

Crucial Role of Ni Point Defects and Sb Doping for Tailoring the Thermoelectric Properties of ZrNiSn Half-Heusler Alloy: An Ab Initio Study


Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
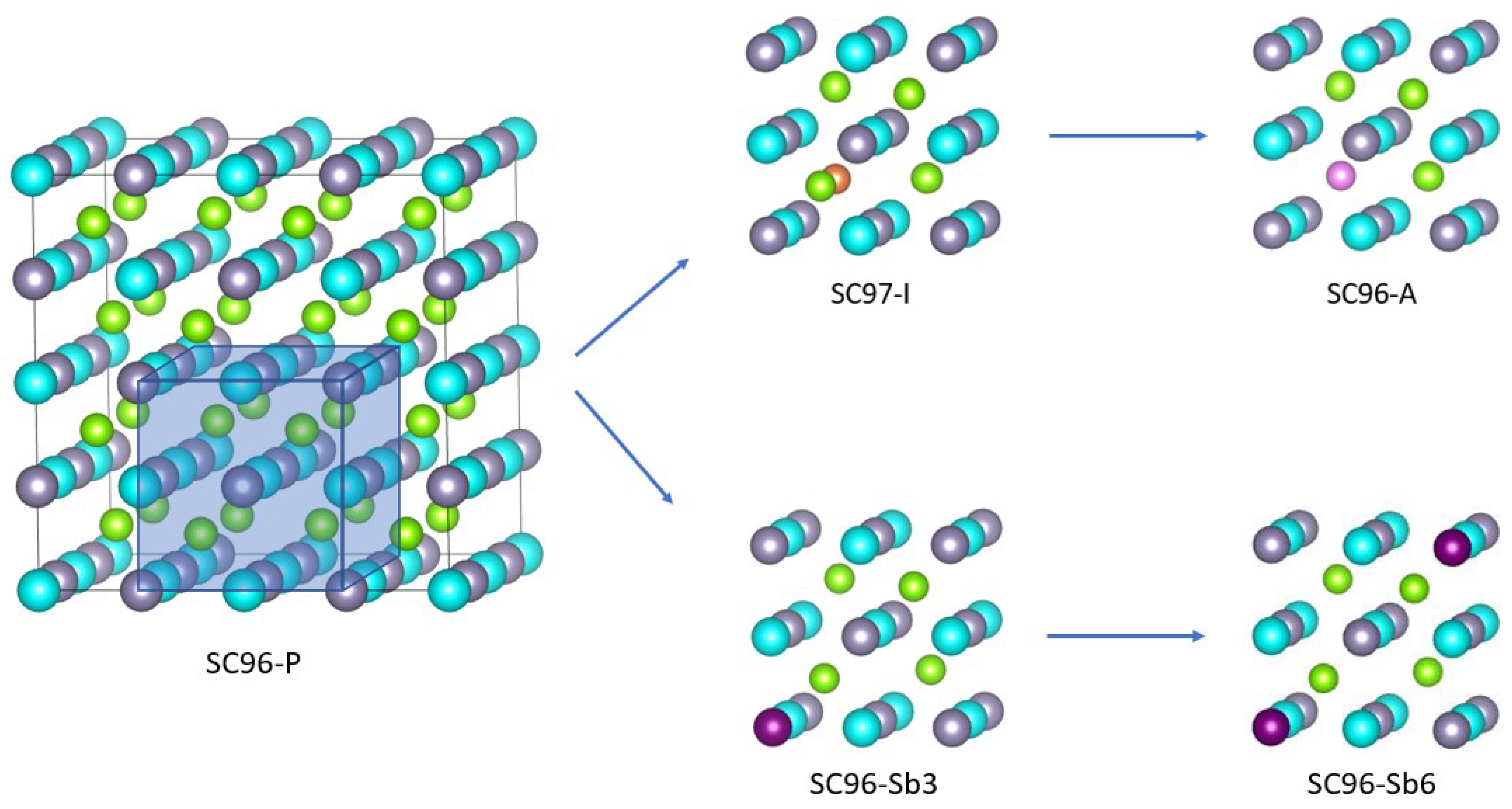
3.1. Models and Structures
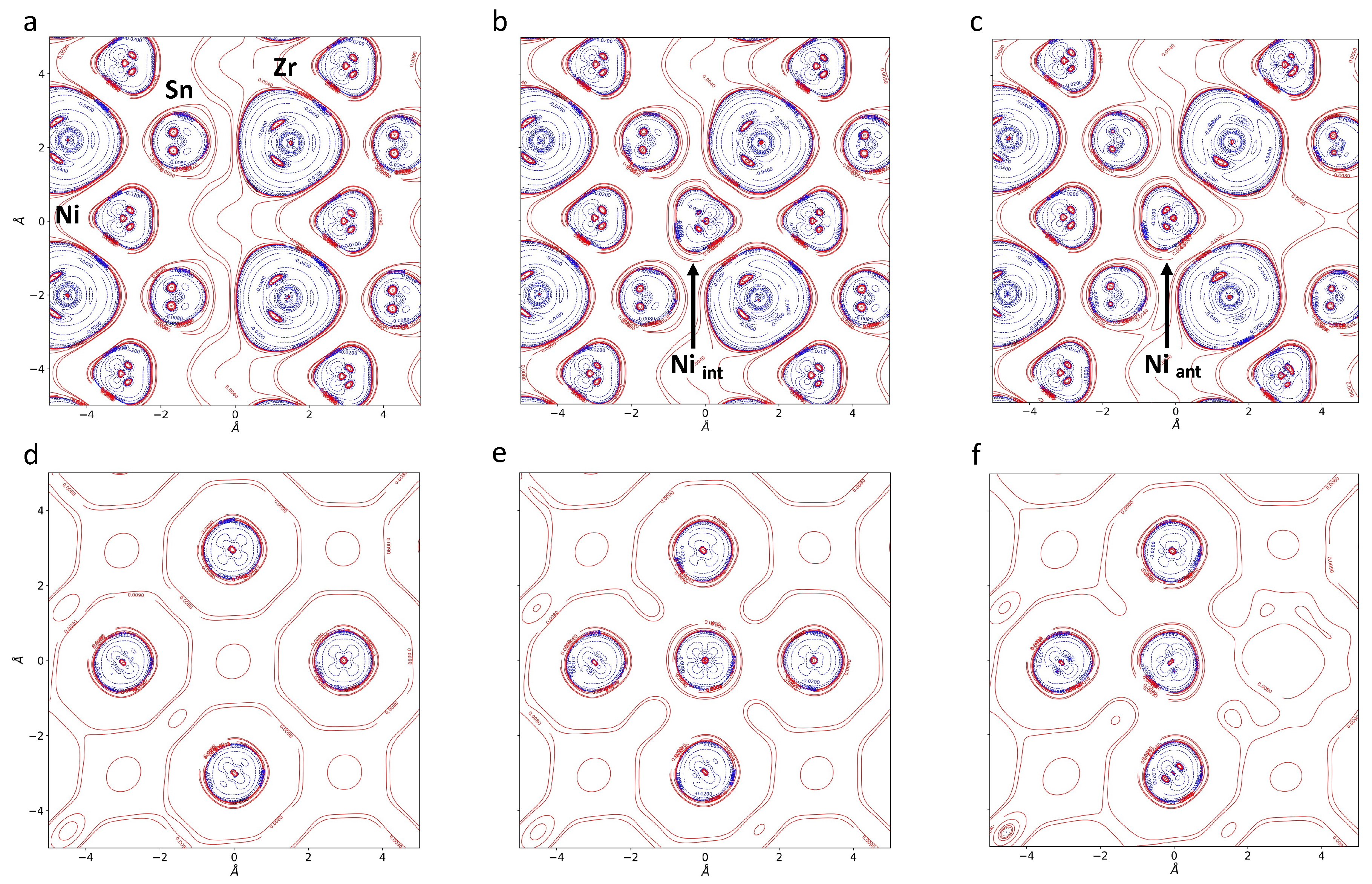
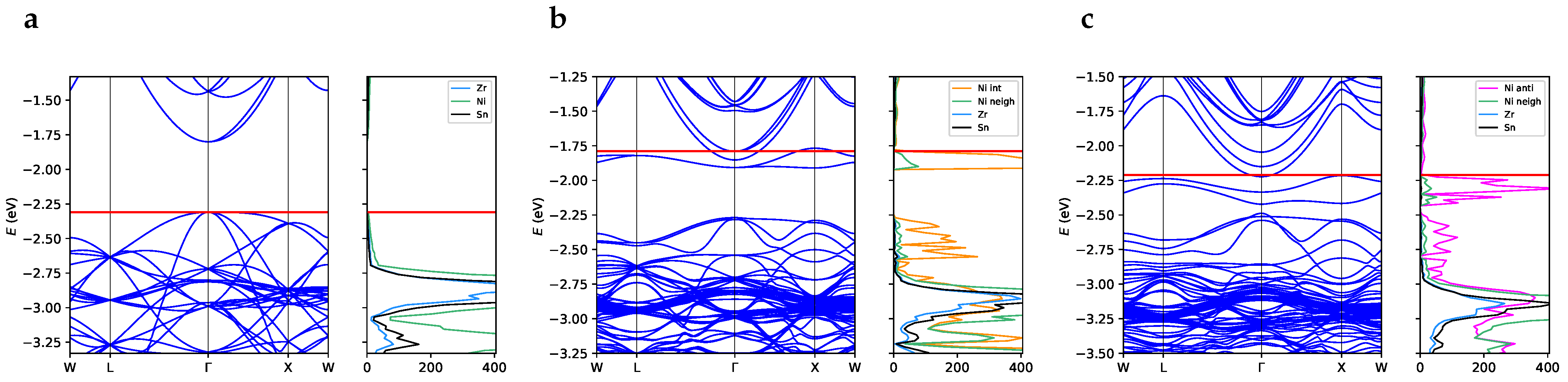
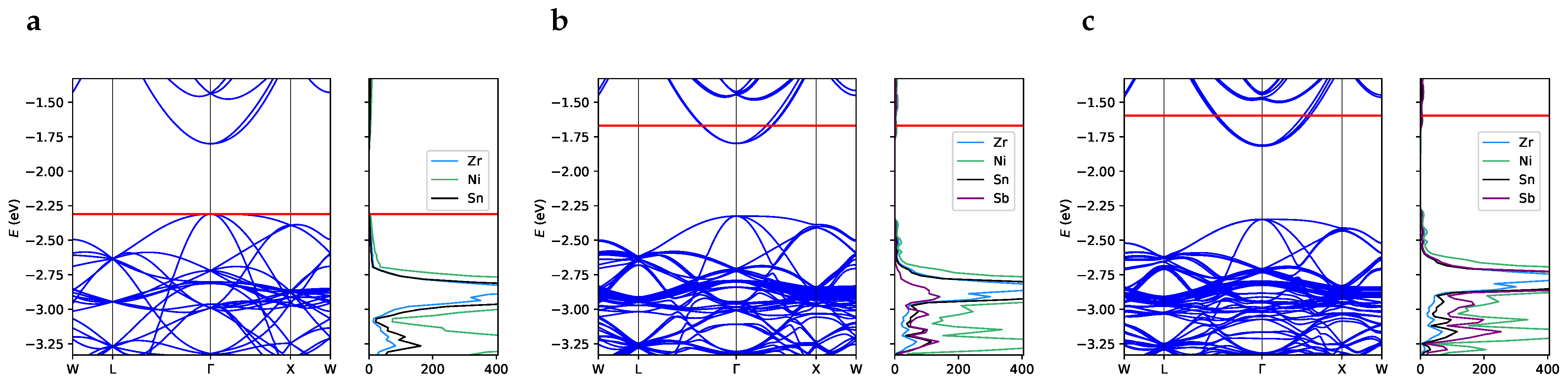
3.2. One-Electron Properties
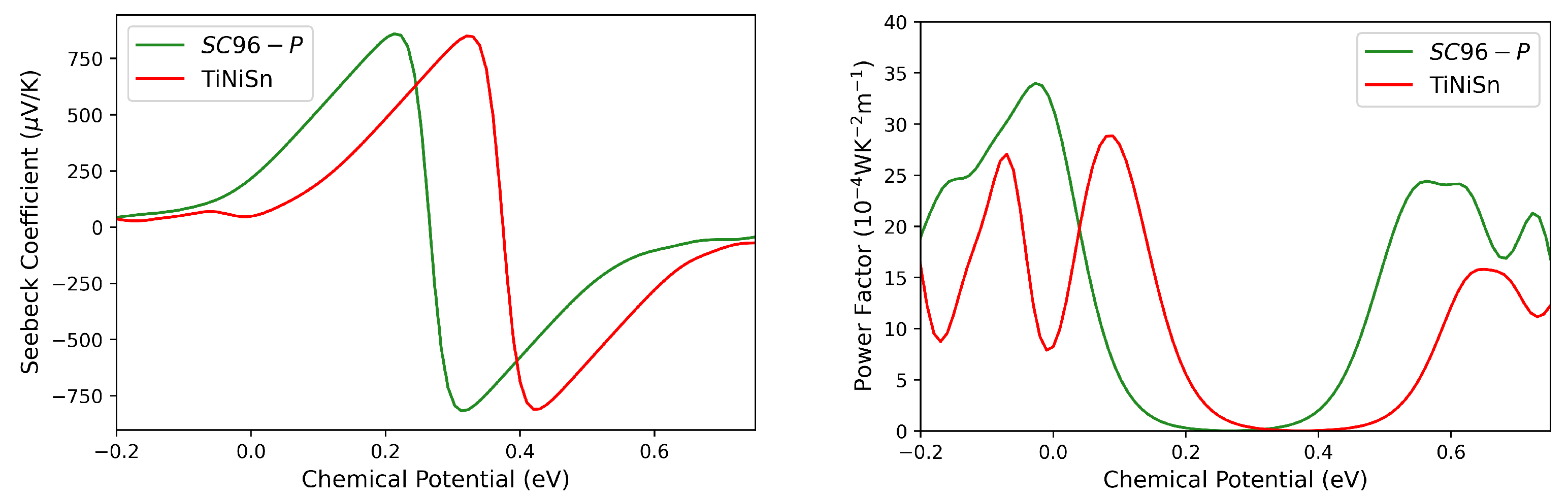
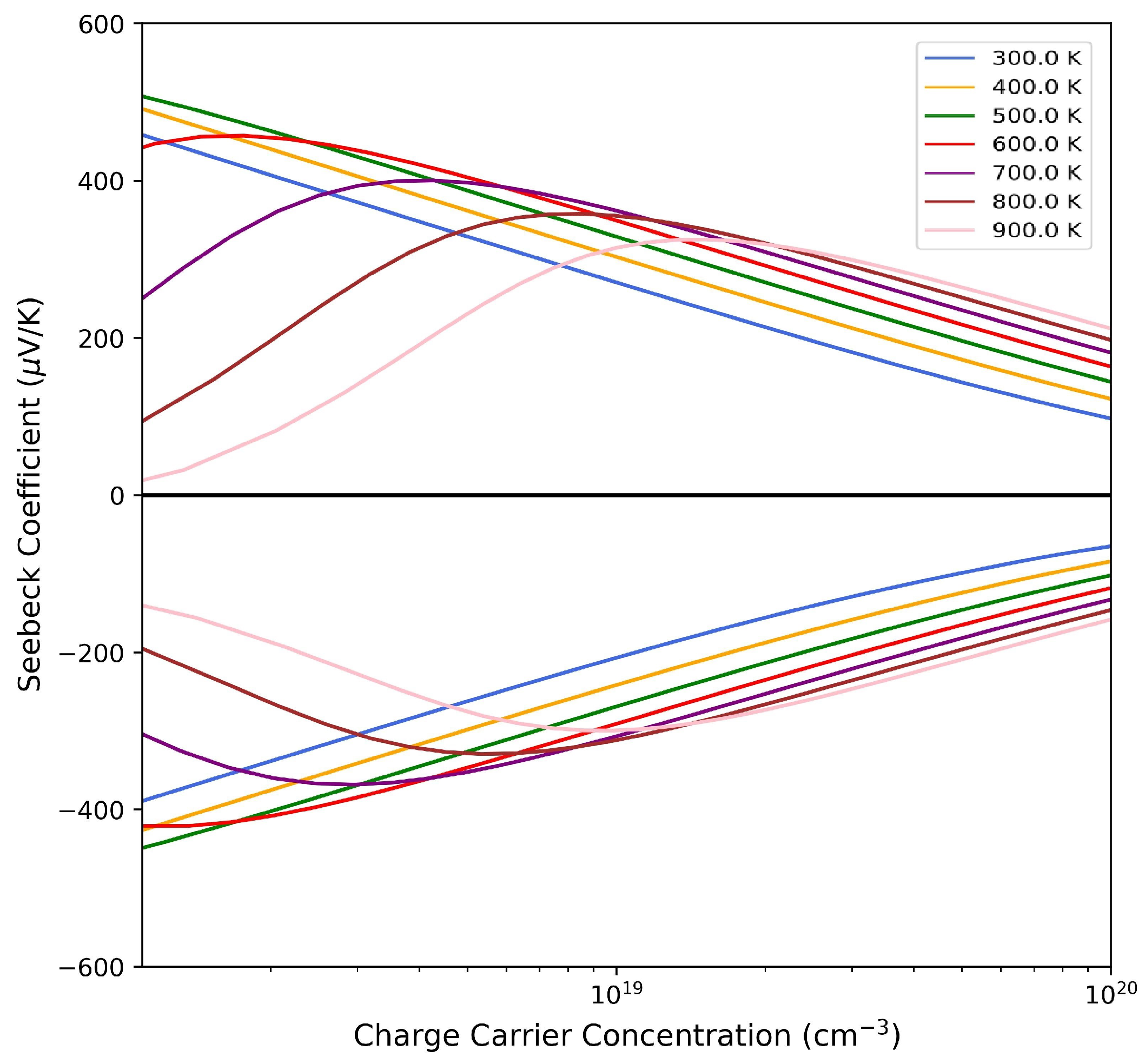
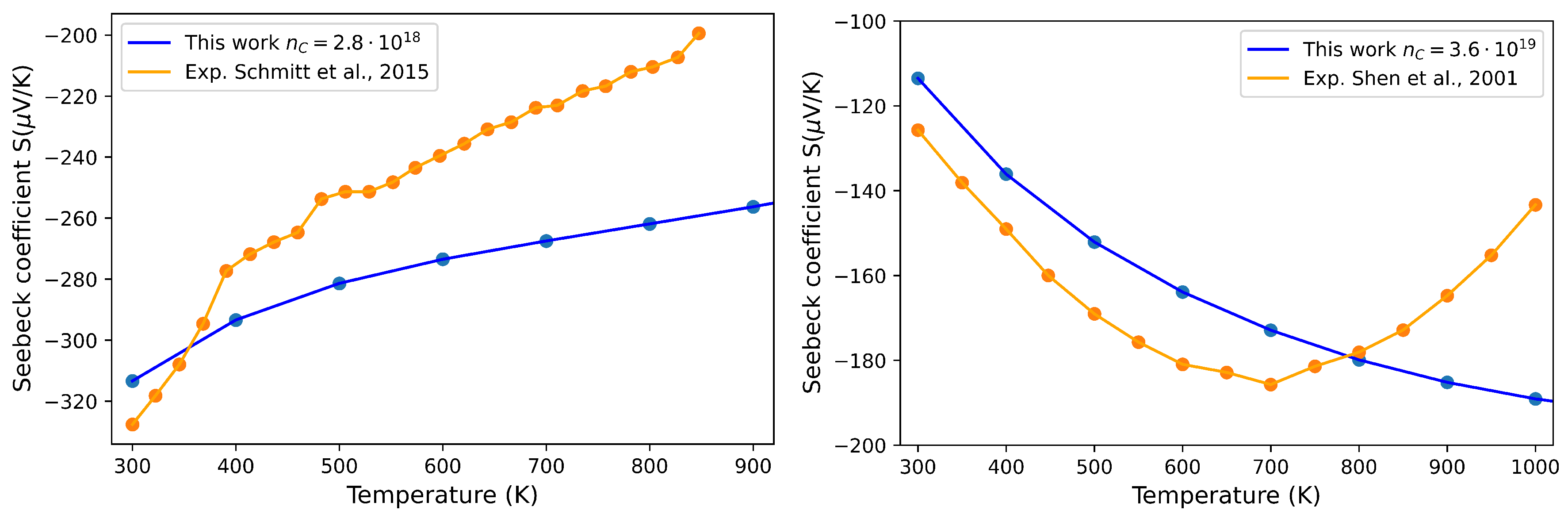
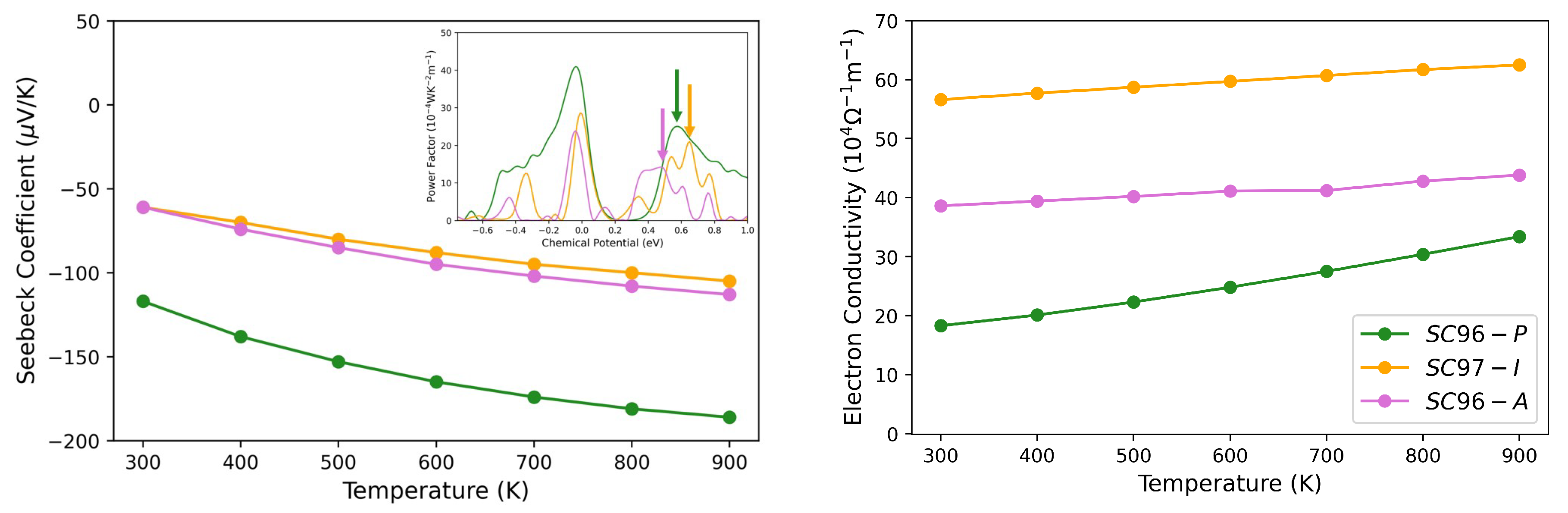
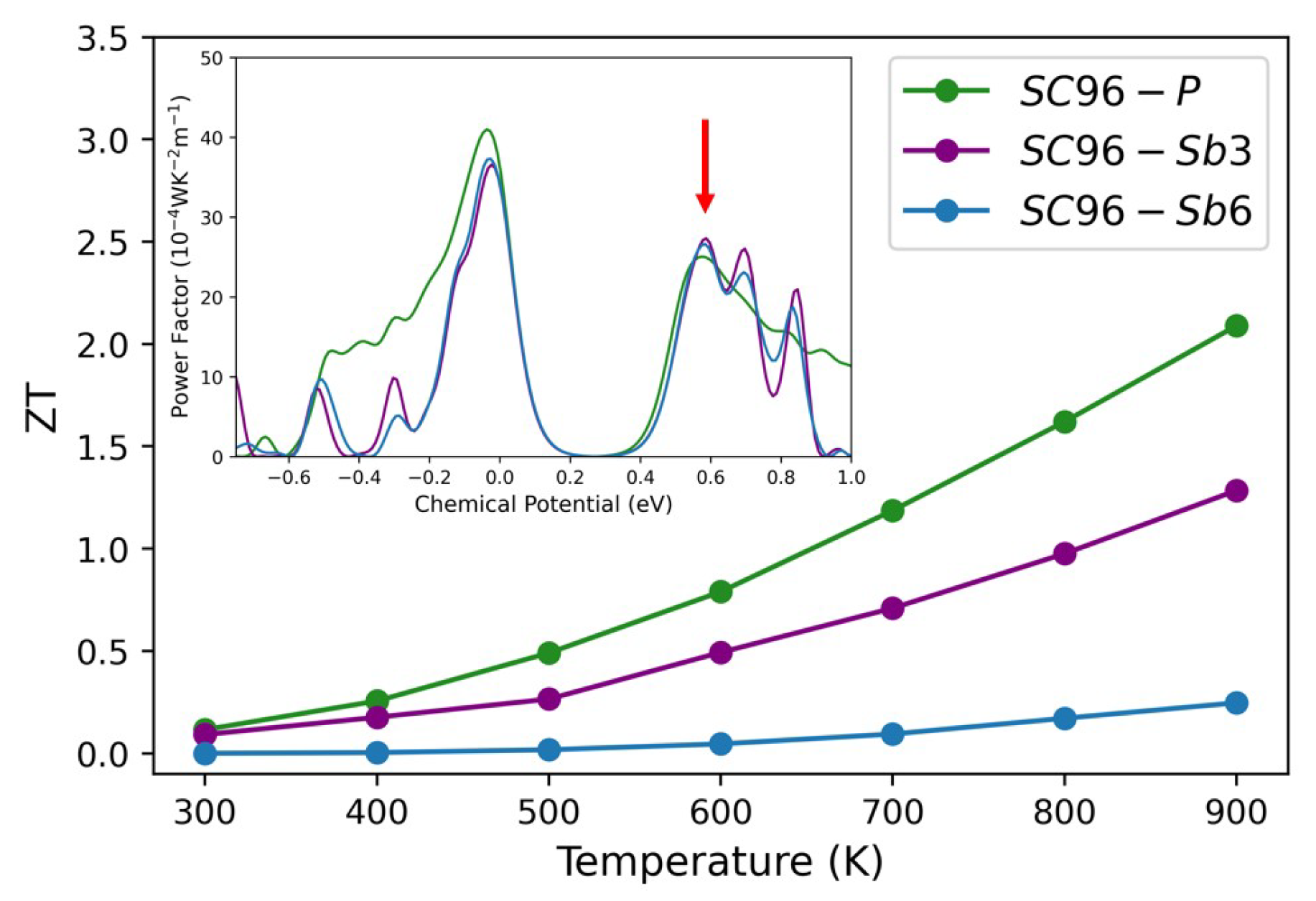
3.3. Thermoelectric Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamid Elsheikh, M.; Shnawah, D.A.; Sabri, M.F.M.; Said, S.B.M.; Haji Hassan, M.; Ali Bashir, M.B.; Mohamad, M. A review on thermoelectric renewable energy: Principle parameters that affect their performance. Renew. Sustain. Energy Rev. 2014, 30, 337–355. [Google Scholar] [CrossRef]
- Fecher, G.H.; Rausch, E.; Balke, B.; Weidenkaff, A.; Felser, C. Half-Heusler materials as model systems for phase-separated thermoelectrics. Phys. Status Solidi A 2016, 213, 716–731. [Google Scholar] [CrossRef]
- Guo, L.; Wang, H. Non-intrusive movable energy harvesting devices: Materials, designs, and their prospective uses on transportation infrastructures. Renew. Sustain. Energy Rev. 2022, 160, 112340. [Google Scholar] [CrossRef]
- Altenkirch, E. Elektrothermische Kälteerzeugung und reversible elektrische Heizung. Phys. Z. 1911, 12, 920–924. [Google Scholar]
- Zheng, J.C. Recent advances on thermoelectric materials. Front. Phys. China 2008, 3, 269–279. [Google Scholar] [CrossRef]
- Vining, C.B. An inconvenient truth about thermoelectrics. Nat. Mater. 2009, 8, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yang, L.; Ma, Z.; Song, P.; Zhang, M.; Ma, J.; Yang, F.; Wang, X. Review of current high-ZT thermoelectric materials. J. Mater. Sci. 2020, 55, 12642–12704. [Google Scholar] [CrossRef]
- Chauhan, N.S.; Bathula, S.; Vishwakarma, A.; Bhardwaj, R.; Johari, K.K.; Gahtori, B.; Saravanan, M.; Dhar, A. Compositional tuning of ZrNiSn half-Heusler alloys: Thermoelectric characteristics and performance analysis. J. Phys. Chem. Solids 2018, 123, 105–112. [Google Scholar] [CrossRef]
- Shi, X.; Chen, L.; Uher, C. Recent advances in high-performance bulk thermoelectric materials. Int. Mater. Rev. 2016, 61, 379–415. [Google Scholar] [CrossRef]
- Xie, W.; Weidenkaff, A.; Tang, X.; Zhang, Q.; Poon, J.; Tritt, T.M. Recent Advances in Nanostructured Thermoelectric Half-Heusler Compounds. Nanomaterials 2012, 2, 379–412. [Google Scholar] [CrossRef]
- Jung, D.; Kurosaki, K.; Kim, C.; Muta, H.; Yamanaka, S. Thermal expansion and melting temperature of the half-Heusler compounds: MNiSn (M = Ti, Zr, Hf). J. Alloys Compd. 2010, 489, 328–331. [Google Scholar] [CrossRef]
- Li, X.; Yang, P.; Wang, Y.; Zhang, Z.; Qin, D.; Xue, W.; Chen, C.; Huang, Y.; Xie, X.; Wang, X.; et al. Phase Boundary Mapping in ZrNiSn Half-Heusler for Enhanced Thermoelectric Performance. Res. Sci. Partn. J. 2020, 2020, 4630948. [Google Scholar] [CrossRef]
- Muta, H.; Kanemitsu, T.; Kurosaki, K.; Yamanaka, S. High-temperature thermoelectric properties of Nb-doped MNiSn (M = Ti, Zr) half-Heusler compound. J. Alloys Compd. 2009, 469, 50–55. [Google Scholar] [CrossRef]
- Gürth, M.; Grytsiv, A.; Vrestal, J.; Romaka, V.V.; Giester, G.; Bauer, E.; Rogl, P. On the constitution and thermodynamic modelling of the system Ti–Ni–Sn. RSC Adv. 2015, 5, 92270–92291. [Google Scholar] [CrossRef]
- Romaka, V.A.; Rogl, P.; Romaka, V.V.; Stadnyk, Y.V.; Hlil, E.K.; Krajovskii, V.Y.; Horyn, A.M. Effect of the accumulation of excess Ni atoms in the crystal structure of the intermetallic semiconductor n-ZrNiSn. Semiconductors 2013, 47, 892–898. [Google Scholar] [CrossRef]
- Fu, C.; Yao, M.; Chen, X.; Maulana, L.Z.; Li, X.; Yang, J.; Imasato, K.; Zhu, F.; Li, G.; Auffermann, G.; et al. Revealing Intrinsic Electron. Struct. 3D Half-Heusler Thermoelectr. Mater. Angle-Resolv. Photoemiss. Spectrosc. Adv. Sci. 2020, 7, 1902409. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.S.; Bathula, S.; Gahtori, B.; Mahanti, S.D.; Bhattacharya, A.; Vishwakarma, A.; Bhardwaj, R.; Singh, V.N.; Dhar, A. Compositional Tailoring for Realizing High Thermoelectric Performance in Hafnium-Free n-Type ZrNiSn Half-Heusler Alloys. ACS Appl. Mater. Interfaces 2019, 11, 47830–47836. [Google Scholar] [CrossRef]
- Miyazaki, H.; Nakano, T.; Inukai, M.; Soda, K.; Izumi, Y.; Muro, T.; Kim, J.; Takata, M.; Matsunami, M.; Kimura, S.; et al. Electronic Local Cryst. Struct. ZrNiSn Half-Heusler Thermoelectr. Mater. Trans. 2014, 55, 1209–1214. [Google Scholar] [CrossRef]
- Shen, Q.; Chen, L.; Goto, T.; Hirai, T.; Yang, J.; Meisner, G.P.; Uher, C. Effects of partial substitution of Ni by Pd on the thermoelectric properties of ZrNiSn-based half-Heusler compounds. Appl. Phys. Lett. 2001, 79, 4165–4167. [Google Scholar] [CrossRef]
- Xie, H.; Wang, H.; Fu, C.; Liu, Y.; Snyder, G.J.; Zhao, X.; Zhu, T. The intrinsic disorder related alloy scattering in ZrNiSn half-Heusler thermoelectric materials. Sci. Rep. 2014, 4, 6888. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Wang, Y. Hf/Sb co-doping induced a high thermoelectric performance of ZrNiSn: First-principles calculation. Sci. Rep. 2017, 7, 14590. [Google Scholar] [CrossRef]
- Sansone, G.; Ferretti, A.; Maschio, L. Ab initio electronic transport and thermoelectric properties of solids from full and range-separated hybrid functionals. J. Chem. Phys. 2017, 147, 114101. [Google Scholar] [CrossRef]
- Linnera, J.; Sansone, G.; Maschio, L.; Karttunen, A.J. Thermoelectric Properties of p-Type Cu2O, CuO, and NiO from Hybrid Density Functional Theory. J. Phys. Chem. C 2018, 122, 15180–15189. [Google Scholar] [CrossRef]
- Dasmahapatra, A.; Daga, L.E.; Karttunen, A.J.; Maschio, L.; Casassa, S. Key Role of Defects in Thermoelectric Performance of TiMSn (M = Ni, Pd, and Pt) Half-Heusler Alloys. J. Phys. Chem. C 2020, 124, 14997–15006. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Erba, A.; Desmarais, J.K.; Casassa, S.; Civalleri, B.; Donà, L.; Bush, I.J.; Searle, B.; Maschio, L.; Edith-Daga, L.; Cossard, A.; et al. CRYSTAL23: A Program for Computational Solid State Physics and Chemistry. J. Chem. Theory Comput. 2022, 19, 6891–6932. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Towler, M.D.; Zupan, A.; Causà, M. Density functional theory in periodic systems using local Gaussian basis sets. Comput. Phys. Commun. 1996, 98, 181–205. [Google Scholar] [CrossRef]
- Valenzano, L.; Civalleri, B.; Chavan, S.; Bordiga, S.; Nilsen, M.H.; Jakobsen, S.; Lillerud, K.P.; Lamberti, C. Disclosing the Complex Structure of UiO-66 Metal Organic Framework: A Synergic Combination of Experiment and Theory. Chem. Mater. 2011, 23, 1700–1718. [Google Scholar] [CrossRef]
- Causà, M.; Dovesi, R.; Roetti, C. Pseudopotential Hartree-Fock study of seventeen III-V and IV-IV semiconductors. Phys. Rev. B 1991, 43, 11937–11943. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL17 User’s Manual; Università di Torino: Torino, Italy, 2017. [Google Scholar]
- Shanno, D.F. Conditioning of quasi-Newton methods for function minimization. Math. Comput. 1970, 24, 647–656. [Google Scholar] [CrossRef]
- Zicovich-Wilson, C.M.; Dovesi, R. On the use of symmetry-adapted crystalline orbitals in SCF-LCAO periodic calculations. I. The construction of the symmetrized orbitals. Int. J. Quantum Chem. 1998, 67, 299–309. [Google Scholar] [CrossRef]
- Erba, A.; Mahmoud, A.; Belmonte, D.; Dovesi, R. High pressure elastic properties of minerals from ab initio simulations: The case of pyrope, grossular and andradite silicate garnets. J. Chem. Phys. 2014, 140, 124703. [Google Scholar] [CrossRef] [PubMed]
- Gatti, C. Chemical bonding in crystals: New directions. Z. Krist. -Cryst. Mater. 2005, 220, 399–457. [Google Scholar] [CrossRef]
- Gatti, C.; Casassa, S. TOPND14 User’s Manual; CNR-ISTM of Milano: Milano, Italy, 2013. [Google Scholar]
- Schrade, M.; Berland, K.; Kosinskiy, A.; Heremans, J.P.; Finstad, T.G. Shallow impurity band in ZrNiSn. J. Appl. Phys. 2020, 127, 045103. [Google Scholar] [CrossRef]
- Schmitt, J.; Gibbs, Z.M.; Snyder, G.J.; Felser, C. Resolving the true band gap of ZrNiSn half-Heusler thermoelectric materials. Mater. Horiz. 2015, 2, 68–75. [Google Scholar] [CrossRef]
- Gong, B.; Li, Y.; Liu, F.; Zhu, J.; Wang, X.; Ao, W.; Zhang, C.; Li, J.; Xie, H.; Zhu, T. Continuously Enhanced Structural Disorder To Suppress the Lattice Thermal Conductivity of ZrNiSn-Based Half-Heusler Alloys by Multielement and Multisite Alloying with Very Low Hf Content. ACS Appl. Mater. Interfaces 2019, 11, 13397–13404. [Google Scholar] [CrossRef]
- Sun, Y.; Qiu, W.; Zhao, L.; He, H.; Yang, L.; Chen, L.; Deng, H.; Shi, X.; Tang, J. Defects engineering driven high power factor of ZrNiSn-based Half-Heusler thermoelectric materials. Chem. Phys. Lett. 2020, 755, 137770. [Google Scholar] [CrossRef]
- Fiedler, G.; Kratzer, P. Ternary semiconductors NiZrSn and CoZrBi with half-Heusler structure: A first-principles study. Phys. Rev. B 2016, 94, 075203. [Google Scholar] [CrossRef]
- Camino, B.; Zhou, H.; Ascrizzi, E.; Boccuni, A.; Bodo, F.; Cossard, A.; Mitoli, D.; Ferrari, A.M.; Erba, A.; Harrison, N.M. CRYSTALpytools: A Python Infrastructure for the Crystal Code. Comput. Phys. Commun. 2023, 292, 108853. [Google Scholar] [CrossRef]
- Kumar, N.; Saini, H.S.; Nisha, S.; Singh, M.; Kashyap, M.K. Enhanced thermoelectric properties of Ta-doped Half-Heusler ZrNiSn. Mater. Today Proc. 2020, 26, 3478–3481. [Google Scholar] [CrossRef]
- Shastri, S.S.; Pandey, S.K. Thermoelectric properties, efficiency and thermal expansion of ZrNiSn half-Heusler by first-principles calculations. J. Phys. Condens. Matter 2020, 32, 355705. [Google Scholar] [CrossRef]
- Öğüt, S.; Rabe, K.M. Band gap and stability in the ternary intermetallic compounds NiSnM (M = Ti, Zr, Hf): A first-principles study. Phys. Rev. B 1995, 51, 10443–10453. [Google Scholar] [CrossRef]
- Aliev, F.G.; Kozyrkov, V.V.; Moshchalkov, V.V.; Scolozdra, R.V.; Durczewski, K. Narrow band in the intermetallic compounds MNiSn (M = Ti, Zr, Hf). Z. Phys. B Condens. Matter 1990, 80, 353–357. [Google Scholar] [CrossRef]
- Do, D.T.; Mahanti, S.D.; Pulikkoti, J.J. Electronic structure of Zr–Ni–Sn systems: Role of clustering and nanostructures in half-Heusler and Heusler limits. J. Phys. Condens. Matter 2014, 26, 275501. [Google Scholar] [CrossRef]
- Yang, J.; Li, H.; Wu, T.; Zhang, W.; Chen, L.; Yang, J. Evaluation of Half-Heusler Compounds as Thermoelectric Materials Based on the Calculated Electrical Transport Properties. Adv. Funct. Mater. 2008, 18, 2880–2888. [Google Scholar] [CrossRef]
- Aversano, F.; Ferrario, A.; Boldrini, S.; Fanciulli, C.; Baricco, M.; Castellero, A. Thermoelectric Properties of TiNiSn Half Heusler Alloy Obtained by Rapid Solidification and Sintering. J. Mater. Eng. Perform. 2018, 27, 6306–6313. [Google Scholar] [CrossRef]
- Barczak, S.A.; Halpin, J.E.; Buckman, J.; Decourt, R.; Pollet, M.; Smith, R.I.; MacLaren, D.A.; Bos, J.G. Grain-by-Grain Compositional Variations and Interstitial Metals-A New Route toward Achieving High Performance in Half-Heusler Thermoelectrics. ACS Appl. Mater. Interfaces 2018, 10, 4786–4793. [Google Scholar] [CrossRef]
- Kirievsky, K.; Shlimovich, M.; Fuks, D.; Gelbstein, Y. An ab initio study of the thermoelectric enhancement potential in nano-grained TiNiSn. Phys. Chem. Chem. Phys. 2014, 16, 20023–20029. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.E.; Birkel, C.S.; Verma, N.; Miller, V.M.; Miao, M.S.; Stucky, G.D.; Pollock, T.M.; Seshadri, R. Phase stability and property evolution of biphasic Ti–Ni–Sn alloys for use in thermoelectric applications. J. Appl. Phys. 2014, 115, 043720. [Google Scholar] [CrossRef]
- Rogl, G.; Grytsiv, A.; Gürth, M.; Tavassoli, A.; Ebner, C.; Wünschek, A.; Puchegger, S.; Soprunyuk, V.; Schranz, W.; Bauer, E.; et al. Mechanical properties of half-Heusler alloys. Acta Mater. 2016, 107, 178–195. [Google Scholar] [CrossRef]
- Liu, X.; He, J.; Xie, H.; Zhao, X.; Zhu, T. Fabrication and thermoelectric properties of Yb-doped ZrNiSn half-Heusler alloys. Int. J. Smart Nano Mater. 2012, 3, 64–71. [Google Scholar] [CrossRef]
- Yousuf, S.; Bhat, T.M.; Singh, S.; Saleem, Z.; Mir, S.A.; Khandy, S.A.; Seh, A.Q.; Sofi, S.A.; Nabi, M.; Sharma, V.K.; et al. Applicability of semi-classical Boltzmann transport theory in understanding the thermoelectric properties of ZrNiSn and ZrNiPb half-heuslers. AIP Conf. Proc. 2019, 2115, 030420. [Google Scholar]
- Laun, J.; Oliveira, D.V.; Bredow, T. Consistent gaussian basis sets of double- and triple-zeta valence with polarization quality of the fifth period for solid-state calculations. J. Comput. Chem. 2018, 39, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Towler, M.D.; Allan, N.L.; Harrison, N.M.; Saunders, V.R.; Mackrodt, W.C.; Aprà, E. Ab initio study of MnO and NiO. Phys. Rev. B 1994, 50, 5041–5054. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Atom | Charge | Coordination | ||
|---|---|---|---|---|---|
| Mulliken | Hirshfeld | Distance | Neighbors | ||
| SC96-P | Ni | −0.981 | −1.262 | 2.6 | 4 Zr, 4 Sn |
| 4.3 | 12 Ni | ||||
| Zr | +2.743 | +2.746 | |||
| Sn | −1.763 | −1.501 | 2.6 | 4 Ni | |
| 3.0 | 6 Zr | ||||
| SC97-I | Niint | −0.679 | −0.980 | 2.6 | 4 Zr, 4 Sn |
| 3.0 | 6 Ni | ||||
| 5.2 | 8 Ni | ||||
| SC96-A | Niant | −0.643 | −0.943 | 2.6 | 4 Zr, 4 Sn |
| 2.9 | 5 Ni | ||||
| 5.2 | 8 Ni | ||||
| Full-Heusler | Ni | +0.380 | +0.455 | 2.7 | 4 Zr, 4 Sn |
| 3.2 | 6 Ni | ||||
| Zr | +0.707 | +0.402 | |||
| Sn | −1.467 | −1.313 | |||
| SC96-Sb | Ni | −1.050 | −1.281 | ||
| Zr | +2.734 | +2.714 | |||
| Sb | −1.390 | −0.957 | 2.5 | 6 Ni | |
| 3.0 | 8 Zr | ||||
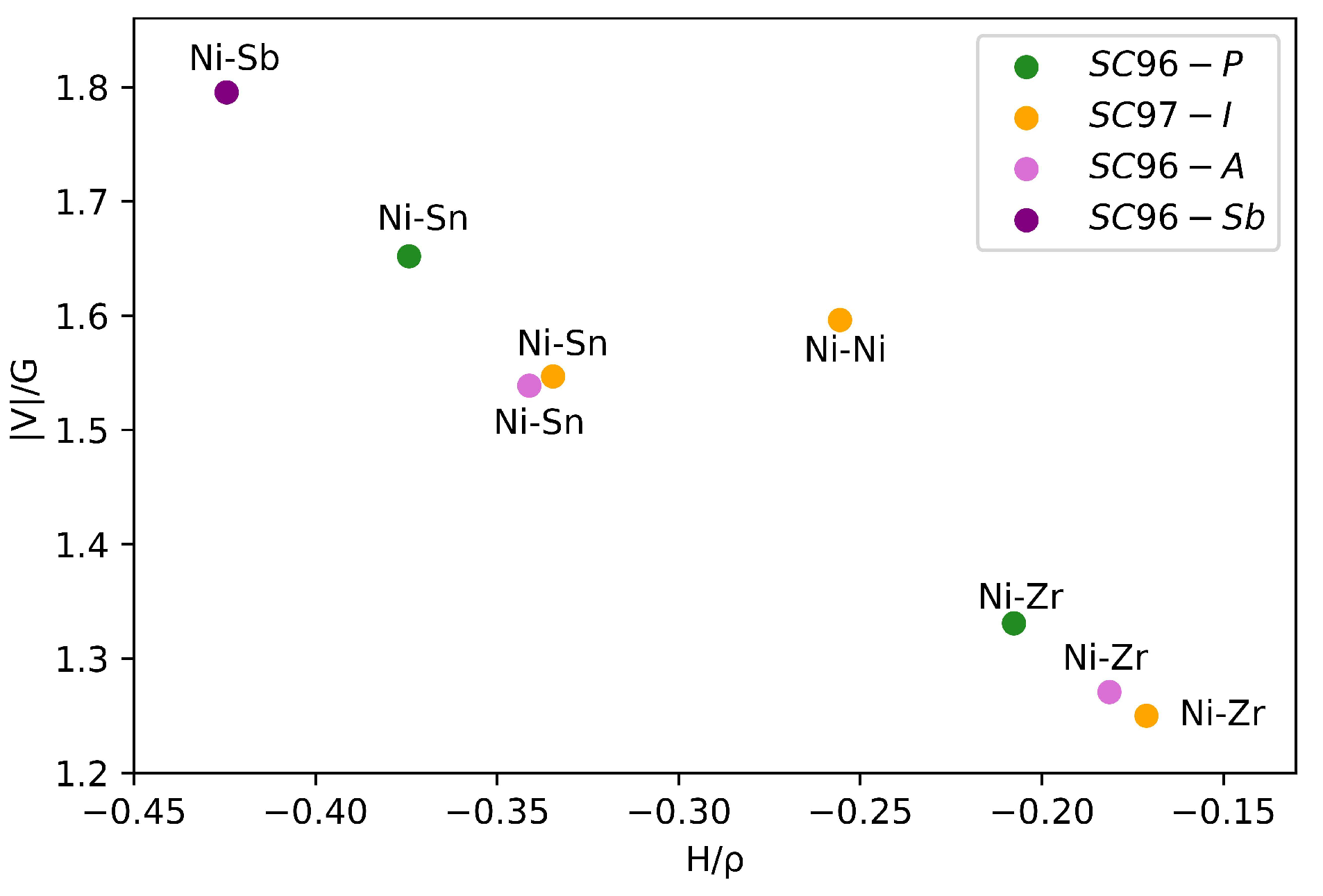
| System | Bond | |||||||
|---|---|---|---|---|---|---|---|---|
| SC96-P | Ni-Sn | 2.579 | 1.186 | 0.058 | 0.046 | 0.091 | −0.3742 | 1.6520 |
| Ni-Zr | 2.579 | 1.276 | 0.050 | 0.084 | 0.166 | −0.2077 | 1.3310 | |
| SC97-I | Niint-Sn | 2.645 | 1.233 | 0.047 | 0.052 | 0.084 | −0.3346 | 1.5468 |
| Niint-Zr | 2.645 | 1.288 | 0.043 | 0.088 | 0.015 | −0.1712 | 1.2501 | |
| Niint-Ni | 3.055 | 1.444 | 0.036 | 0.025 | 0.043 | −0.2556 | 1.5963 | |
| SC96-A | Niant-Sn | 2.600 | 1.212 | 0.050 | 0.058 | 0.095 | −0.3411 | 1.5388 |
| Niant-Zr | 2.610 | 1.281 | 0.045 | 0.087 | 0.163 | −0.1814 | 1.2709 | |
| SC96-Sb | Ni-Sb | 2.530 | 1.164 | 0.063 | 0.027 | 0.082 | −0.4245 | 1.7953 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ascrizzi, E.; Ribaldone, C.; Casassa, S. Crucial Role of Ni Point Defects and Sb Doping for Tailoring the Thermoelectric Properties of ZrNiSn Half-Heusler Alloy: An Ab Initio Study. Materials 2024, 17, 1061. https://doi.org/10.3390/ma17051061
Ascrizzi E, Ribaldone C, Casassa S. Crucial Role of Ni Point Defects and Sb Doping for Tailoring the Thermoelectric Properties of ZrNiSn Half-Heusler Alloy: An Ab Initio Study. Materials. 2024; 17(5):1061. https://doi.org/10.3390/ma17051061
Chicago/Turabian StyleAscrizzi, Eleonora, Chiara Ribaldone, and Silvia Casassa. 2024. "Crucial Role of Ni Point Defects and Sb Doping for Tailoring the Thermoelectric Properties of ZrNiSn Half-Heusler Alloy: An Ab Initio Study" Materials 17, no. 5: 1061. https://doi.org/10.3390/ma17051061
APA StyleAscrizzi, E., Ribaldone, C., & Casassa, S. (2024). Crucial Role of Ni Point Defects and Sb Doping for Tailoring the Thermoelectric Properties of ZrNiSn Half-Heusler Alloy: An Ab Initio Study. Materials, 17(5), 1061. https://doi.org/10.3390/ma17051061