Modification of Collagen Derivatives with Water-Soluble Polymers for the Development of Cross-Linked Hydrogels for Controlled Release


 ,
,  ,
, 

Abstract

1. Introduction
2. Materials and Methods
2.1. Synthesis of P(SSNa-co-GMAx) Copolymers
2.2. Modification of Collagen Hydrolysate with P(SSNa-co-GMAx)
2.3. Modification of Collagen Hydrolysate with Encapsulated Nutrients and Polymeric Materials
2.4. Characterization Techniques
2.4.1. Proton Nuclear Magnetic Resonance (1H NMR)
2.4.2. Thermal Analysis
2.4.3. Size Exclusion Chromatography (SEC)
2.4.4. Attenuated Total Reflection Fourier Transform Infrared Spectroscopy (ATR-FTIR)
2.4.5. Energy Dispersive X-ray analysis-Scanning Electron Microscope (EDAX-SEM)
2.5. Toxicological Assessment of the Copolymer P(SSNa-co-GMA20)
2.5.1. Cell Lines and Cell Culture Conditions
2.5.2. Cell Viability
2.6. Water-Uptake Measurements
2.7. Release Degree of Oxidable Compounds in Water
3. Results
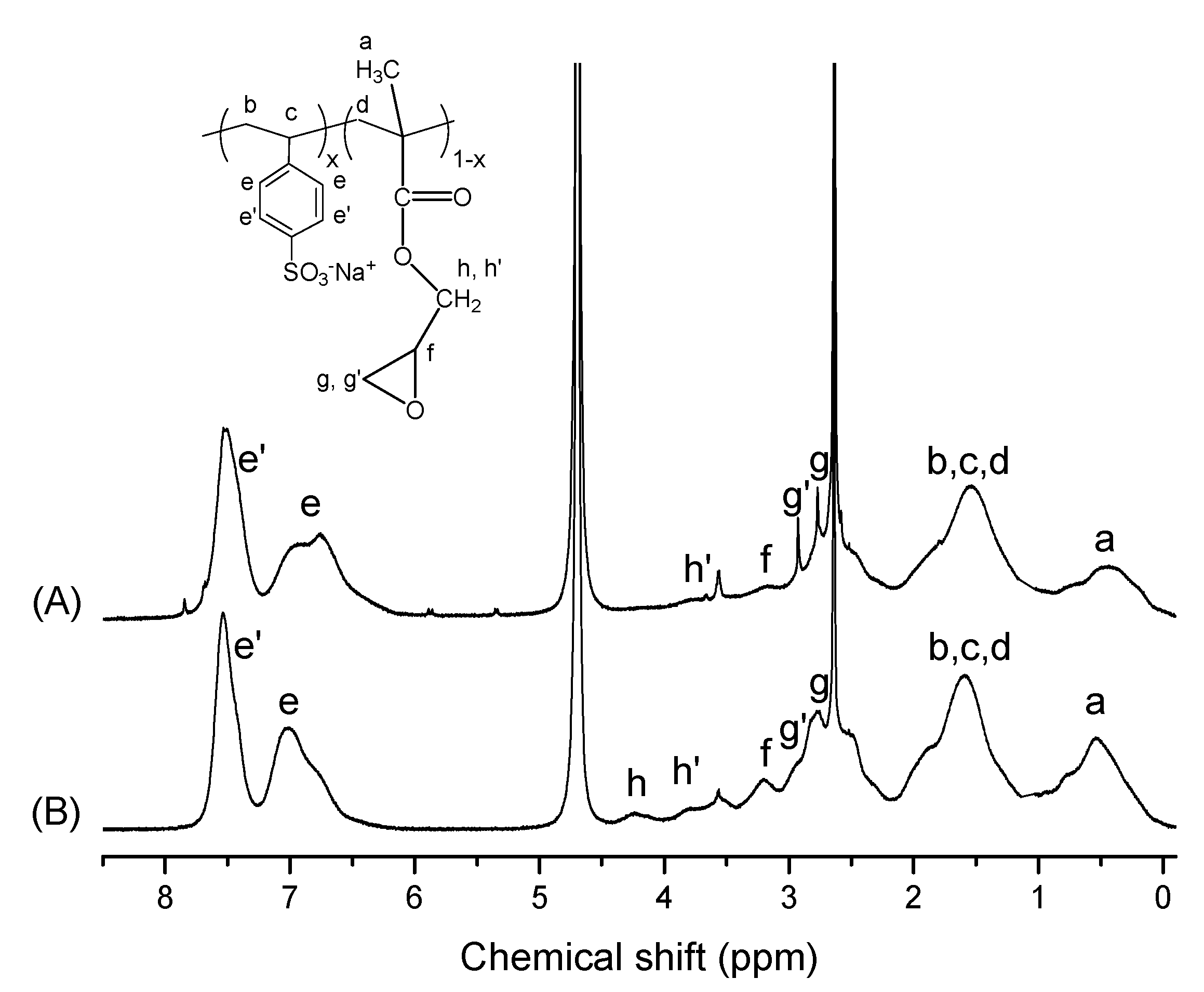
3.1. Synthesis and Characterization of P(SSNa-co-GMAx) Copolymers
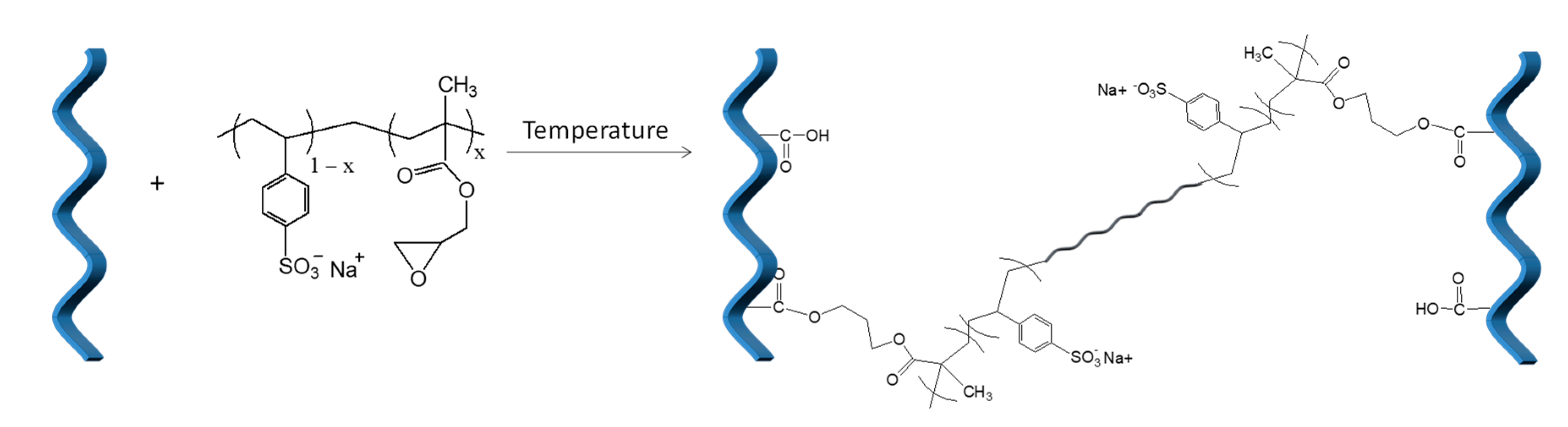
3.2. Preparation of the Collagen-P(SSNa-co-GMAx) Cross-Linked Network Hydrogels
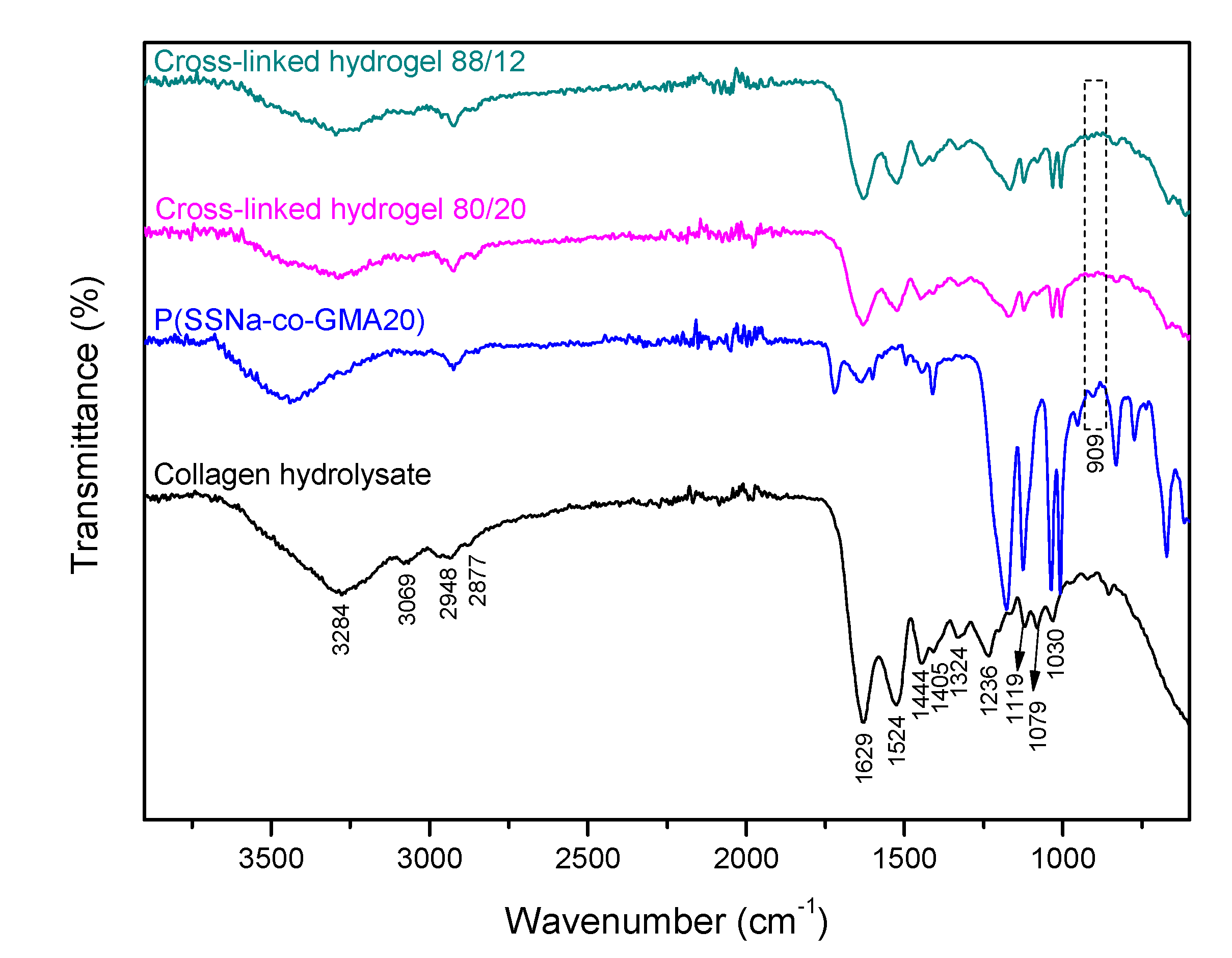
3.3. Characterization of Collagen Hydrogel Fertilizers
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stefan, D.S.; Constantinescu, R.R.; Meghea, A.; Anghel, R.; Stefan, M.; Tudosie, M.S. Obtaining of Biofertilisers Using Pelt Skin Wastes. Rev. Chim.-Bucharest. 2016, 67, 1401–1405. [Google Scholar]
- Dang, X.; Shan, Z. Dust pollution and control with leather waste. Envin. Chem. Lett. 2017, 16, 427–437. [Google Scholar] [CrossRef]
- Dang, X.; Yang, M.; Zhang, B.; Chen, H.; Wang, Y. Recovery and utilization of collagen protein powder extracted from chromium leather scrap waste. Environ. Sci. Pollut. Res. 2019, 26, 7277–7283. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Li, X.; Wang, Y.; Hu, Z.; Wang, R. Characterization of slow-release collagen-g-poly(acrylic acid-co-2-acrylamido-2-methyl-1-propane sulfonic acid)–iron(III) superabsorbent polymer containing fertilizer. J. Appl. Polym. Sci. 2019, 136, 47178. [Google Scholar] [CrossRef]
- Strauss, K.; Chmielewski, J. Advances in the design and higher-order assembly of collagenmimetic peptides for regenerativemedicine. Curr. Opin. Biotechnol. 2017, 46, 34–41. [Google Scholar] [CrossRef]
- Sionkowska, A.; Skrzyński, S.; Śmiechowski, K.; Kołodziejczak, A. The review of versatile application of collagen. Pol. Adv. Technol. 2016, 28, 4–9. [Google Scholar] [CrossRef]
- John Sundar, V.; Gnanamani, A.; Muralidharan, C.; Chandrababu, N.K.; Mandal, A.B. Recovery and utilization of proteinous wastes of leather making: A review. Rev. Environ. Sci. Biotechnol. 2011, 10, 151–163. [Google Scholar] [CrossRef]
- Wang, X.; Chen, K.; Li, W.; Hao, D.; Guo, P. A paper sizing agent based on leather collagen hydrolysates modified by glycol diglycidyl ether and its compound performance. Int. J. Biol. Macromol. 2019, 124, 1205–1212. [Google Scholar] [CrossRef]
- Masilamani, D.; Srinivasan, V.; Ramachandran, R.K.; Gopinath, A.; Madhan, B.; Saravanan, P. Sustainable packaging materials from tannery trimming solid waste: A new paradigm in wealth from waste approaches. J. Clean. Prod. 2017, 164, 885–891. [Google Scholar] [CrossRef]
- Catalina, M.; Cot, J.; Borras, M.; Lapuente, J.D.; González, J.; Balu, A.M.; Luque, R. From Waste to Healing Biopolymers: Biomedical Applications of Bio-Collagenic Materials Extracted from Industrial Leather Residues in Wound Healing. Materials 2013, 6, 1599–1607. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, P.K.; Dandge, P.B. Collagen and collagenolytic proteases: A review. Biocatal. Agric. Biotechnol. 2018, 15, 43–55. [Google Scholar] [CrossRef]
- Ferreira, A.M.; Gentile, P.; Chiono, V.; Ciardelli, G. Collagen for bone tissue regeneration. Acta Biomater. 2012, 8, 3191–3200. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, N.; Hayashi, A.; Yamanaka, K.; Sakiyama, A.; Nakano, A.; Shibata, M. Preparation and Mechanical Properties of Photo-Crosslinked Fish Gelatin/Imogolite Nanofiber Composite Hydrogel. Materials 2012, 5, 2573–2585. [Google Scholar] [CrossRef]
- Liguori, A.; Uranga, J.; Panzavolta, S.; Guerrero, P.; de la Caba, K.; Focarete, M.L. Electrospinning of Fish Gelatin Solution Containing Citric Acid: An Environmentally Friendly Approach to Prepare Crosslinked Gelatin Fibers. Materials 2019, 12, 2808. [Google Scholar] [CrossRef]
- Philippova, O.E.; Rumyantsev, A.M.; Kramarenko, E.Y.; Khokhlov, A.R. New Type of Swelling Behavior upon Gel Ionization: Theory vs. Experiment. Macromolecules 2013, 46, 9359–9367. [Google Scholar] [CrossRef]
- Tanaka, Y.; Gong, J.P.; Osada, Y. Novel Hydrogels with Excellent Mechanical Performance. Prog. Polym. Sci. 2005, 30, 1–9. [Google Scholar] [CrossRef]
- Essawy, H.A.; Ghazy, M.B.M.; El-Hai, F.A.; Mohamed, M.F. Superabsorbent hydrogels via graft polymerization of acrylic acid from chitosan-cellulose hybrid and their potential in controlled release of soil nutrients. Int. J. Biol. Macromol 2016, 89, 144–151. [Google Scholar] [CrossRef]
- Li, X.; Li, Q.; Xu, X.; Su, Y.; Yue, Q.; Gao, B. Characterization, swelling and slow-release properties of a new controlled release fertilizer based on wheat straw cellulose hydrogel. J. Taiwan Inst. Chem. Eng. 2016, 60, 564–572. [Google Scholar] [CrossRef]
- Calcagnile, P.; Sibillano, T.; Giannini, C.; Sannino, A.; Demitri, C. Biodegradable poly(lactic acid)/cellulose-based superabsorbent hydrogel composite material as water and fertilizer reservoir in agricultural applications. J. Appl. Polym. Sci. 2019, 136, 47546. [Google Scholar] [CrossRef]
- Liu, M.; Liang, R.; Zhan, F.; Liu, Z.; Niu, A. Synthesis of a slow-release and superabsorbent nitrogen fertilizer and its properties. Polym. Adv. Technol. 2006, 17, 430–438. [Google Scholar] [CrossRef]
- Guo, M.; Liu, M.; Zhan, F.; Wu, L. Preparation and Properties of a Slow-Release Membrane-Encapsulated Urea Fertilizer with Superabsorbent and Moisture Preservation. Ind. Eng. Chem. Res. 2005, 44, 4206–4211. [Google Scholar] [CrossRef]
- Zainescu, G.; Constantinescu, R.R.; Sirbu, C. Smart hydrogels with collagen structure made of pelt waste. Rev. Chim.-Bucharest. 2017, 68, 393–395. [Google Scholar]
- Lainioti, G.C.; Bounos, G.; Voyiatzis, G.A.; Kallitsis, J.K. Enhanced Water Vapor Transmission through Porous Membranes Based on Melt Blending of Polystyrene Sulfonate with Polyethylene Copolymers and Their CNT Nanocomposites. Polymers 2016, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- Druvari, D.; Koromilas, N.D.; Lainioti, G.C.; Bokias, G.; Vasilopoulos, G.; Vantarakis, A.; Baras, I.; Dourala, N.; Kallitsis, J.K. Polymeric Quaternary Ammonium-Containing Coatings with Potential Dual Contact-Based and Release-Based Antimicrobial Activity. ACS Appl. Mater. Interface 2016, 8, 35593–35605. [Google Scholar] [CrossRef] [PubMed]
- Zainescu, G.A.; Albu, L.; Constantinescu, R.R. Study of collagen hydrogel biodegradability over time. Rev. Chim.-Bucharest. 2018, 69, 101–104. [Google Scholar]
- Aletras, A.J.; Trilivas, I.; Christopoulou, M.E.; Drakouli, S.; Georgakopoulos, C.D.; Pharmakakis, N. UVB-mediated down-regulation of proteasome in cultured human primary pterygium fibroblasts. BMC Ophthalmol. 2018, 18, 328. [Google Scholar] [CrossRef]
- Cory, A.H.; Owen, T.C.; Barltrop, J.A.; Cory, J.G. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991, 3, 207–212. [Google Scholar] [CrossRef]
- Lubkowski, K.; Grzmil, B. Controlled release fertilizers. Pol. J. Chem. Tech. 2007, 9, 81–84. [Google Scholar] [CrossRef]
- Kim, C.H.; Cho, K.Y.; Park, J.K. Grafting of glycidyl methacrylate onto polycaprolactone: Preparation and characterization. Polymer 2001, 42, 5135–5142. [Google Scholar] [CrossRef]
- De, R.; Ray, D.; Das, B. Influence of temperature, added electrolyte, and polymer molecular weight on the counterion condensation phenomenon in aqueous solution of sodium polystyrenesulfonate: A scaling theory approach. RSC Adv. 2015, 5, 54890–54898. [Google Scholar] [CrossRef]
- De, R.; Lee, H.; Das, B. Exploring the interactions in binary mixtures of polyelectrolytes: Influence of mixture composition, concentration, and temperature on counterion condensation. J. Mol. Liq. 2018, 251, 94–99. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, W.; Lu, Z.; Li., M.C. Photografted poly(methyl methacrylate)-based high performance protein microarray for hepatitis B virus biomarker detection in human serum. Med. Chem. Commun. 2010, 1, 132–135. [Google Scholar] [CrossRef]
- Tapeinos, C.; Efthimiadou, E.K.; Boukos, N.; Charitidis, C.A.; Koklioti, M.; Kordas, G. Microspheres as therapeutic delivery agents: Synthesis and biological evaluation of pH responsiveness. J. Mater. Chem. B 2013, 1, 194–203. [Google Scholar] [CrossRef]
- Ozaki, T.; Koto, T.; Nguyen, T.V.; Nakanishi, H.; Norisuye, T.; Tran-Cong-Miyata, Q. The roles of the Trommsdorff–Norrish effect in phase separation of binary polymer mixtures induced by photopolymerization. Polymer 2014, 55, 1809–1816. [Google Scholar] [CrossRef]
- Li, C.; Liao, H.; Zhang, X.; Yu, X.; Tong, M. Preparation of cationic modified collagen extracted from leather wastes and their application in dye flocculation. J. Appl. Polym. Sci. 2017, 134, 45363. [Google Scholar] [CrossRef]
- Vyskočilovα, G.; Ebersbach, M.; Kopeckα, R.; Prokeš, L.; Přνhoda, J. Model study of the leather degradation by oxidation and hydrolysis. Herit. Sci. 2019, 7, 26. [Google Scholar] [CrossRef]
- Sobhanian, P.; Khorram, M.; Hashemi, S.S.; Mohammadi, A. Development of nanofibrous collagen-grafted poly (vinyl alcohol)/gelatin/alginate scaffolds as potential skin substitute. Int. J. Biol. Macromol. 2019, 130, 977–987. [Google Scholar] [CrossRef]
- Saruchi; Kumar, V.; Mittal, H.; Alhassan, S.M. Biodegradable hydrogels of tragacanth gum polysaccharide to improve water retention capacity of soil and environment-friendly controlled release of agrochemicals. Int. J. Biol. Macromol. 2019, 132, 1252–1261. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymers | Code | Feed Composition (mol% GMA) | 1H NMR Composition (mol% GMA) | TGA Composition (mol% GMA) |
|---|---|---|---|---|
| P(SSΝa-co-GMA2) | IN2 | 2 | 2 | 2 |
| P(SSΝa-co-GMA5) | IN5 | 5 | 6 | 6 |
| P(SSΝa-co-GMA20) | IN20a | 20 | 20 | 20 |
| P(SSΝa-co-GMA20) | IN20b | 20 | 15 | 20 |
| P(SSΝa-co-GMA20) | IN20c | 20 | 13 | 20 |
| P(SSΝa-co-GMA20) | IN20d | 20 | 15 | 19 |
| P(SSΝa-co-GMA20) | IN20e | 20 | 12 | 19 |
| P(SSΝa-co-GMA30) | IN30 | 30 | 29 | 30 |
| P(SSΝa-co-GMA40) | IN40 | 40 | 42 | 40 |
| Sample | P(SSNa-co-GMAx) | Solubility in Water | Mn | Mw | DPn |
|---|---|---|---|---|---|
| IN20a | 20 | +++ | 83,000 | 360,000 | 4.3 |
| IN20b | 20 | +++ | 71,000 | 398,000 | 5.6 |
| IN20c | 20 | +++ | 69,900 | 1,100,000 | 15.6 |
| IN20d | 20 | +++ | 63,000 | 371,000 | 5.9 |
| IN30 | 30 | ++ | 41,500 | 147,000 | 3.5 |
| IN40 | 40 | ++ | 30,000 | 372,000 | 12.2 |
| GMA (mol% A) | Residue, R (%) | Water-Uptake Capacity, Q (%) |
|---|---|---|
| 2 | 77 | Cloudy mass |
| 5 | 70 | Cloudy mass |
| 20 | 87 | 476 |
| 30 | 100 | 416 |
| 40 | 71 | 438 |
| Collagen Hydrolysate (wt%) | P(SSNa-co-GMA20) (wt%) | Water-Uptake Capacity, Q (%) |
|---|---|---|
| 99 | 1 | Cloudy mass |
| 93 | 7 | 157% |
| 88 | 12 | 213% |
| 80 | 20 | 476% |
| Thermal Treatment of the Residue | Residue (%) | Water-Uptake Capacity, Q (%) |
|---|---|---|
| 15 min | 82 | 946 |
| 30 min | 79 | 833 |
| 45 min | 82 | 1230 |
| 60 min | 79 | 1300 |
| 90 min | 81 | 1430 |
| 120 min | 82 | 696 |
| 4 h | 81 | 830 |
| 20h | 82 | 476 |
| Thermal Treatment of the Residue | Residue (%) | Water-Uptake Capacity, Q (%) |
|---|---|---|
| 15 min | 81 | 1874 |
| 30 min | 81 | 1834 |
| 45 min | 82 | 1312 |
| 60 min | 83 | 1661 |
| 90 min | 82 | 1058 |
| 120 min | 81 | 996 |
| Characteristics | Sample Code | Standard Used | ||
|---|---|---|---|---|
| CH-Ref | CH-IN | CH-Starch | ||
| Dry Substance, % | 61.22 | 65.80 | 21.59 | SR EN ISO 4684: 2006 |
| Ash, % | 22.36 | 25.18 | 17.23 | SR EN ISO 4047: 2002 |
| Total nitrogen, % | 10.55 | 10.14 | 8.29 | SR ISO 5397: 1996 |
| pH | 7.20 | 6.87 | 6.76 | STAS 8619/3: 1990 |
| Characteristics | Sample Code | Standard Used | ||
|---|---|---|---|---|
| CH-Ref | CH-IN | CH-Starch | ||
| Total N, % | 10.55 | 10.14 | 8.29 | SR ISO 5397: 1996 |
| P2O5, % | 7.67 | 6.75 | 5.54 | SR EN 15959/2012 |
| K2O, % | 10.07 | 8.21 | 10.05 | SR ISO 5397: 1996 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzoumani, I.; Lainioti, G.C.; Aletras, A.J.; Zainescu, G.; Stefan, S.; Meghea, A.; Kallitsis, J.K. Modification of Collagen Derivatives with Water-Soluble Polymers for the Development of Cross-Linked Hydrogels for Controlled Release. Materials 2019, 12, 4067. https://doi.org/10.3390/ma12244067
Tzoumani I, Lainioti GC, Aletras AJ, Zainescu G, Stefan S, Meghea A, Kallitsis JK. Modification of Collagen Derivatives with Water-Soluble Polymers for the Development of Cross-Linked Hydrogels for Controlled Release. Materials. 2019; 12(24):4067. https://doi.org/10.3390/ma12244067
Chicago/Turabian StyleTzoumani, Ioanna, Georgia Ch. Lainioti, Alexios J. Aletras, Gabriel Zainescu, Simina Stefan, Aurelia Meghea, and Joannis K. Kallitsis. 2019. "Modification of Collagen Derivatives with Water-Soluble Polymers for the Development of Cross-Linked Hydrogels for Controlled Release" Materials 12, no. 24: 4067. https://doi.org/10.3390/ma12244067
APA StyleTzoumani, I., Lainioti, G. C., Aletras, A. J., Zainescu, G., Stefan, S., Meghea, A., & Kallitsis, J. K. (2019). Modification of Collagen Derivatives with Water-Soluble Polymers for the Development of Cross-Linked Hydrogels for Controlled Release. Materials, 12(24), 4067. https://doi.org/10.3390/ma12244067