Methodologies in Spectral Tuning of DSSC Chromophores through Rational Design and Chemical-Structure Engineering




Abstract
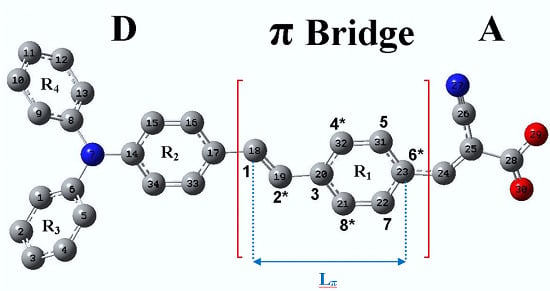
1. Introduction
2. Working Principles of Dye-Sensitized Solar Cells (DSSCs)
3. Computational Methods
4. Results and Discussion
4.1. New Dyes Based on the TA-St-CA Dye—Modification of the π-Conjugated Bridge
4.2. CARD for Enhanced Structures from Carbz-PAHTDDT Dye Sensitizer
4.3. CARD for Enhanced Structures from Zinc Porphyrin Macrocyclic Dyes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jacoby, M. The future of low-cost solar cells. Am. Chem. Soc. 2016, 94, 30–35. [Google Scholar]
- Nazeeruddin, M.K.; Splivallo, R.; Liska, P.; Comte, P.; Grätzel, M. A swift dye uptake procedure for dye sensitized solar cells. Chem. Commun. 2003, 12, 1456–1457. [Google Scholar] [CrossRef] [PubMed]
- Nazeeruddin, M.K.; De Angelis, F.; Fantacci, S.; Selloni, A.; Viscardi, G.; Liska, P.; Ito, S.; Takeru, B.; Grätzel, M. Combined experimental and DFT-TDDFT computational study of photoelectrochemical cell ruthenium sensitizers. J. Am. Chem. Soc. 2005, 127, 16835–16847. [Google Scholar] [CrossRef] [PubMed]
- Philipps, S.; Fraunhofer ISE; Warmuth, W. Photovoltaics Report; Fraunhofer Institute for Solar Energy Systems: Freiburg, Germany, 2018. [Google Scholar]
- Einstein, A. Über einen die Erzeugung und Verwandlung des Lichtes betreffenden heuristischen Gesichtspunkt. Ann. Der Phys. 1905, 322, 132–148. [Google Scholar] [CrossRef]
- Przewoźnik, J.; Kowalik, M.; Kołodziejczyk, A.; Gritzner, G.; Kapusta, C. Magnetic and magnetotransport properties of the (La0.67Pb0.33)(Mn1−xFex)O3 (0≤ x≤ 0.1) compounds. J. Alloys Compd. 2010, 497, 17–23. [Google Scholar] [CrossRef]
- Becquerel, A.E. Memoire sur les Effects d’Electriques Produits Sous l’Influence des Rayons Solaires. C. R. De L’academie Des Sci. 1839, 9, 561–567. [Google Scholar]
- Green, M.A. Third Generation Photovoltaics: Advanced Solar Energy Conversion; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Kay, A.; Rodicio, I.; Humphry-Baker, R.; Müller, E.; Liska, P.; Vlachopoulos, N.; Grätzel, M. Conversion of light to electricity by cis-X2bis (2,2′-bipyridyl-4,4′-dicarboxylate) ruthenium (II) charge-transfer sensitizers (X= Cl-, Br-, I-, CN-, and SCN-) on nanocrystalline titanium dioxide electrodes. J. Am. Chem. Soc. 1993, 115, 6382–6390. [Google Scholar] [CrossRef]
- Grätzel, M. Conversion of sunlight to electric power by nanocrystalline dye-sensitized solar cells. J. Photochem. Photobiol. A Chem. 2004, 164, 3–14. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Pechy, P.; Renouard, T.; Zakeeruddin, S.M.; Humphry-Baker, R.; Comte, P.; Liska, P.; Cevey, L.; Costa, E.; Shklover, V. Engineering of efficient panchromatic sensitizers for nanocrystalline TiO2-based solar cells. J. Am. Chem. Soc. 2001, 123, 1613–1624. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Pechy, P.; Grätzel, M. Efficient panchromatic sensitization of nanocrystallineTiO2 films by a black dye based on atrithiocyanato–ruthenium complex. Chem. Commun. 1997, 18, 1705–1706. [Google Scholar] [CrossRef]
- Chiba, Y.; Islam, A.; Watanabe, Y.; Komiya, R.; Koide, N.; Han, L. Dye-sensitized solar cells with conversion efficiency of 11.1%. Jpn. J. Appl. Phys. 2006, 45, L638. [Google Scholar] [CrossRef]
- Pastore, M.; Mosconi, E.; De Angelis, F.; Grätzel, M. A computational investigation of organic dyes for dye-sensitized solar cells: Benchmark, strategies, and open issues. J. Phys. Chem. C 2010, 114, 7205–7212. [Google Scholar] [CrossRef]
- Li, S.L.; Jiang, K.J.; Shao, K.F.; Yang, L.M. Novel organic dyes for efficient dye-sensitized solar cells. Chem. Commun. 2006, 26, 2792–2794. [Google Scholar] [CrossRef] [PubMed]
- Heredia, D.; Natera, J.; Gervaldo, M.; Otero, L.; Fungo, F.; Lin, C.Y.; Wong, K.T. Spirobifluorene-bridged donor/acceptor dye for organic dye-sensitized solar cells. Org. Lett. 2009, 12, 12–15. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef]
- Mishra, A.; Fischer, M.K.; Bäuerle, P. Metal-free organic dyes for dye-sensitized solar cells: From structure: Property relationships to design rules. Angew. Chem. Int. Ed. 2009, 48, 2474–2499. [Google Scholar] [CrossRef]
- Grätzel, M. Dye-sensitized solar cells. J. Photochem. Photobiol. C Photochem. Rev. 2003, 4, 145–153. [Google Scholar] [CrossRef]
- Peter, L.M. The gratzel cell: Where next? J. Phys. Chem. Lett. 2011, 2, 1861–1867. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.B.; Sun, S.L.; Geng, Y.; Wu, Y.; Su, Z.M. Density functional theory characterization and design of high-performance diarylamine-fluorene dyes with different π spacers for dye-sensitized solar cells. J. Mater. Chem. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Chen, C.H.; Hsu, Y.C.; Chou, H.H.; Thomas, K.J.; Lin, J.T.; Hsu, C.P. Dipolar Compounds Containing Fluorene and a Heteroaromatic Ring as the Conjugating Bridge for High-Performance Dye-Sensitized Solar Cells. Chem. A Eur. J. 2010, 16, 3184–3193. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, N.; Mahon, P.J.; Wang, F. Toward rational design of organic dye sensitized solar cells (DSSCs): An application to the TA-St-CA dye. J. Mol. Graph. Model. 2013, 40, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, N.; Wang, F. First-principles study of Carbz-PAHTDDT dye sensitizer and two Carbz-derived dyes for dye sensitized solar cells. J. Mol. Model. 2014, 20, 2177. [Google Scholar] [CrossRef] [PubMed]
- Arooj, Q.; Wilson, G.; Wang, F. Shifting UV-vis absorption spectrum through rational structural modifications of zinc porphyrin photoactive compounds. RSC Adv. 2016, 6, 15345–15353. [Google Scholar] [CrossRef]
- Liu, Z.; Xiong, D.; Xu, X.; Arooj, Q.; Wang, H.; Yin, L.; Li, W.; Wu, H.; Zhao, Z.; Chen, W. Modulated charge injection in p-type dye-sensitized solar cells using fluorene-based light absorbers. ACS Appl. Mater. Interfaces 2014, 6, 3448–3454. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, W.; Topa, S.; Xu, X.; Zeng, X.; Zhao, Z.; Wang, M.; Chen, W.; Wang, F.; Cheng, Y.-B. Fine tuning of fluorene-based dye structures for high-efficiency p-type dye-sensitized solar cells. ACS Appl. Mater. Interfaces 2014, 6, 10614–10622. [Google Scholar] [CrossRef]
- Roughley, S.D.; Jordan, A.M. The medicinal chemist’s toolbox: An analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef]
- Ooms, F. Molecular modeling and computer aided drug design. Examples of their applications in medicinal chemistry. Curr. Med. Chem. 2000, 7, 141–158. [Google Scholar] [CrossRef]
- Silverman, R.B.; Holladay, M.W. The Organic Chemistry of Drug Design and Drug Action; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Lee, M.W.; Kim, J.Y.; Son, H.J.; Kim, J.Y.; Kim, B.; Kim, H.; Lee, D.K.; Kim, K.; Lee, D.H.; Ko, M.J. Tailoring of energy levels in D-π-A organic dyes via fluorination of acceptor units for efficient dye-sensitized solar cells. Sci. Rep. 2015, 5, 7711. [Google Scholar] [CrossRef]
- Geiger, T.; Kuster, S.; Yum, J.H.; Moon, S.J.; Nazeeruddin, M.K.; Grätzel, M.; Nüesch, F. Molecular design of unsymmetrical squaraine dyes for high efficiency conversion of low energy photons into electrons using TiO2 nanocrystalline films. Adv. Funct. Mater. 2009, 19, 2720–2727. [Google Scholar] [CrossRef]
- Dewar, M.J.S. 478. Colour and constitution. Part I. Basic dyes. J. Chem. Soc. (Resumed) 1950, 2329–2334. [Google Scholar] [CrossRef]
- Tributsch, H.; Gerischer, H. The use of semiconductor electrodes in the study of photochemical reactions. Ber. Der Bunsenges. Für Phys. Chem. 1969, 73, 850–854. [Google Scholar]
- Hauffe, K.; Danzmann, H.; Pusch, H.; Range, J.; Volz, H. New experiments on the sensitization of zinc oxide by means of the electrochemical cell technique. J. Electrochem. Soc. 1970, 117, 993–999. [Google Scholar] [CrossRef]
- Chen, S.N.; Deb, S.K.; Witzke, H. Dye-Titanium Dioxide Photogalvanic Cell. U.S. Patent 4,080,488, 21 March 1978. [Google Scholar]
- Hardin, B.; Snaith, H.; McGehee, M. The renaissance of dye-sensitized solar cells. Nat. Photonics 2012, 6, 162. [Google Scholar] [CrossRef]
- Hamann, T.W.; Jensen, R.A.; Martinson, A.B.; Van Ryswyk, H.; Hupp, J.T. Advancing beyond current generation dye-sensitized solar cells. Energy Environ. Sci. 2008, 1, 66–78. [Google Scholar] [CrossRef]
- Kroon, J.; Bakker, N.; Smit, H.; Liska, P.; Thampi, K.; Wang, P.; Zakeeruddin, S.; Grätzel, M.; Hinsch, A.; Hore, S. Nanocrystalline dye-sensitized solar cells having maximum performance. Prog. Photovolt. Res. Appl. 2007, 15, 1–18. [Google Scholar] [CrossRef]
- Yella, A.; Lee, H.-W.; Tsao, H.N.; Yi, C.; Chandiran, A.K.; Nazeeruddin, M.K.; Diau, E.W.-G.; Yeh, C.-Y.; Zakeeruddin, S.M.; Grätzel, M. Porphyrin-sensitized solar cells with cobalt (II/III)–based redox electrolyte exceed 12 percent efficiency. Science 2011, 334, 629–634. [Google Scholar] [CrossRef]
- Burschka, J.; Pellet, N.; Moon, S.J.; Humphry-Baker, R.; Gao, P.; Nazeeruddin, M.K.; Grätzel, M. Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature 2013, 499, 316. [Google Scholar] [CrossRef]
- Snaith, H.J. Perovskites: The emergence of a new era for low-cost, high-efficiency solar cells. J. Phys. Chem. Lett. 2013, 4, 3623–3630. [Google Scholar] [CrossRef]
- Wang, J.T.W.; Ball, J.M.; Barea, E.M.; Abate, A.; Alexander-Webber, J.A.; Huang, J.; Saliba, M.; Mora-Sero, I.; Bisquert, J.; Snaith, H.J. Low-temperature processed electron collection layers of graphene/TiO2 nanocomposites in thin film perovskite solar cells. Nano Lett. 2013, 14, 724–730. [Google Scholar] [CrossRef]
- Kamat, P.V. Evolution of Perovskite Photovoltaics and Decrease in Energy Payback Time; ACS Publications: Washington, DC, USA, 2013. [Google Scholar]
- Niu, G.; Guo, X.; Wang, L. Review of recent progress in chemical stability of perovskite solar cells. J. Mater. Chem. A 2015, 3, 8970–8980. [Google Scholar] [CrossRef]
- Grätzel, M. The light and shade of perovskite solar cells. Nat. Mater. 2014, 13, 838. [Google Scholar] [CrossRef]
- Singh, V.K.; Kanaparthi, R.K.; Giribabu, L. Emerging molecular design strategies of unsymmetrical phthalocyanines for dye-sensitized solar cell applications. RSC Adv. 2014, 4, 6970–6984. [Google Scholar] [CrossRef]
- Boschloo, G.; Hagfeldt, A. Characteristics of the iodide/triiodide redox mediator in dye-sensitized solar cells. Acc. Chem. Res. 2009, 42, 1819–1826. [Google Scholar] [CrossRef]
- Qin, C.; Peng, W.; Zhang, K.; Islam, A.; Han, L. A novel organic sensitizer combined with a cobalt complex redox shuttle for dye-sensitized solar cells. Org. Lett. 2012, 14, 2532–2535. [Google Scholar] [CrossRef]
- Nusbaumer, H.; Moser, J.E.; Zakeeruddin, S.M.; Nazeeruddin, M.K.; Grätzel, M. CoII(dbbip)22+ Complex Rivals Tri-iodide/Iodide Redox Mediator in Dye-Sensitized Photovoltaic Cells. J. Phys. Chem. B 2001, 105, 10461–10464. [Google Scholar] [CrossRef]
- Geetha, M.; Kumar, P.S.; Vasudevan, K.; Prakasam, A.; Meenakshi, G.; Anbarasan, P. Molecular modeling of 3, 4-pyridinedicarbonitrile dye sensitizer for solar cells using quantum chemical calculations. J. Saudi Chem. Soc. 2010, 14, 399–407. [Google Scholar] [CrossRef]
- Labat, F.d.; Le Bahers, T.; Ciofini, I.; Adamo, C. First-principles modeling of dye-sensitized solar cells: Challenges and perspectives. Acc. Chem. Res. 2012, 45, 1268–1277. [Google Scholar] [CrossRef]
- Ding, W.L.; Wang, D.M.; Geng, Z.Y.; Zhao, X.L.; Xu, W.B. Density functional theory characterization and verification of high-performance indoline dyes with D–A–π–A architecture for dye-sensitized solar cells. Dye. Pigment. 2013, 98, 125–135. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Ma, N.N.; Yan, L.K.; Su, Z.M. Theoretical studies on organoimido-substituted hexamolybdates dyes for dye-sensitized solar cells (DSSC). Dye. Pigment. 2013, 99, 440–446. [Google Scholar] [CrossRef]
- Oprea, C.I.; Panait, P.; Cimpoesu, F.; Ferbinteanu, M.; Gîrţu, M.A. Density functional theory (DFT) study of coumarin-based dyes adsorbed on TiO2 nanoclusters—Applications to dye-sensitized solar cells. Materials 2013, 6, 2372–2392. [Google Scholar] [CrossRef]
- Pastore, M.; De Angelis, F. Modeling Materials and Processes in Dye-Sensitized Solar Cells: Understanding the Mechanism, Improving the Efficiency. In Multiscale Modelling of Organic and Hybrid Photovoltaics; Springer: Berlin/Heidelberg, Germany, 2013; pp. 151–236. [Google Scholar]
- Meng, S.; Kaxiras, E.; Nazeeruddin, M.K.; Grätzel, M. Design of dye acceptors for photovoltaics from first-principles calculations. J. Phys. Chem. C 2011, 115, 9276–9282. [Google Scholar] [CrossRef]
- Fan, W.; Tan, D.; Deng, W.Q. Acene-Modified Triphenylamine Dyes for Dye-Sensitized Solar Cells: A Computational Study. ChemPhysChem 2012, 13, 2051–2060. [Google Scholar] [CrossRef]
- Gu, X.; Zhou, L.; Li, Y.; Sun, Q.; Jena, P. Design of new metal-free dyes for dye-sensitized solar cells: A first-principles study. Phys. Lett. A 2012, 376, 2595–2599. [Google Scholar] [CrossRef]
- Zhang, J.; Kan, Y.H.; Li, H.B.; Geng, Y.; Wu, Y.; Su, Z.M. How to design proper π-spacer order of the D-π-A dyes for DSSCs? A density functional response. Dye Pigment 2012, 95, 313–321. [Google Scholar] [CrossRef]
- Sánchez-de-Armas, R.; San-Miguel, M.A.; Oviedo, J.; Sanz, J.F. Molecular modification of coumarin dyes for more efficient dye sensitized solar cells. J. Chem. Phys. 2012, 136, 05B615. [Google Scholar] [CrossRef]
- Sánchez-de-Armas, R.; San Miguel, M.Á.; Oviedo, J.; Sanz, J.F. Coumarin derivatives for dye sensitized solar cells: A TD-DFT study. Phys. Chem. Chem. Phys. 2012, 14, 225–233. [Google Scholar] [CrossRef]
- Yang, L.; Guo, L.; Chen, Q.; Sun, H.; Liu, J.; Zhang, X.; Pan, X.; Dai, S. Theoretical design and screening of panchromatic phthalocyanine sensitizers derived from TT1 for dye-sensitized solar cells. J. Mol. Graph. Model. 2012, 34, 1–9. [Google Scholar] [CrossRef]
- Wang, J.; Cong, S.; Wen, S.; Yan, L.; Su, Z. A rational design for dye sensitizer: Density functional theory study on the electronic absorption spectra of organoimido-substituted hexamolybdates. J. Phys. Chem. C 2013, 117, 2245–2251. [Google Scholar] [CrossRef]
- Feng, J.; Jiao, Y.; Ma, W.; Nazeeruddin, M.K.; Grätzel, M.; Meng, S. First principles design of dye molecules with ullazine donor for dye sensitized solar cells. J. Phys. Chem. C 2013, 117, 3772–3778. [Google Scholar] [CrossRef]
- Tarsang, R.; Promarak, V.; Sudyoadsuk, T.; Namuangruk, S.; Jungsuttiwong, S. Tuning the electron donating ability in the triphenylamine-based D-π-A architecture for highly efficient dye-sensitized solar cells. J. Photochem. Photobiol. A Chem. 2014, 273, 8–16. [Google Scholar] [CrossRef]
- Dong, Z.; Ren, H.; Hessel, C.M.; Wang, J.; Yu, R.; Jin, Q.; Yang, M.; Hu, Z.; Chen, Y.; Tang, Z. Quintuple-shelled SnO2 hollow microspheres with superior light scattering for high-performance dye-sensitized solar cells. Adv. Mater. 2014, 26, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; Boschloo, G.; Mukhtar, E.; Hagfeldt, A. Ultrafast studies of electron injection in Ru dye sensitized SnO2 nanocrystalline thin film. Int. J. Photoenergy 2002, 4, 17–20. [Google Scholar] [CrossRef]
- Gubbala, S.; Russell, H.B.; Shah, H.; Deb, B.; Jasinski, J.; Rypkema, H.; Sunkara, M.K. Surface properties of SnO2 nanowires for enhanced performance with dye-sensitized solar cells. Energy Environ. Sci. 2009, 2, 1302–1309. [Google Scholar] [CrossRef]
- Sadoughi, G.; Sivaram, V.; Gunning, R.; Docampo, P.; Bruder, I.; Pschirer, N.; Irajizad, A.; Snaith, H.J. Enhanced electronic contacts in SnO2–dye–P3HT based solid state dye sensitized solar cells. Phys. Chem. Chem. Phys. 2013, 15, 2075–2080. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Liang, J.; Sumathy, K. Review on dye-sensitized solar cells (DSSCs): Fundamental concepts and novel materials. Renew. Sustain. Energy Rev. 2012, 16, 5848–5860. [Google Scholar] [CrossRef]
- Quintana, M.; Edvinsson, T.; Hagfeldt, A.; Boschloo, G. Comparison of dye-sensitized ZnO and TiO2 solar cells: Studies of charge transport and carrier lifetime. J. Phys. Chem. C 2007, 111, 1035–1041. [Google Scholar] [CrossRef]
- Saito, M.; Fujihara, S. Large photocurrent generation in dye-sensitized ZnO solar cells. Energy Environ. Sci. 2008, 1, 280–283. [Google Scholar] [CrossRef]
- Ou, J.Z.; Rani, R.A.; Ham, M.H.; Field, M.R.; Zhang, Y.; Zheng, H.; Reece, P.; Zhuiykov, S.; Sriram, S.; Bhaskaran, M. Elevated temperature anodized Nb2O5: A photoanode material with exceptionally large photoconversion efficiencies. ACS Nano 2012, 6, 4045–4053. [Google Scholar] [CrossRef]
- Ghosh, R.; Brennaman, M.; Uher, T.; Ok, M.; Samulski, E.; McNeil, L.; Meyer, T.; Lopez, R.; Nb, N. O5 photoanodes for dye-sensitized solar cells by pulsed laser deposition. ACS Appl. Mater. Interfaces 2011, 3, 3929. [Google Scholar] [CrossRef]
- Le Viet, A.; Jose, R.; Reddy, M.; Chowdari, B.; Ramakrishna, S. Nb2O5 photoelectrodes for dye-sensitized solar cells: Choice of the polymorph. J. Phys. Chem. C 2010, 114, 21795–21800. [Google Scholar] [CrossRef]
- Lee, J.J.; Rahman, M.M.; Sarker, S.; Nath, N.D.; Ahammad, A.S.; Lee, J.K. Metal Oxides and their Composites for the Photoelectrode of Dye Sensitized Solar Cells. In Advances in Composite Materials for Medicine and Nanotechnology; InTech: Rotterdam, The Netherlands, 2011. [Google Scholar]
- Li, C.T.; Lee, C.P.; Lee, C.T.; Li, S.R.; Sun, S.S.; Ho, K.C. Iodide-Free Ionic Liquid with Dual Redox Couples for Dye-Sensitized Solar Cells with High Open-Circuit Voltage. ChemSusChem 2015, 8, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.C.D.; Lee, H.J.; Choi, W.Y.; Lee, J.J. Electrochemical approach to enhance the open-circuit voltage (Voc) of dye-sensitized solar cells (DSSCs). Electrochim. Acta 2013, 109, 39–45. [Google Scholar] [CrossRef]
- Rani, S.; Suri, P.; Mehra, R.M. Mechanism of charge recombination and IPCE in ZnO dye-sensitized solar cells having I−/I and Br−/Br redox couple. Prog. Photovolt. Res. Appl. 2011, 19, 180–186. [Google Scholar] [CrossRef]
- Kato, F.; Kikuchi, A.; Okuyama, T.; Oyaizu, K.; Nishide, H. Nitroxide Radicals as Highly Reactive Redox Mediators in Dye-Sensitized Solar Cells. Angew. Chem. 2012, 124, 10324–10327. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, C.; Lee, Y.M.; Cho, K.Y.; Choi, J.W.; Park, J.K. Thiophene–nitroxide radical as a novel combination of sensitizer–redox mediator for dye-sensitized solar cells. J. Solid State Electrochem. 2012, 16, 657–663. [Google Scholar] [CrossRef]
- Gryn’Ova, G.; Barakat, J.M.; Blinco, J.P.; Bottle, S.E.; Coote, M.L. Computational Design of Cyclic Nitroxides as Efficient Redox Mediators for Dye-Sensitized Solar Cells. Chem. A Eur. J. 2012, 18, 7582–7593. [Google Scholar] [CrossRef]
- Li, D.; Li, H.; Luo, Y.; Li, K.; Meng, Q.; Armand, M.; Chen, L. Non-Corrosive, Non-Absorbing Organic Redox Couple for Dye-Sensitized Solar Cells. Adv. Funct. Mater. 2010, 20, 3358–3365. [Google Scholar] [CrossRef]
- Tian, H.; Jiang, X.; Yu, Z.; Kloo, L.; Hagfeldt, A.; Sun, L. Efficient organic-dye-sensitized solar cells based on an iodine-free electrolyte. Angew. Chem. (Int. Ed. Engl.) 2010, 49, 7328–7331. [Google Scholar] [CrossRef]
- Daeneke, T.; Mozer, A.J.; Kwon, T.-H.; Duffy, N.W.; Holmes, A.B.; Bach, U.; Spiccia, L. Dye regeneration and charge recombination in dye-sensitized solar cells with ferrocene derivatives as redox mediators. Energy Environ. Sci. 2012, 5, 7090–7099. [Google Scholar] [CrossRef]
- Daeneke, T.; Kwon, T.H.; Holmes, A.B.; Duffy, N.W.; Bach, U.; Spiccia, L. High-efficiency dye-sensitized solar cells with ferrocene-based electrolytes. Nat. Chem. 2011, 3, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Hamann, T.W.; Farha, O.K.; Hupp, J.T. Outer-Sphere Redox Couples as Shuttles in Dye-Sensitized Solar Cells. Performance Enhancement Based on Photoelectrode Modification via Atomic Layer Deposition. J. Phys. Chem. C 2008, 112, 19756–19764. [Google Scholar] [CrossRef]
- Bai, Y.; Yu, Q.; Cai, N.; Wang, Y.; Zhang, M.; Wang, P. High-efficiency organic dye-sensitized mesoscopic solar cells with a copper redox shuttle. Chem. Commun. (Camb. Engl.) 2011, 47, 4376–4378. [Google Scholar] [CrossRef]
- Ye, M.; Wen, X.; Wang, M.; Iocozzia, J.; Zhang, N.; Lin, C.; Lin, Z. Recent advances in dye-sensitized solar cells: From photoanodes, sensitizers and electrolytes to counter electrodes. Mater. Today 2015, 18, 155–162. [Google Scholar] [CrossRef]
- Yum, J.-H.; Baranoff, E.; Kessler, F.; Moehl, T.; Ahmad, S.; Bessho, T.; Marchioro, A.; Ghadiri, E.; Moser, J.-E.; Yi, C.; et al. A cobalt complex redox shuttle for dye-sensitized solar cells with high open-circuit potentials. Nat. Commun. 2012, 3, 631. [Google Scholar] [CrossRef] [PubMed]
- Spokoyny, A.M.; Li, T.C.; Farha, O.K.; Machan, C.W.; She, C.; Stern, C.L.; Marks, T.J.; Hupp, J.T.; Mirkin, C.A. Electronic tuning of nickel-based bis(dicarbollide) redox shuttles in dye-sensitized solar cells. Angew. Chem. (Int. Ed. Engl.) 2010, 49, 5339–5343. [Google Scholar] [CrossRef]
- Li, T.C.; Spokoyny, A.M.; She, C.; Farha, O.K.; Mirkin, C.A.; Marks, T.J.; Hupp, J.T. Ni(III)/(IV) bis(dicarbollide) as a fast, noncorrosive redox shuttle for dye-sensitized solar cells. J. Am. Chem. Soc. 2010, 132, 4580–4582. [Google Scholar] [CrossRef]
- Singh, R.; Kumar, S.; Bedi, R.K.; Saxena, V.; Aswal, D.K.; Mahajan, A. Optimization of / Ni2+Ni3+ ratio in reduced graphene oxide/nickel oxide nanohybrids for platinum free dye sensitized solar cells. J. Phys. Chem. Solids 2018, 123, 191–197. [Google Scholar] [CrossRef]
- Yanagida, S.; Yu, Y.; Manseki, K. Iodine/Iodide-Free Dye-Sensitized Solar Cells. Acc. Chem. Res. 2009, 42, 1827–1838. [Google Scholar] [CrossRef]
- Hamann, T.W.; Ondersma, J.W. Dye-sensitized solar cell redox shuttles. Energy Environ. Sci. 2011, 4, 370–381. [Google Scholar] [CrossRef]
- Rong, Y.; Liu, G.; Wang, H.; Li, X.; Han, H. Monolithic all-solid-state dye-sensitized solar cells. Front. Optoelectron. 2013, 6, 359–372. [Google Scholar] [CrossRef]
- Balanay, M.P.; Kim, D.H. DFT/TD-DFT molecular design of porphyrin analogues for use in dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2008, 10, 5121–5127. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.-J.; Song, C.; Ren, X.F. Theoretical study of zinc porphyrin-based dyes for dye-sensitized solar cells. J. Photochem. Photobiol. A Chem. 2017, 333, 200–207. [Google Scholar] [CrossRef]
- Teng, C.; Yang, X.; Yang, C.; Li, S.; Cheng, M.; Hagfeldt, A.; Sun, L. Molecular Design of Anthracene-Bridged Metal-Free Organic Dyes for Efficient Dye-Sensitized Solar Cells. J. Phys. Chem. C 2010, 114, 9101–9110. [Google Scholar] [CrossRef]
- Selopal, G.S.; Wu, H.P.; Lu, J.; Chang, Y.C.; Wang, M.; Vomiero, A.; Concina, I.; Diau, E.W.G. Metal-free organic dyes for TiO2 and ZnO dye-sensitized solar cells. Sci. Rep. 2016, 6, 18756. [Google Scholar] [CrossRef]
- Nagarajan, B.; Kushwaha, S.; Elumalai, R.; Mandal, S.; Ramanujam, K.; Raghavachari, D. Novel ethynyl-pyrene substituted phenothiazine based metal free organic dyes in DSSC with 12% conversion efficiency. J. Mater. Chem. A 2017, 5, 10289–10300. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Gryff-Keller, A.; Szczeciński, P. An efficient DFT method of predicting the one-, two- and three-bond indirect spin–spin coupling constants involving a fluorine nucleus in fluoroalkanes. RSC Adv. 2016, 6, 82783–82792. [Google Scholar] [CrossRef][Green Version]
- Plumley, J.A.; Dannenberg, J.J. A Comparison of the Behavior of Functional/Basis Set Combinations for Hydrogen-Bonding in the Water Dimer with Emphasis on Basis Set Superposition Error. J. Comput. Chem. 2011, 32, 1519–1527. [Google Scholar] [CrossRef]
- Tao, J.; Tretiak, S.; Zhu, J.X. Absorption spectra of blue-light-emitting oligoquinolines from time-dependent density functional theory. J. Phys. Chem. B 2008, 112, 13701–13710. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Accurate excitation energies from time-dependent density functional theory: Assessing the PBE0 model for organic free radicals. Chem. Phys. Lett. 1999, 314, 152–157. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. TD-DFT Performance for the Visible Absorption Spectra of Organic Dyes: Conventional versus Long-Range Hybrids. J. Chem. Theory Comput. 2008, 4, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Chibani, S.; Charaf-Eddin, A.; Le Guennic, B.; Jacquemin, D. Boranil and Related NBO Dyes: Insights From Theory. J. Chem. Theory Comput. 2013, 9, 3127–3135. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2009; citeulike-article-id:9096580. [Google Scholar]
- Hwang, S.; Lee, J.H.; Park, C.; Lee, H.; Kim, C.; Park, C.; Lee, M.H.; Lee, W.; Park, J.; Kim, K.; et al. A highly efficient organic sensitizer for dye-sensitized solar cells. Chem. Commun. (Camb. Engl.) 2007, 4887–4889. [Google Scholar] [CrossRef]
- Hung, J.; Liang, W.; Luo, J.; Shi, Z.; Jen, A.K.Y.; Li, X. Rational Design Using Dewar’s Rules for Enhancing the First Hyperpolarizability of Nonlinear Optical Chromophores. J. Phys. Chem. C 2010, 114, 22284–22288. [Google Scholar] [CrossRef]
- Ruiz-Morales, Y. HOMO−LUMO Gap as an Index of Molecular Size and Structure for Polycyclic Aromatic Hydrocarbons (PAHs) and Asphaltenes: A Theoretical Study. I. J. Phys. Chem. A 2002, 106, 11283–11308. [Google Scholar] [CrossRef]
- Zhang, C.R.; Liu, Z.J.; Chen, Y.H.; Chen, H.S.; Wu, Y.Z.; Feng, W.; Wang, D.B. DFT and TD-DFT study on structure and properties of organic dye sensitizer TA-St-CA. Curr. Appl. Phys. 2010, 10, 77–83. [Google Scholar] [CrossRef]
- Kwon, T.H.; Armel, V.; Nattestad, A.; Macfarlane, D.R.; Bach, U.; Lind, S.J.; Gordon, K.C.; Tang, W.; Jones, D.J.; Holmes, A.B. Dithienothiophene (DTT)-based dyes for dye-sensitized solar cells: Synthesis of 2,6-dibromo-DTT. J. Org. Chem. 2011, 76, 4088–4093. [Google Scholar] [CrossRef] [PubMed]
- Koumura, N.; Wang, Z.S.; Mori, S.; Miyashita, M.; Suzuki, E.; Hara, K. Alkyl-functionalized organic dyes for efficient molecular photovoltaics. J. Am. Chem. Soc. 2006, 128, 14256–14257. [Google Scholar] [CrossRef] [PubMed]
- Dreuw, A.; Head-Gordon, M. Failure of Time-Dependent Density Functional Theory for Long-Range Charge-Transfer Excited States: the Zincbacteriochlorin−Bacteriochlorin and Bacteriochlorophyll−Spheroidene Complexes. J. Am. Chem. Soc. 2004, 126, 4007–4016. [Google Scholar] [CrossRef] [PubMed]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef]
- Mohammadi, N. Computational Study of Compounds with Application in Dye Sensitized Solar Cells; Swinburne University of Technology: Melbourne, Australia, 2014. [Google Scholar]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, M.K.; Gratzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef]
- Schmidt-Mende, L.; Campbell, W.M.; Wang, Q.; Jolley, K.W.; Officer, D.L.; Nazeeruddin, M.K.; Gratzel, M. Zn-porphyrin-sensitized nanocrystalline TiO2 heterojunction photovoltaic cells. ChemPhysChem 2005, 6, 1253–1258. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Gouterman, M. Spectra of porphyrins. J. Mol. Spectrosc. 1961, 6, 138–163. [Google Scholar] [CrossRef]
- Imahori, H.; Umeyama, T.; Ito, S. Large π-Aromatic Molecules as Potential Sensitizers for Highly Efficient Dye-Sensitized Solar Cells. Acc. Chem. Res. 2009, 42, 1809–1818. [Google Scholar] [CrossRef]
- Han, L.H.; Zhang, C.R.; Zhe, J.W.; Jin, N.Z.; Shen, Y.L.; Wang, W.; Gong, J.J.; Chen, Y.H.; Liu, Z.J. Understanding the Electronic Structures and Absorption Properties of Porphyrin Sensitizers YD2 and YD2-o-C8 for Dye-Sensitized Solar Cells. Int. J. Mol. Sci. 2013, 14, 20171–20188. [Google Scholar] [CrossRef] [PubMed]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Campbell, W.M.; Burrell, A.K.; Officer, D.L.; Jolley, K.W. Porphyrins as light harvesters in the dye-sensitised TiO2 solar cell. Coord. Chem. Rev. 2004, 248, 1363–1379. [Google Scholar] [CrossRef]
- Ding, Y.; Li, X.; Hill, J.P.; Ariga, K.; Ågren, H.; Andréasson, J.; Zhu, W.; Tian, H.; Xie, Y. Acid/Base Switching of the Tautomerism and Conformation of a Dioxoporphyrin for Integrated Binary Subtraction. Chem. A Eur. J. 2014, 20, 12910–12916. [Google Scholar] [CrossRef] [PubMed]
- Allegrucci, A.; Lewcenko, N.A.; Mozer, A.J.; Dennany, L.; Wagner, P.; Officer, D.L.; Sunahara, K.; Mori, S.; Spiccia, L. Improved performance of porphyrin-based dye sensitised solar cells by phosphinic acid surface treatment. Energy Environ. Sci. 2009, 2, 1069–1073. [Google Scholar] [CrossRef][Green Version]
- Xiang, N.; Huang, X.; Feng, X.; Liu, Y.; Zhao, B.; Deng, L.; Shen, P.; Fei, J.; Tan, S. The structural modification of thiophene-linked porphyrin sensitizers for dye-sensitized solar cells. Dye Pigment 2011, 88, 75–83. [Google Scholar] [CrossRef]
- Huang, X.; Nakanishi, K.; Berova, N. Porphyrins and metalloporphyrins: Versatile circular dichroic reporter groups for structural studies. Chirality 2000, 12, 237–255. [Google Scholar] [CrossRef]
- Wu, S.L.; Lu, H.P.; Yu, H.T.; Chuang, S.H.; Chiu, C.L.; Lee, C.W.; Diau, E.W.G.; Yeh, C.Y. Design and characterization of porphyrin sensitizers with a push-pull framework for highly efficient dye-sensitized solar cells. Energy Environ. Sci. 2010, 3, 949–955. [Google Scholar] [CrossRef]
- Hashimoto, T.; Choe, Y.K.; Nakano, H.; Hirao, K. Theoretical Study of the Q and B Bands of Free-Base, Magnesium, and Zinc Porphyrins, and Their Derivatives. J. Phys. Chem. A 1999, 103, 1894–1904. [Google Scholar] [CrossRef]
- Arooj, Q.; Wang, F. Switching on optical properties of D-π-A DSSC sensitizers from π-spacers towards machine learning. Sol. Energy 2019, 188, 1189–1200. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | Structure * | Substitution Type | Chemical Fragment | Position |
|---|---|---|---|---|
| ED-I |  | Electron Donating | NH2 | 2* |
| ED-II |  | 4* | ||
| ED-III |  | 8* | ||
| ED-IV |  | N(CH3)2 | 2* | |
| ED-V |  | 4* | ||
| ED-VI |  | 8* | ||
| EW-I |  | Electron Withdrawing | CN | 1 |
| EW-II |  | 5 | ||
| EW-III |  | 7 |
| Carbz-PAHTDTT (S9) | S9-D1 | S9-D2 | |||||
|---|---|---|---|---|---|---|---|
| Method (a) | TD-B3LYP | TD-BHandH | Exp.(b) | TD-BHandH | Δλ (c) | TD-BHandH | Δλ (d) |
| λ1 (nm) | 668 | 490 | 491 | 662 | 172 | 535 | 45 |
| λ2 (nm) | 540 | 402 | 426 | 528 | 126 | 394 | −8 |
| λ3 (nm) | 488 | 360 | 330 | 440 | 80 | 374 | 14 |
| Zn-Porphyrin | VACUUM | CCl4 |
|---|---|---|
| Soret band, Q-band | Soret band, Q-band | |
| Pzn-EDOT (exp) | - | 423(2.55), 550(0.17) |
| Pzn-EDOT(theo) | 415(1.98), 570(0.22) | 432(2.26), 585(0.32) |
| Group I | ||
| Zn-P-NH2 | 420 (1.72), 409 (0.77), 622(0.21) | 438(1.84), 429(0.94), 642(0.28) |
| Zn-P-OCH3 | 393(1.11), 372(1.00), 585(0.23) | 433(2.33), 411(1.46), 601(0.33) |
| Zn-P-N(CH3)2 | 429(0.97), 405(0.87), 658(0.19) | 449(1.04), 422(1.01), 685(0.25) |
| Group II | ||
| Zn-P_π1 | 413(2.07), 392(0.90), 643(0.34) | 427(2.36), 407(1.40), 629(0.43) |
| Zn-P_π2 | 416(1.03), 401(1.03), 687(0.31) | 422(2.29), 410(1.38), 666(0.38) |
| Zn-P_π3 | 437(1.11), 392(0.77), 637(0.18) | 451(1.56), 409(1.39), 650(0.24) |
| Zn-P_π4 | 419(1.97), 390(1.05), 680(0.20) | 432(2.22), 409(1.28), 686(0.30) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arooj, Q.; Wilson, G.J.; Wang, F. Methodologies in Spectral Tuning of DSSC Chromophores through Rational Design and Chemical-Structure Engineering. Materials 2019, 12, 4024. https://doi.org/10.3390/ma12244024
Arooj Q, Wilson GJ, Wang F. Methodologies in Spectral Tuning of DSSC Chromophores through Rational Design and Chemical-Structure Engineering. Materials. 2019; 12(24):4024. https://doi.org/10.3390/ma12244024
Chicago/Turabian StyleArooj, Qudsia, Gregory J. Wilson, and Feng Wang. 2019. "Methodologies in Spectral Tuning of DSSC Chromophores through Rational Design and Chemical-Structure Engineering" Materials 12, no. 24: 4024. https://doi.org/10.3390/ma12244024
APA StyleArooj, Q., Wilson, G. J., & Wang, F. (2019). Methodologies in Spectral Tuning of DSSC Chromophores through Rational Design and Chemical-Structure Engineering. Materials, 12(24), 4024. https://doi.org/10.3390/ma12244024