hydrogen-bonding network. Water molecules likely form strong, directional hydrogen bonds not only with the sulfonic acid group but also with the polar O and N linker atoms, thereby partially constraining the wagging fragment in semi-extended conformations. Upon desolvation (MC(W)), this restriction is lifted, resulting in a broader and flatter distribution that indicates increased conformational freedom.
In summary, the average wagging angle distribution reflects a delicate interplay between solvent-induced hydrogen bonding and conformational deformation. Strong hydrogen-bonding solvents such as water and methanol initially stabilize specific wagging orientations, while desolvation tends to release these constraints, allowing MC molecules to explore a wider conformational landscape. Acetonitrile, by contrast, exerts minimal influence in both solvated and desolvated states, permitting maximum flexibility throughout.
The average angular distributions between the average plane normal of the rocking fragment and the orientation vector of the wagging fragment in solvated MC systems highlight solvent-specific influences on molecular conformational preferences. In acetonitrile (MCA), the angle distribution is relatively broad, though it displays a subtle bias toward a specific angular range. This suggests that while the rocking–wagging relationship retains flexibility in solution, there may be weak stabilizing interactions or packing preferences. Upon desolvation (MC(A)), the distribution sharpens significantly, with a distinct peak emerging, indicating a pronounced preference for a specific conformation. This reflects enhanced intramolecular ordering driven by the absence of competing solvent interactions.
For the ethanol-solvated system (MCE), the distribution is bimodal, with peaks clustered in two angular regions: 25–80° and 90–120°. This pattern suggests the coexistence of multiple stable rocking–wagging configurations, likely stabilized by ethanol’s moderate hydrogen-bonding interactions. Desolvation (MC(E)) causes the angular distribution to consolidate into a narrower range (55–95°), reflecting a convergence toward a more defined and stable conformation once solvent constraints are removed.
The methanol-solvated system (MCM) presents a broad and shallow angular distribution spanning approximately 60–140°, with no pronounced maxima. This suggests a high degree of rocking–wagging flexibility in solution, consistent with methanol’s strong hydrogen-bonding capacity and small size, which facilitate dynamic conformational sampling. Following desolvation (MC(M)), the distribution narrows and becomes modestly peaked, with a maximum centered around ~70°, indicating a reduction in conformational diversity and the emergence of a preferred geometry.
Among the solvated systems, water (MCW) produces the broadest range of angular distribution with five groups of peaks. This is attributed to water’s strong and dynamic hydrogen bonding with the sulfonic acid group and polar O and N linker atoms, which may temporarily disrupt intramolecular alignment. However, upon desolvation (MC(W)), the angle distribution contracts and becomes highly peaked, pointing to a rigid and well-defined geometry between the rocking and wagging fragments in the absence of water.
Overall, these results reveal a common trend: all solvents allow for significant rocking–wagging flexibility in solution, with the degree of freedom varying by solvent strength and polarity. Desolvation universally promotes angular restriction and conformational ordering, with the most pronounced shift observed in systems initially solvated with water.
3.2. The MC Packing Analysis
The analyses of the densities of the equilibrated solvated systems reveal an ordering that corresponds to the polarity of the solvents: MCE < MCA < MCM < MCW. Upon desolvation, the densities increase in the same order MC(E) < MC(A) < MC(W) < MC(M) (see
Table 1). The radial distribution function (RDF) of the centers of the pentagonal pyrrole-type C
4N ring of MC molecules provides insight into how the MC molecules are spatially distributed relative to one another in the presence and absence of solvent. The low limits of RDF (see
Figure S5 in Supplementary Materials), which indicates the shortest distances between the centers of MC molecules (d
min), are 3.88, 4.88, 5.13 and 5.63 Å for solvated MC molecules in MCW, MCE, MCM, and MCA, respectively, (see
Table 1). The order changes for the desolvated MC systems, where the inter-MC distances are 3.88, 4.38, 4.38, and 4.88 Å, following the sequence MC(A) < MC(E) = MC(W) < MC(M). While a reduction in inter-MC distances was expected upon solvent removal, an increase was observed for MC(W).
The presence of a strong first peak of RDF denotes a strong MC—solvent interactions and the presence of some successive well determined peaks suggest the regular spatial arrangement of the solute molecules. The position of the first RDF peak, its height, and width indicate preferred intermolecular distances and degrees of structural organization or compactness. The profile of RDF for MCA has a flattened slope, with a first peak at distances of about 7.8 Å, which indicates a delayed structuring. The desolvated MC(A) exhibits a pronounced first peak at ~4.5 Å, which is significantly left-shifted compared to MCA, indicating a more defined and structured organization. The MCE presents a relatively intense RDF first-peak at about 6.13 Å, implying that ethanol molecules partially clustered the MC molecules. The desolvation affects this trend of clusterization of the MC molecules determining a flattener profile for RDF, with a first maximum at 6.88 Å. Both distributions for MCM and MC(M) are relatively broad, but MCM exhibits a modest first peak at 5.63 Å compared to the more diffused profile of MC(M) with a well determined first peak at 6.38 Å. This behavior is possibly due to methanol-induced clustering. MC(W) first peak located at ~4.88 Å, while MCW shows a first RDF peak at ~4.38 Å—the lowest among all solvated systems. The MCW shows several other distinct peaks for large distances. The RDF profiles of the investigated systems clearly show that solvent polarity and hydrogen-bonding strength govern MC molecular packing. Desolvation always results in compaction (shorter g(r) peak distance), except for methanol, which shows possible swelling or reorganization. Water stands out as the most structuring solvent, followed by ethanol, methanol, and finally acetonitrile.
We characterized the packing of MC molecules by examining the unoccupied space available near the MC molecules, known as the solvent surface free volume, which reflects the potential interaction space for solvent molecules around the MC molecules. In the case of MCA and MCM, a very small space is calculated (see
Table 1). However, MCE and MCW show some empty spaces, particularly near the C
10NH
6 groups of the fixed and rocking fragments of MC molecules, which repel the OH- groups from ethanol and water. Analyzing the systems, we observe that the methanol molecules do not disperse between the MC molecules but instead accumulate in the pockets formed by the MC molecules. The MC(M) system remained after the methanol removal from the simulation box is the most stable desolvated system, being characterized by the lowest average formation energy (see
Table 1). The formation energies reported in
Table 1 reflect the contributions from van der Waals, electrostatics, H-bonding, and conformational strain. Acetonitrile and ethanol stabilize MC systems mainly through favorable packing and solvent–MC H-bonding or dipole interactions. Methanol and water, despite high H-bonding potential, show modest values of the formation energy gains due to dynamic and competing H-bonding environments. Solvation limits MC–MC interactions (higher formation energies), while desolvation release the constraints of MC fragments, but often at the expense of stability.
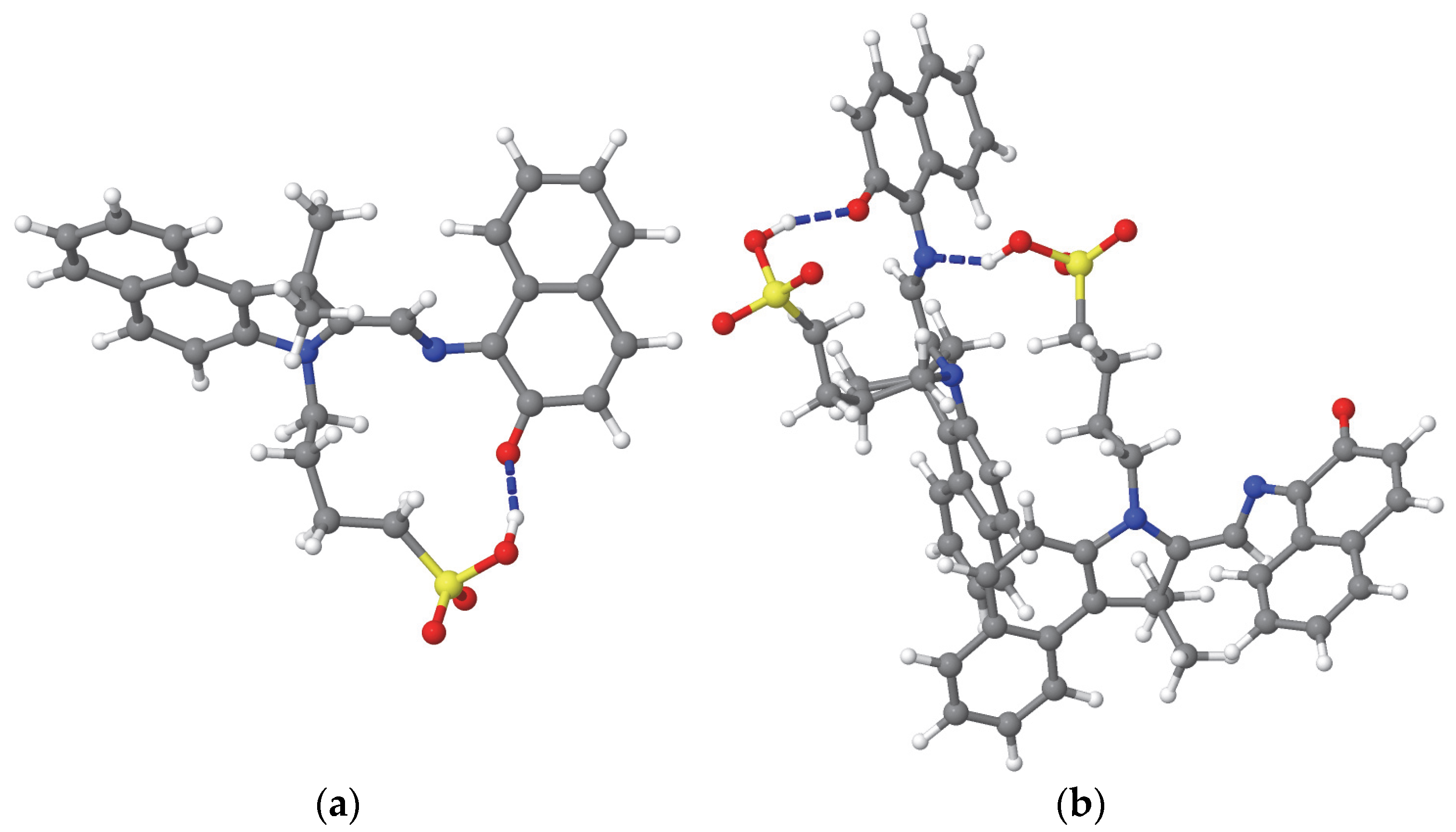
The MC molecule has a hydrophilic sulfonic acid (–SO
3H) terminus on a flexible wagging fragment. The nitrogen and oxygen atoms in the central region of the MC molecule can form intramolecular hydrogen bonds with the hydrogen atom of the sulfonyl group through bending of the wagging fragment within the same molecule (see
Figure 3a,b) or from another MC molecule (see
Figure 3b). Additionally, the sulfonic end and these central nitrogen and oxygen atoms, along with the oxygen and hydrogen atoms of the sulfonyl group, can form intermolecular hydrogen bonds with the OH groups of the solvent molecules. In contrast, the fixed and rocking fragments are hydrophobic, tending to repel the OH groups of the solvent.
Table 2 presents the number of the intra-MC and inter-MC hydrogen bonds, as well as those formed with the solvent molecules. It is noted that, except for methanol, where the number of the hydrogen bonds between MC molecules remain unchanged, the number of hydrogens increase after desolvation in all other cases. The system MC(M) is characterized by a high number of intramolecular hydrogen bonds (see
Table 2) that are specific to MC-HB
O, the most stable isomer of MC. Thus, the highest stability of MC(M) compared to other desolvated systems MC(X) can be justified.
Acetonitrile, with its moderate hydrogen bond acceptor capacity, primarily forms hydrogen bonds with the sulfonic acid group of the MC molecules. In contrast, the protic solvents (ethanol, methanol, and water) engage in more extensive hydrogen bonding: they act both as hydrogen bond acceptors interacting with the sulfonic group, and as hydrogen donors forming hydrogen bonds with the sulfonic groups as well as with the central nitrogen and oxygen atoms along the MC backbone (see
Table 2).
The average distributions of angles formed by normal vectors to the average planes containing the fixed fragments of neighboring MC molecules show monomodal peak around 70–100°, indicating a somewhat preferred but flexible orientation between fixed fragments (see
Figure S6 in Supplementary Materials). This suggests a similar distribution pattern in angle preference, though slight variations in intensity and slight shifts between the solvated and desolvated systems. Slight flattening of the curves for MC(M) and MC(W) implies greater angular freedom. MCW shows a strongly modulated, trimodal distribution, with pronounced features at ~25°, ~82°, and ~115°, which indicates distinct preferred orientations, suggesting specific packing geometries or multi-mode interactions and possible longer-range order or hydrophobic interlocking of fixed fragments. The distribution sharpens and becomes centered near 80° by water removing, consistent with other desolvated systems. Water removal allows the MC molecules to regain intrinsic structural order, previously masked by strong solvent interactions. The presence of a low angles peak for MCE and MCW suggests the stacking of MC molecules with similar orientation of the fixed fragments.
The hydrophobic rocking fragments, being partially flexible, respond sensitively to solvent interactions and hydrogen bonding, providing insight into conformational stability and solvent-induced distortion or ordering. The rocking fragments of neighboring MC molecules in acetonitrile exhibit an average angle distribution with a fairly broad and slightly skewed profile (see
Figure S7 in Supplementary Materials). The peak lies near ~90°, but the tails extend toward both lower and higher angles. The angle distribution of the desolvated MC(A) becomes narrower and more symmetrical, peaking around at 88°. This tightening suggests that in the absence of solvent, intrinsic molecular interactions stabilize as a preferred conformation. Acetonitrile allows free rotation in solutions. Upon desolvation, the MC molecules adopt a more ordered conformation dominated by intramolecular forces.
For MCE, the rocking angle average-distribution is bi-modal, with a prominent peak around 108–140°, which indicates that ethanol’s hydrogen bonding dampens rocking fragment flexibility. The angle distribution for the desolvated MC(E) becomes even narrower and more peaked, centered at ~73°, which demonstrates that ethanol pre-organizes MC conformations.
The rocking angle average-distribution for MCM is sharply defined, peaking near 85°, with narrow spread, which suggests that methanol’s strong hydrogen bonding and polarity constrain the rocking fragment efficiently. The rocking angle distribution for MC(M) is very similar to MCM, showing minimal change in angle distribution. This highlights significant pre-organization, where methanol stabilizes a geometry close to that of the desolvated system.
The MCW rocking angle average-distribution is bimodal, with fluctuations in intensity across the range, indicating a disrupted conformational behavior due to extensive hydrogen bonding with water molecules. This behavior suggests that water interacts dynamically with the rocking fragment, preventing a stable geometry from dominating. The distribution for desolvated MC(W) becomes significantly more ordered, with a strong central peak near 70°. The water molecules destabilize the rocking geometry in solution, but MC regains order in the dry state.
In conclusion, the rocking fragment is more sensitive than the fixed fragment to solvent-induced disorder or stabilization. The protic solvents like methanol and ethanol strongly restrict rocking motion, whereas aprotic acetonitrile allow high flexibility. Water causes dynamic structural noise, which resolves into ordered geometry after desolvation. These results support the idea that preorganization in solution, especially by methanol, may promote more ordered assembly in bulk phases or upon drying.
The wagging fragments of neighboring MC molecules, both in solvated and desolvated systems exhibit similar fairly broad and diffused wagging–wagging angle average-distributions, centered around 90°, with a symmetrical shape around this central peak, which correspond to a perpendicular orientation of the wagging fragments of different MC molecules. This uniform, bell-like distribution is reminiscent of an uncorrelated or weakly correlated angular relationship, typically seen in systems where interactions are non-specific or driven mainly by thermal fluctuations. This behavior highlights the notable flexibility of the wagging fragments.
3.3. FT-IR Analysis
In
Figure 4a, naked-eye pictures and the color of the studied MC desolvated powders can be seen. The FTIR—ATR spectra of these powders are presented in
Figure 4b and the observed vibrational frequencies of the desolvated samples are listed in
Table 3. From left to right in
Figure 4a, the colors of the powders range from navy to green to dark red, corresponding to the increasing polarity of the solvent. Thus, the chromatic shift in the MC polymorphs is attributed to the polarity of the solvent through solute-solvent interactions, resulting in changes such as shifts in the vibrational frequencies of certain functional groups due to modifications of bond angles and dihedral angles between atoms within the MC. By comparing the FTIR spectra of the obtained MC isomers, insignificant differences were observed for MC(A), MC(E), and MC(M), while some differences are evident for MC(W). Water provides a medium with increased hydrogen bonding capabilities; thus, the stronger interaction between water molecules and MC may result in more pronounced changes in the FTIR spectrum compared to when the molecule is in a less polar solvent environment.
For the studied samples, the associated frequencies of (C-O)
spiro structure appear in the spectra shifted to lower wavenumbers than usual (969 cm
−1 for MC(W) and 972 cm
−1 for the other MC samples), which means a lengthening of the C-O bond in
spiro structures [
21]. After the ring-opening process, the spatial orientation of the butyl sulfonic moiety shown by the position of the peaks ascribed to the hydrocarbon chain (in
Table 3) was affected, especially for MC(W). As shown in
Figure 4, the wide characteristic band in the range of 3700 cm
−1 to 3100 cm
−1 (including 3399 cm
−1, 3390 cm
−1, 3400 cm
−1 and 3402 cm
−1 for MC(A), MC(W), MC(E) and MC(M), respectively) could be attributed to the –OH stretching vibration due to -SO
3H group and of the physical adsorption of water [
22].
The characteristic band at 1646 cm
−1 only for MC(W) could be attributed to the stretching vibration of water when hydrogen bond is present. The typical band related to C=O (carbonyl stretching vibration) at 1769 cm
−1 in MC(A), MC(E), MC(M) appear at a relatively high frequency in the IR spectra while for the MC(W) sample, the carboxyl group appears at lower frequency in IR spectrum (1765 cm
−1). This shift occurs due to the resonance effects between the C=C or phenyl group and the C=O group, which affects the bond strength and frequency of vibration. Only in MC(W) spectrum, the pre-eminent peak at 1688 cm
−1 and the other two faint peaks (~1765 cm
−1 and 1646 cm
−1) indicating the occurrence of intermolecular hydrogen bonds with a combination of phenomena related to the presence of bonded water molecules [
23]. The peak at 1646 cm
−1 in MC(W) typically could be attributed to the stretching vibration of water when a hydrogen bond is present. The specific interpretation may be related to the presence of the oxygen atom in the carbonyl group that can act as a hydrogen bond acceptor, forming hydrogen bonds with hydrogen atoms attached to electronegative atoms like oxygen or nitrogen [
24,
25].
In the range of 1750 cm
−1 to 1650 cm
−1, the spectra of the MC(A), MC(E), MC(M) isomers present a small broad peak, which arise from vibrations involving multiple bonds or a complex molecular structure caused by a possible remain solvent. The group of three separated bands at 1621 cm
−1 (1623 cm
−1 in MC(W)), 1591 cm
−1 and the shoulder at 1574 cm
−1 (1580 cm
−1 in MC(W) are related to C=N bond and indicate that the nitrogen in the MC molecules is interacting with solute molecules [
22,
26]. Moreover, the presence of amide I bands (peaks at about 1621 cm
−1 and 1623 cm
−1) suggest the possibility of hydrogen bonds and a possible twist of naphtho-moiety of the MC molecule.
3.4. DSC/TG Thermal Analysis
Figure 5 shows the thermal analysis results for the solid powders of the MC samples, obtained by desolvation from different solvents. The thermogravimetric parameters determined from the analysis of the thermogravimetric curves are presented in
Table 4.
The DSC curves of desolvated MC powders are a complex combination of superposed thermal events. It can be seen that there are no distinctly observable phase transitions. The components resulting from the successive degradation (in steps) of the component fragments can be considered as impurities in the remaining sample mass and appear as impurities in the degradation of the following fragments of MC [
27]. Thus, in the presented thermograms, enlarged endotherms can be observed for the analyzed samples. For such an endotherm, the mathematically calculated peak is very different from the observed peak [
28]. MC samples present complicated thermal behavior. Therefore, the thermal stability of MC is strongly affected by its molecular conformation and by interactions with the solvent. A perspective is that the MC samples could be seen as a combination of multiple organic function groups. MC structural function groups could have their own distinct behavior concerning thermal stability. In MC, such function groups may fulfill their own function independently or in cooperation with neighboring functions. Thus, when multiple functional groups are present in a molecule, the individual thermal transitions can overlap or occur at different temperatures, resulting in multiple events on the DSC thermograms [
29,
30].
Another point of discussion is the polarity of the solvent, which contributes to the polymorphic selectivity of MC. This may be possible because at a molecular level the solvent selectively absorbs onto MC, respecting the presence of the function groups present in the molecule [
31]. Generally, longer alkyl chains can increase the hydrophobic interactions, leading to an increase in the melting temperatures [
32], but if an electrically charged group is attached to the alkyl group, the behavior upon heating will change [
33,
34]. Thus, the presence of an alkyl sulfate chain linked to an organic structure can affect the melting temperature by influencing the overall molecular structure of MC as well as the interactions within the MC isomer. Moreover, it enhances MC water solubility and affects the stability of merocyanine polymorph in aqueous environments [
35,
36]. As MC exhibited, a similar thermal behavior has been noted in the literature for ionic liquids, i.e., the melting temperature of ionic liquids with short alkyl chains decreases as the chain length increases [
37]. In particular, it can be seen in
Figure 5a that two solid–solid phase transitions are mainly detected in temperature ranges from 25 °C to 100 °C.
In the presented thermograms, for MC(A) the endothermic transition at about 37 °C is related to the solid–solid polymorphic transition due to the length of the hydrocarbon chain of the butyl sulfonic moiety of MC. This thermal event is followed by another transition almost without loss of mass starting at about 59 °C which corresponds to the solid–liquid phase transition of the MC. In the temperature range of 150 °C to 350 °C, the curve shows four endotherms for MC(A) due to the thermal transformations of the MC(A) fragments. The components resulting from the mass loss occurs in a wide temperature range and can be clearly observed in
Figure 5a up to around the temperature of 460 °C.
Table 5 presents the DSC parameters for the thermal events occurring within the temperature range corresponding to the start of heating (25 °C) until the modification of the baseline of the thermograms, over which the degradation of the samples occurs through the detachment of component fragments. The thermal events within the considered temperature range correspond to the solid–solid polymorphic transition, the solid–liquid phase transition of the MC, and the release of remaining solvent molecules, which overlap in this range. It was observed that such degradation temperature is influenced by the solvent that was used in the preparations. Further, a deconvolution of endotherms was performed in order to separate the overlapping signals of the MC and solvent located in the same temperature domain. The overlapping transitions of the heating thermograms were deconvoluted into individual constituent peaks (results are in
Figure S9 and
Table S2) [
38]. It was observed that MC desolvates do not have a specific melting point but rather soften over a range of temperatures depending on solvent nature. The results show that the solid–solid transition at about 37 °C is clearly evidenced for MC(A) and MC(W), while for MC(M) and MC(E) samples the solvent presence slightly lower the temperature of this transition. For all the samples, a stepwise elimination of the solvent took place, possible along with another polymorphic transition. Nevertheless, several aspects, such as boiling point of the solvents and also their different abilities to form physical bonds with function groups of MC must be considered in the differentiation of such thermal behavior. In MC(A) and MC(W) samples, the solvents ethanol and methanol strongly interact with hydrophobic fragments of MC, resulting in endotherms shifting to higher temperatures in MC(M) and MC(E) samples. Considering the different hydrophobicity and hydrophilicity of the fragments of MC molecule, the obtained results suggest that methanol exhibits a balance between polar and nonpolar characteristics, thus extending the range of interactions of methanol with MC molecule more than the other solvents in this study [
39]. However, in the case of the MCW sample, a particularity of the thermal behavior can be highlighted. As can be seen in
Figure 5b, the rate of change in the mass of sample, as a function of temperature (dTG curves) more clearly reveals the evolution of thermal events. It is observed that the dTG curve of the MCW sample is clearly different from the other samples. Compared to the other samples, MCW shows a significant mass loss (24.61%, as shown in
Table 4) at temperatures above 400 °C. This suggests that the presence of water has played a role in forming a polymorph with a more thermally resistant arrangement of MC molecules. The FTIR data and the results in
Table 2 indicate that the presence of hydrogen bonds leads to stronger interactions between water molecules and MC, which may lead to more pronounced changes in desolvated MC.
In
Figure 5, SEM images are shown only for the MCA sample (
Figure 5a
1,a
2) as these were the most appropriate to present. Obtaining the SEM images was challenging due to a number of factors necessary for the accurate recording of the SEM images, which caused real-time modifications of the sample surface. Although we used reasonable recording conditions, consistent with those for sensitive organic substances, parameters such as the chamber pressure (0.08–1.5 Torr) and the energy of the electron beam used for SEM, induced changes to the sample itself (e.g., heating and charging effects), which to some extent altered the morphology during scan/imaging. However, the SEM instrument we used does not have the capability to capture high-speed events, thus limiting the ability to observe dynamic processes in sensitive samples such as MC desolvates. Nevertheless, we found that MC is highly sensitive to very fine modifications caused by different stimuli, which serves as a premise for future investigations. The fact that MC undergoes phase transitions triggered by various stimuli at physiological temperature (37 °C) makes this material ideal for biological applications.
The solvation of MC involves the interaction between the solvent molecules and its functional groups. Further, the presence of the first solvation sphere, which consists of arrangement the solvent molecules in immediate contact with the MC different function groups can affect the conformation of the MC by stabilizing certain conformations through hydrogen bonding or electrostatic interactions. The solvent polarity and its capability to form hydrogen bonds can influence the conformational preferences of the MC molecule. When the solvent molecules are removed, the intermolecular interactions between the solvent and the MC molecule are disrupted. This can lead to a change in the conformational state of the MC, as it seeks to minimize its energy and stabilize itself in the absence of solvent interactions [
40,
41]. It is important to note that the conformational change resulting from solvent removing is not always predictable and may vary depending on the MC—solvent system. Moreover, the Molecular Dynamics simulations suggest that solvents influence the conformation of MC molecules by causing the flexible rocking and wagging fragments to fold due to steric hindrance and hydrogen bonding. The interactions between solvent molecules, particularly through hydrogen bonds, constrain the MC molecules, reducing their spatial occupancy. Upon solvent removal, the MC molecules relax into a configuration resembling their solvated state, which in turn shapes the microstructure of the desolvated system.
The hydrogen bonding and conformational preferences of the MC molecules observed in simulations (e.g., orientation of rocking and wagging fragments, intra-/intermolecular H-bond formation, density) are supported by experimental FTIR spectra and thermal analyses (DSC/TG), which show solvent-dependent polymorphism, molecular ordering, and stability in the desolvated MC powders. The increase in hydrogen bonding upon desolvation seen in simulations is reflected in FTIR shifts associated with NH/OH groups. The conformational stabilization and packing predicted by MD (especially for methanol and water) correlate with enhanced thermal stability and specific phase transitions in DSC/TG. Solvent-induced differences in molecular arrangement in simulations match experimental observations of morphological and structural differences in the solid MC phases.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}