
Theoretical Investigations of Si-Ge Alloys in P42/ncm Phase: First-Principles Calculations

Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
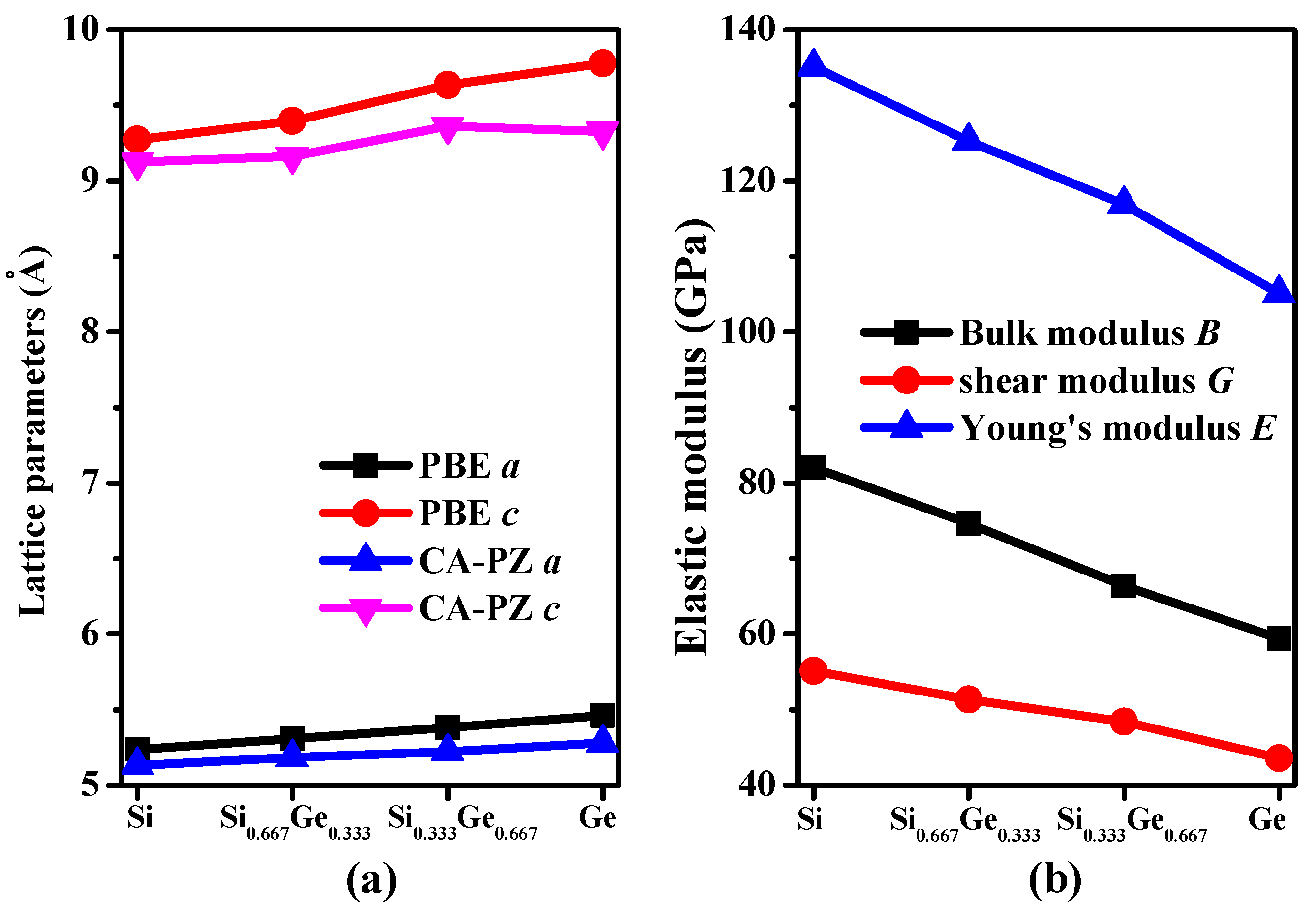
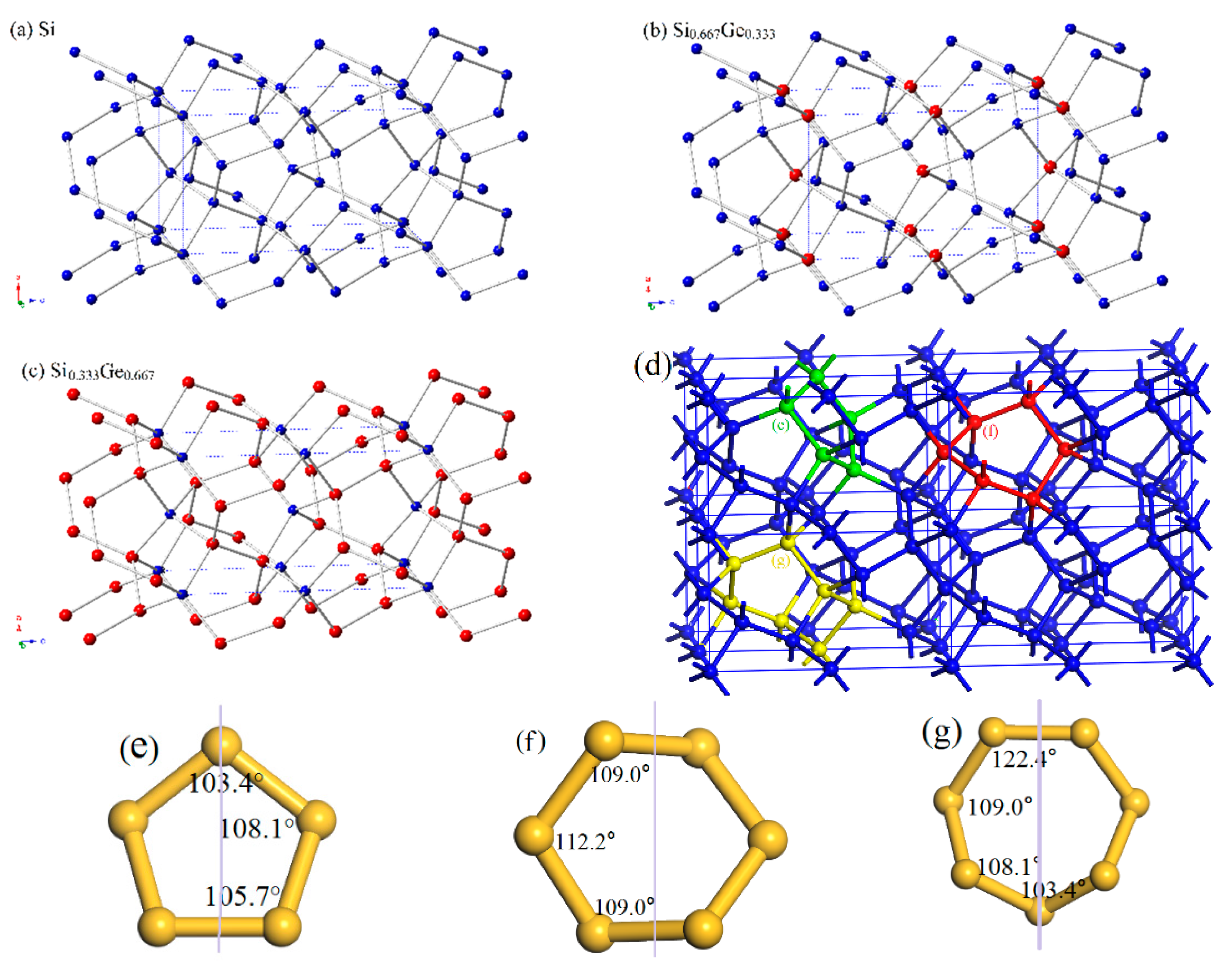
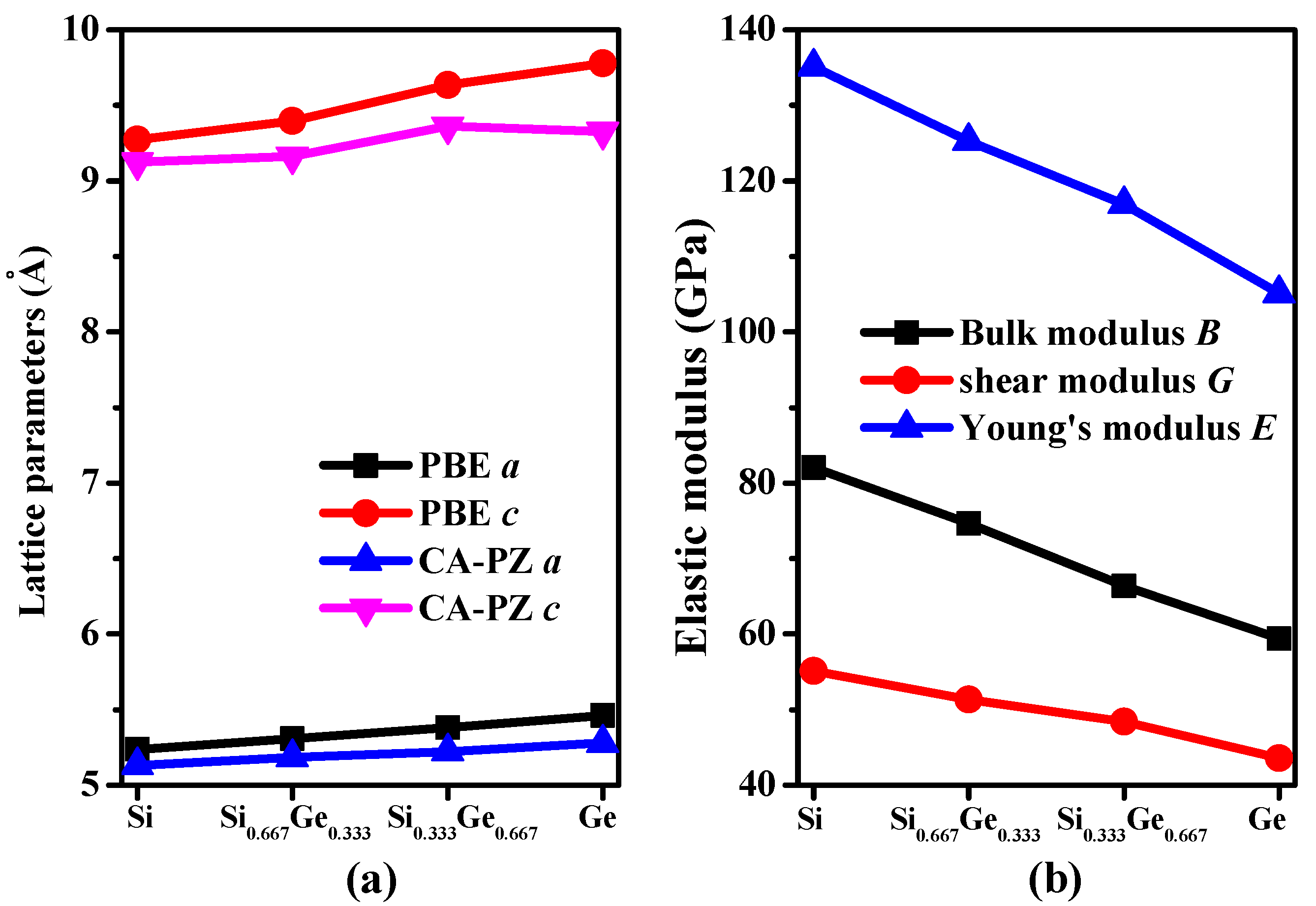
3.1. Structural Properties
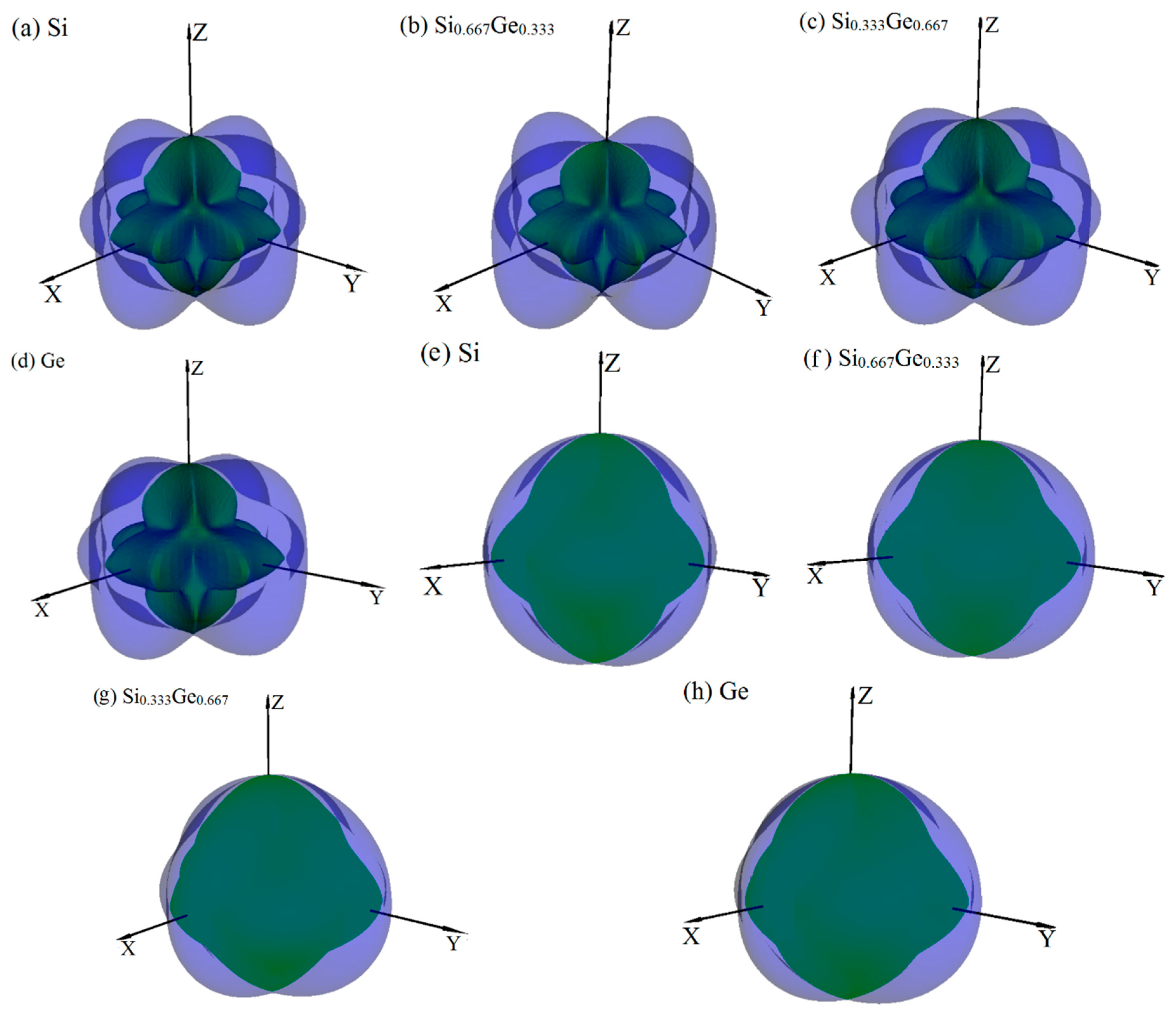
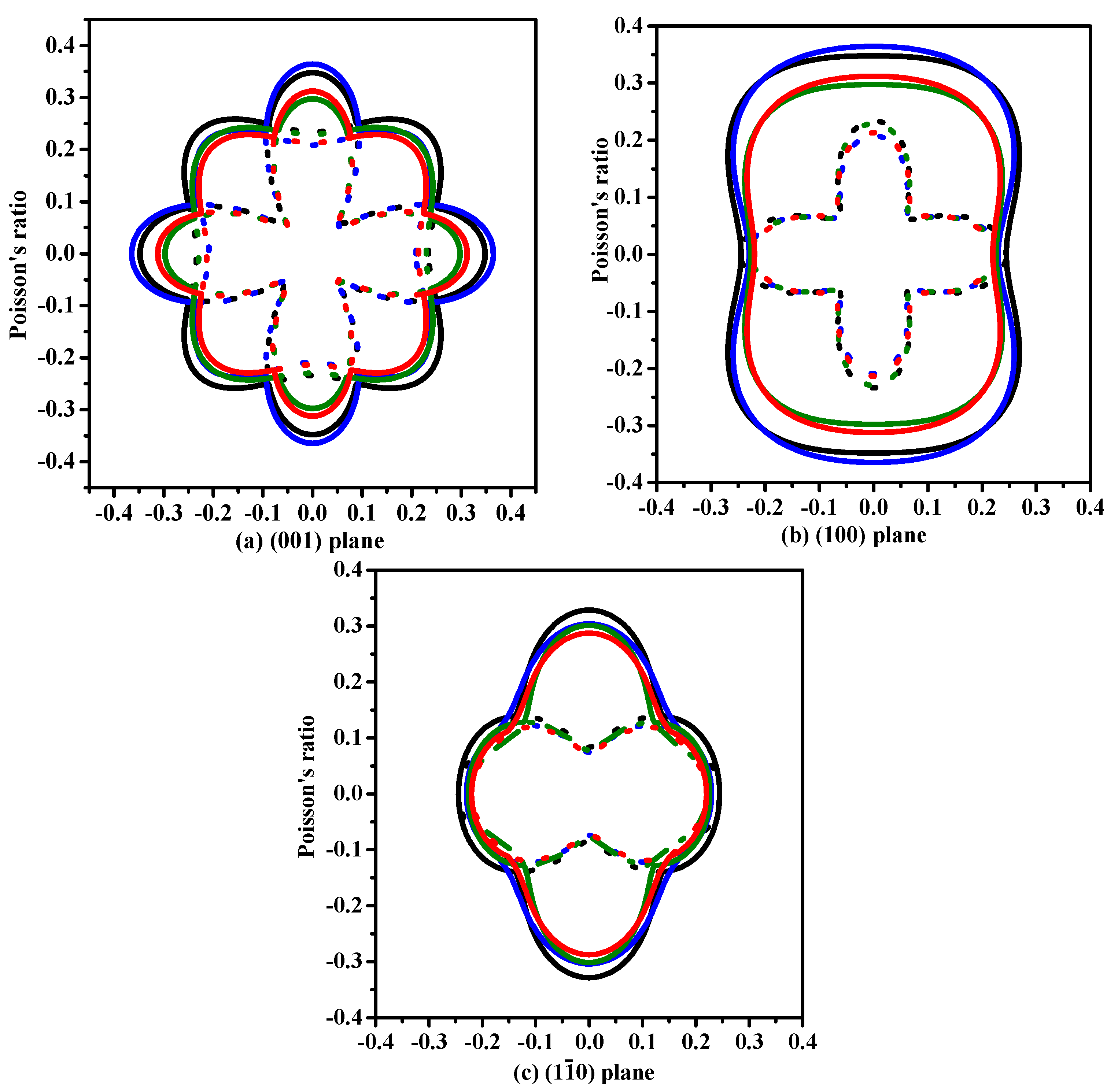
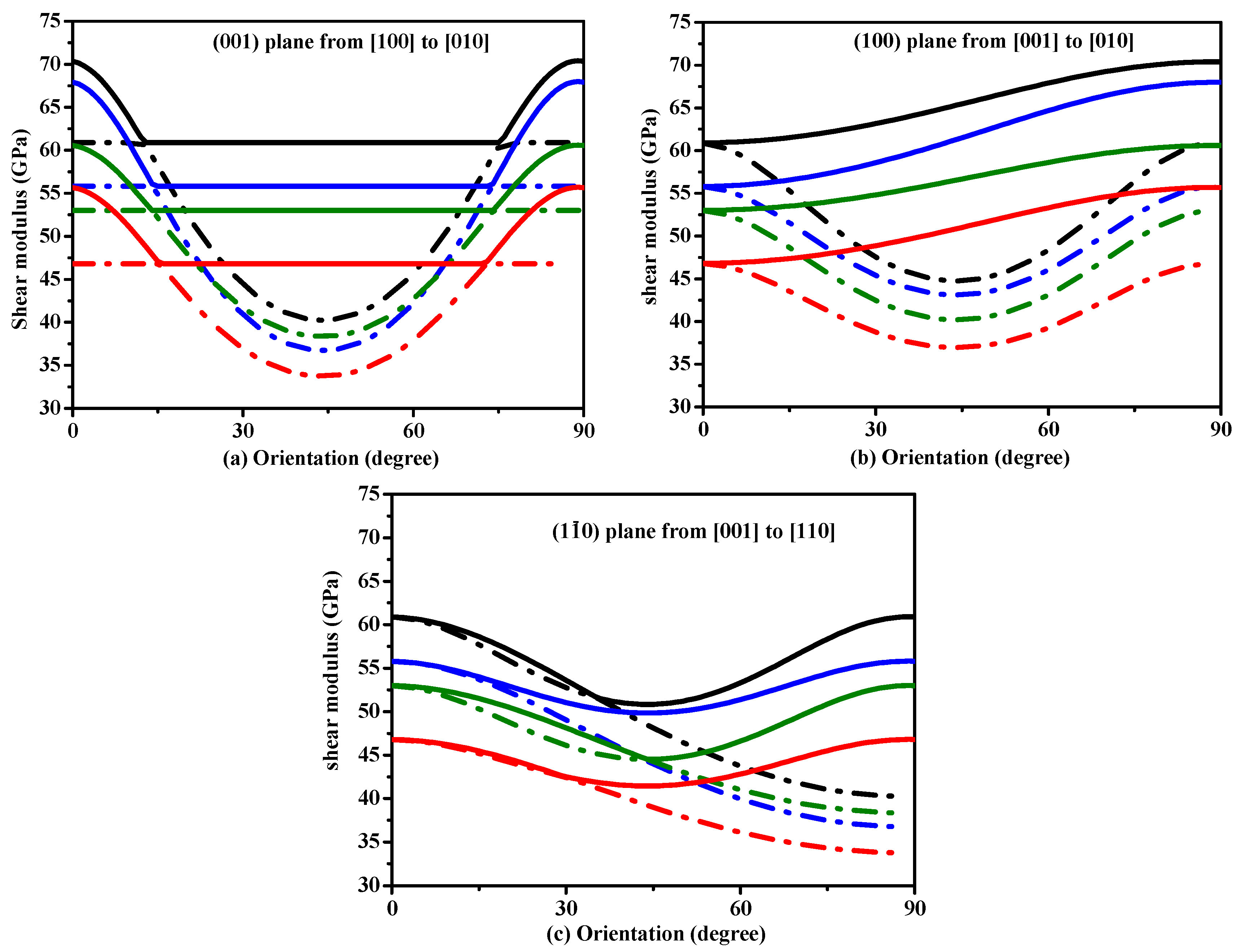
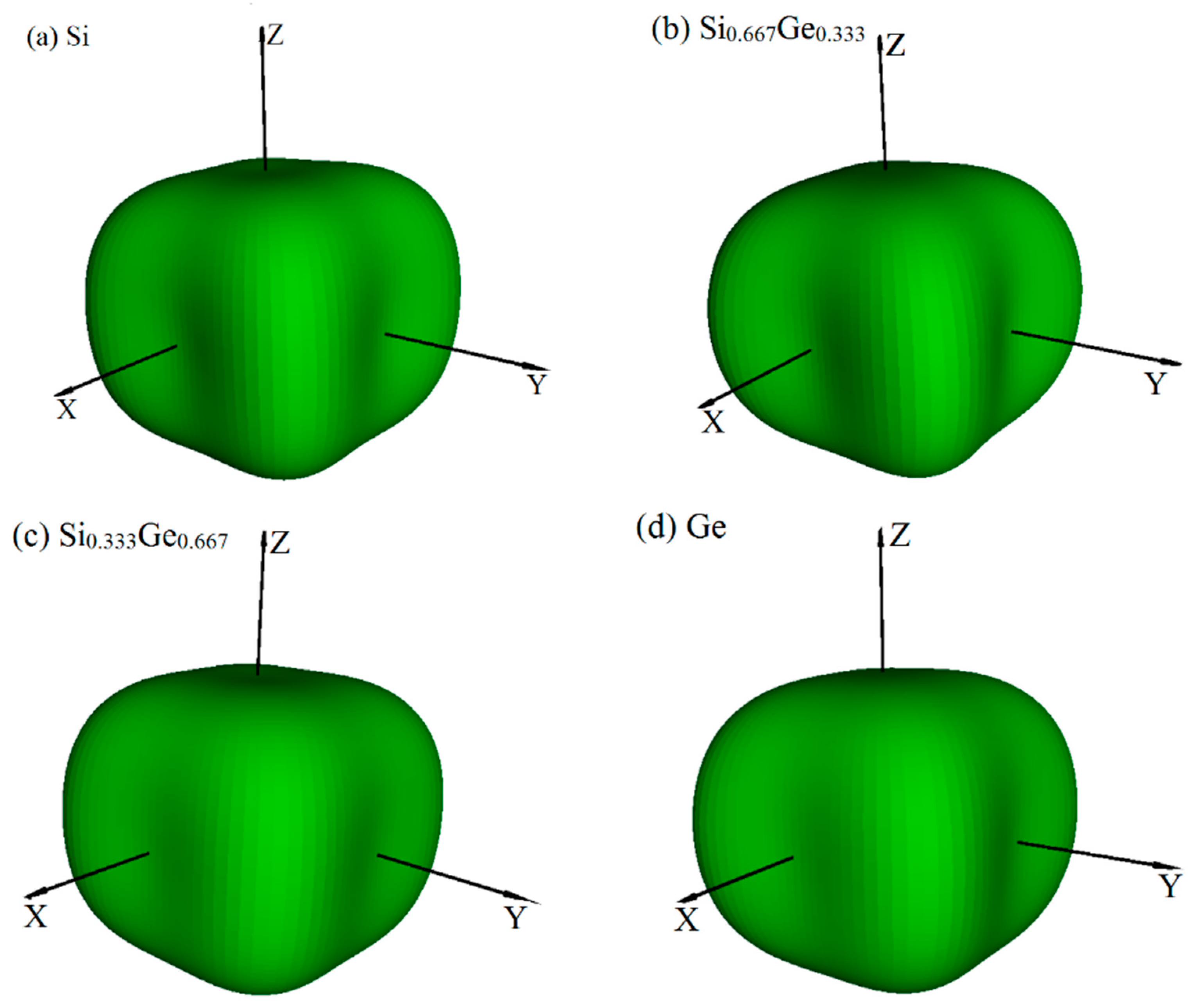
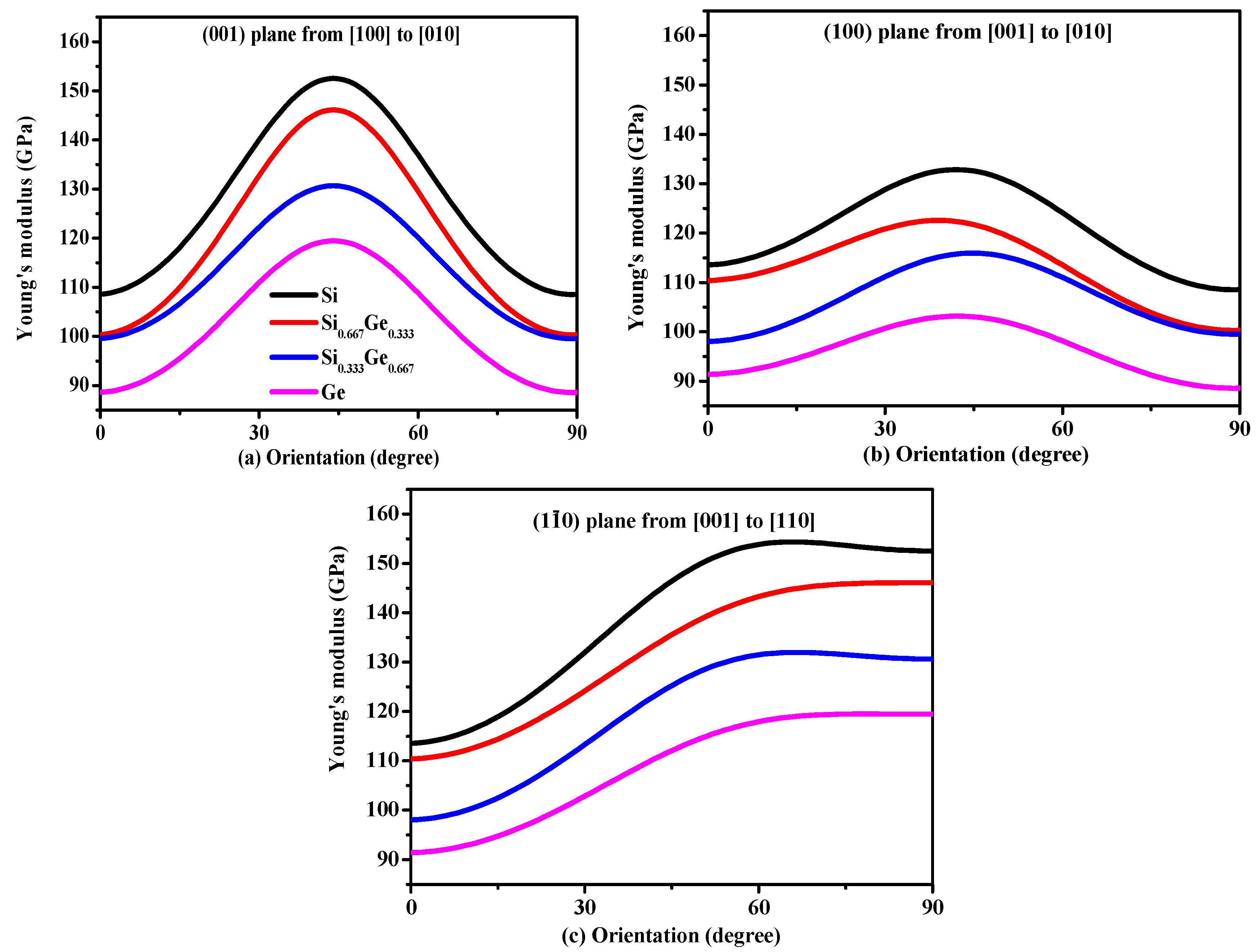
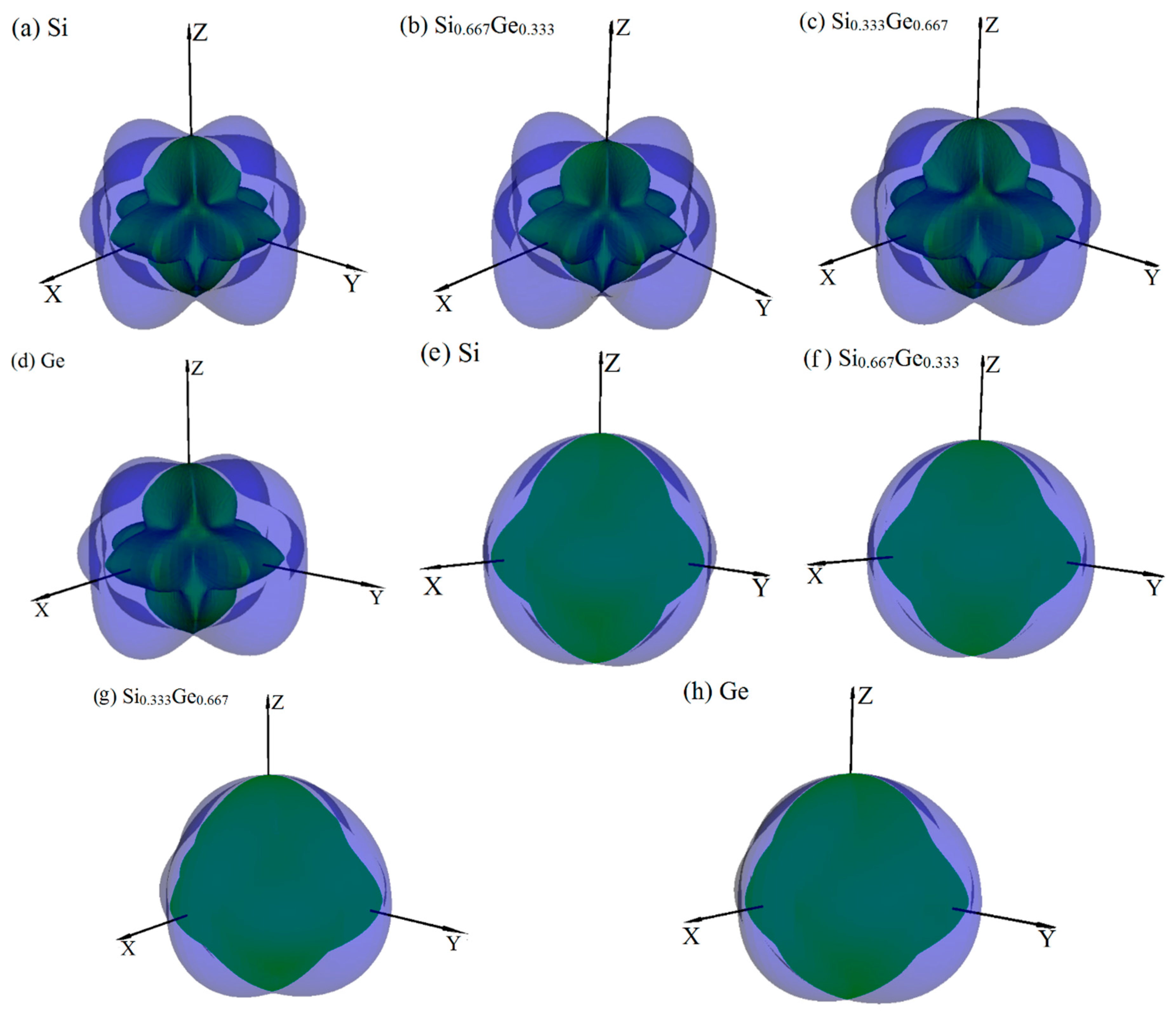
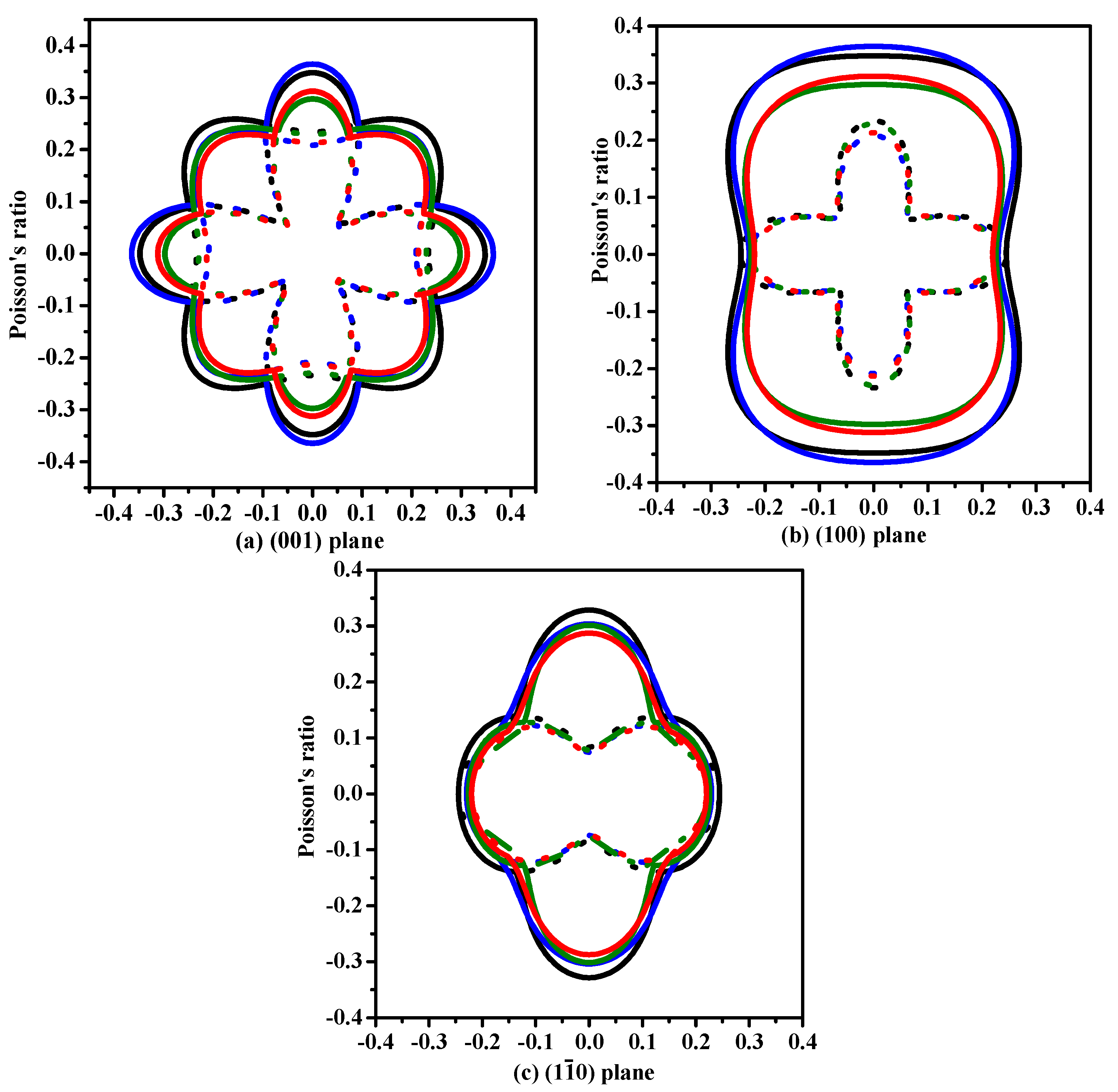
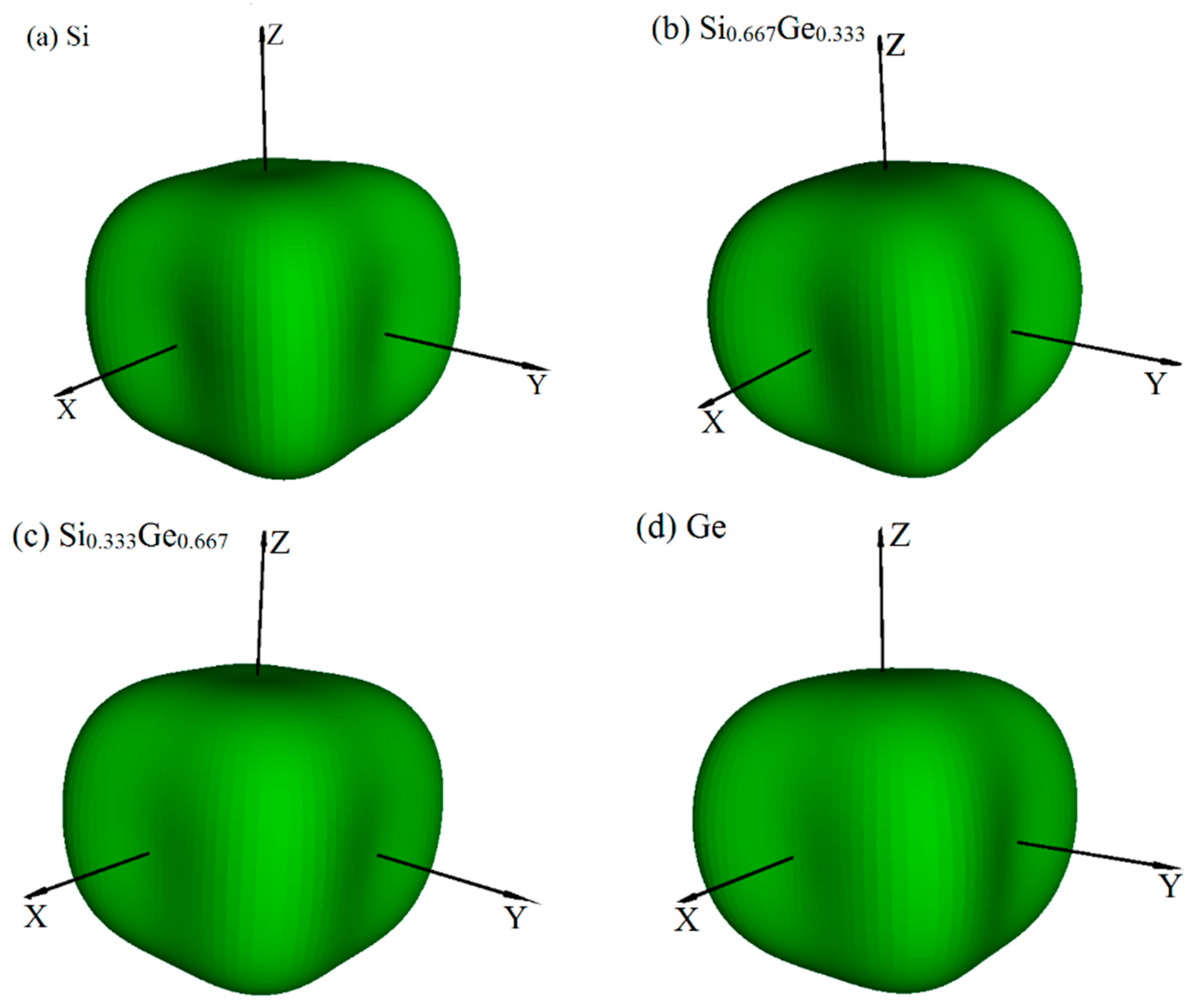
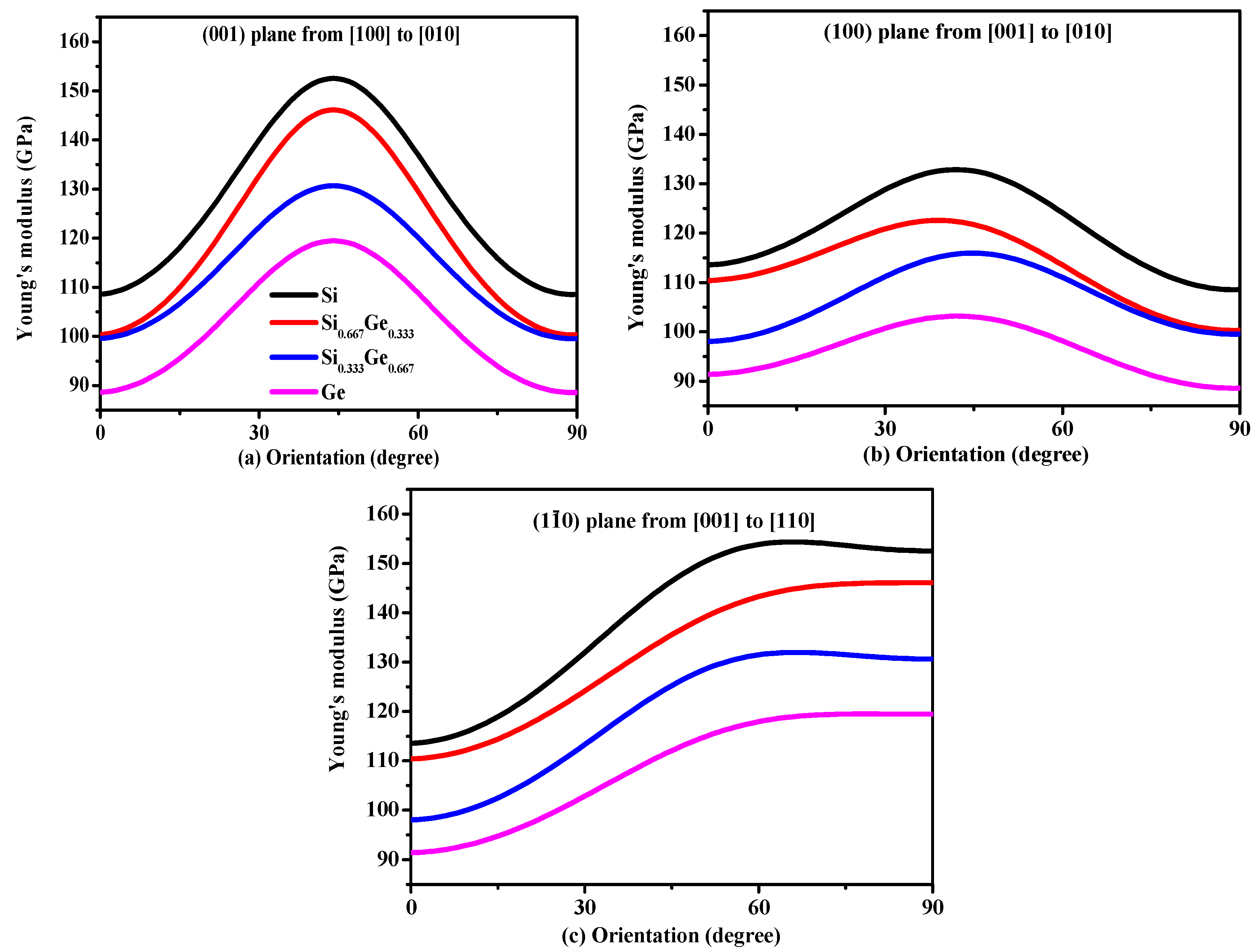
3.2. Mechanical and Anisotropic Properties
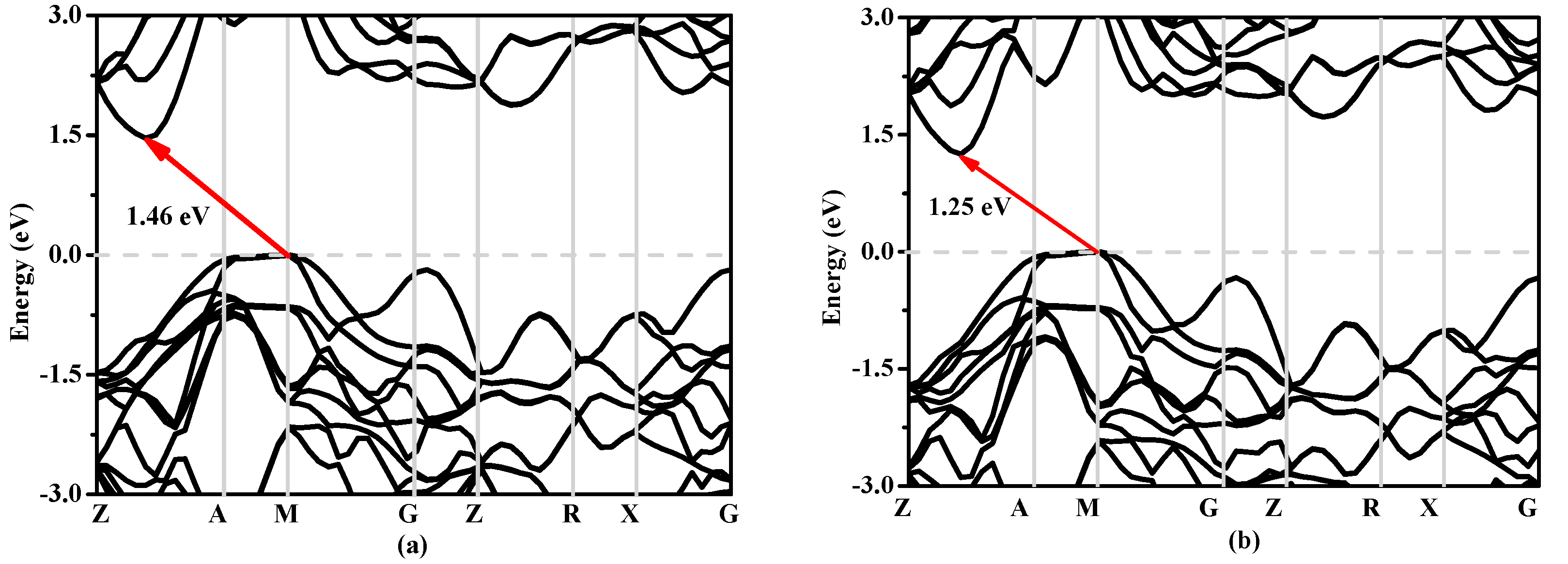
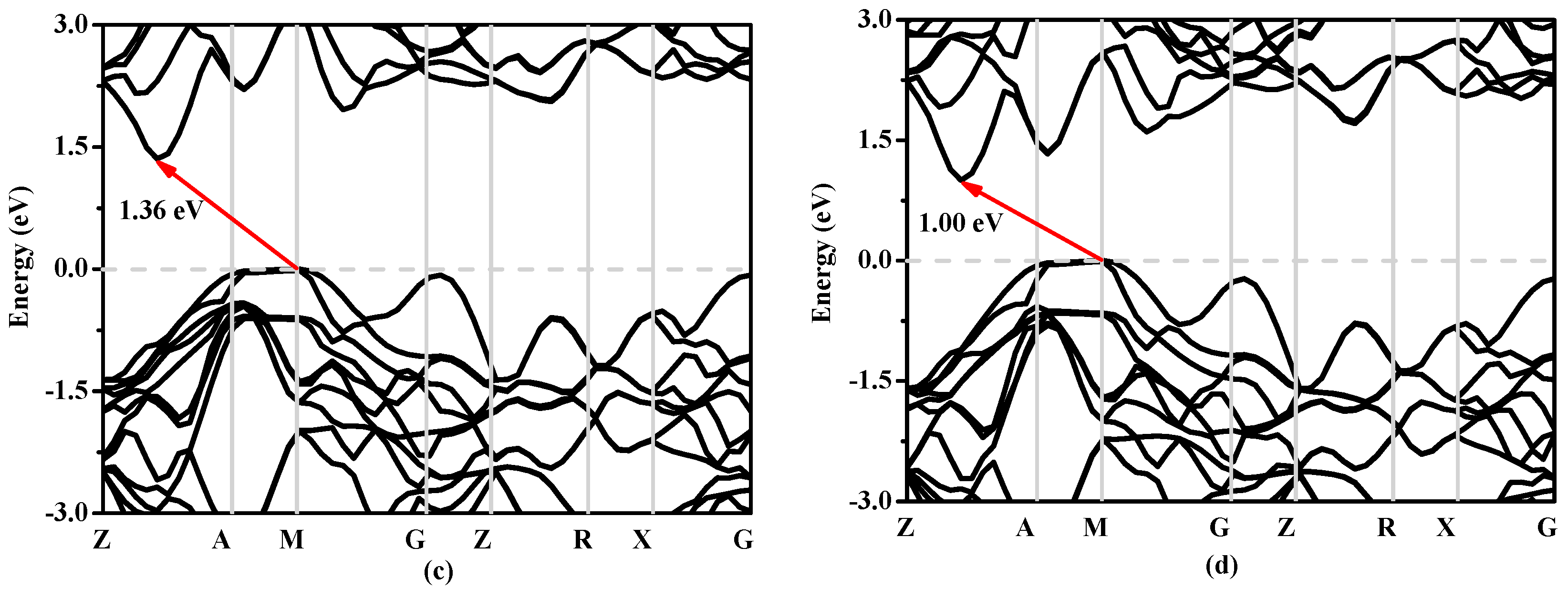
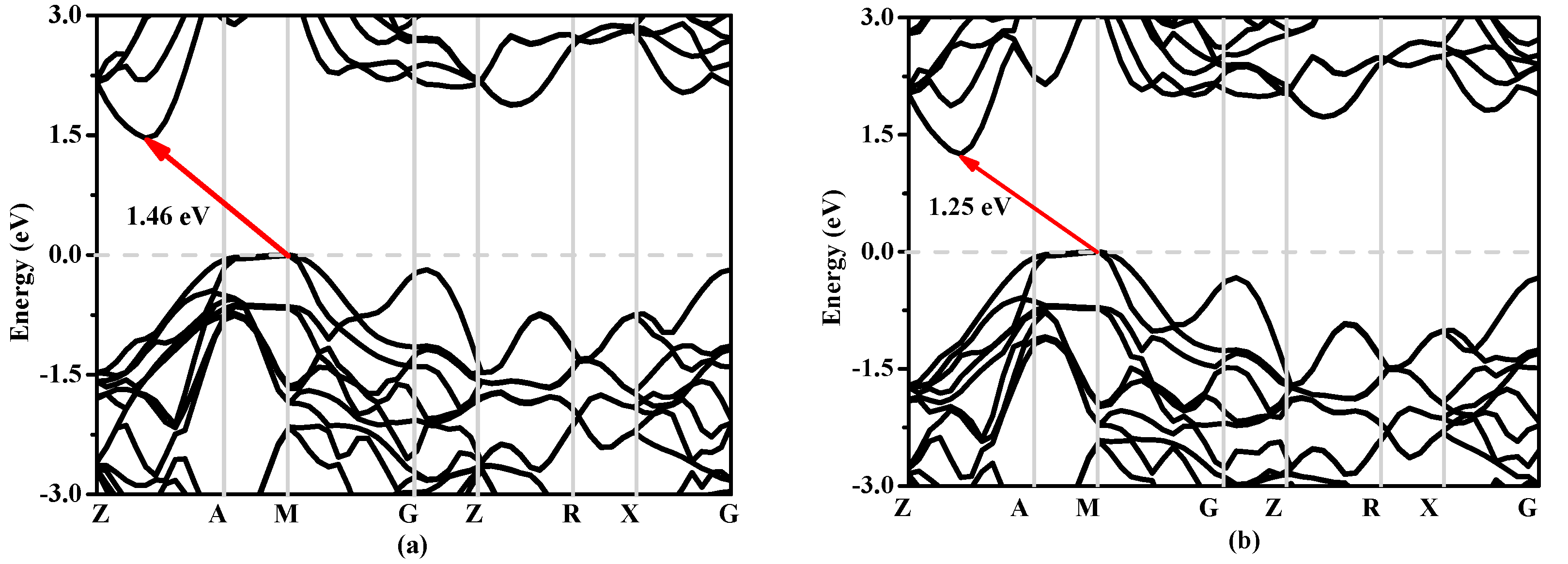
3.3. Electronic Properties
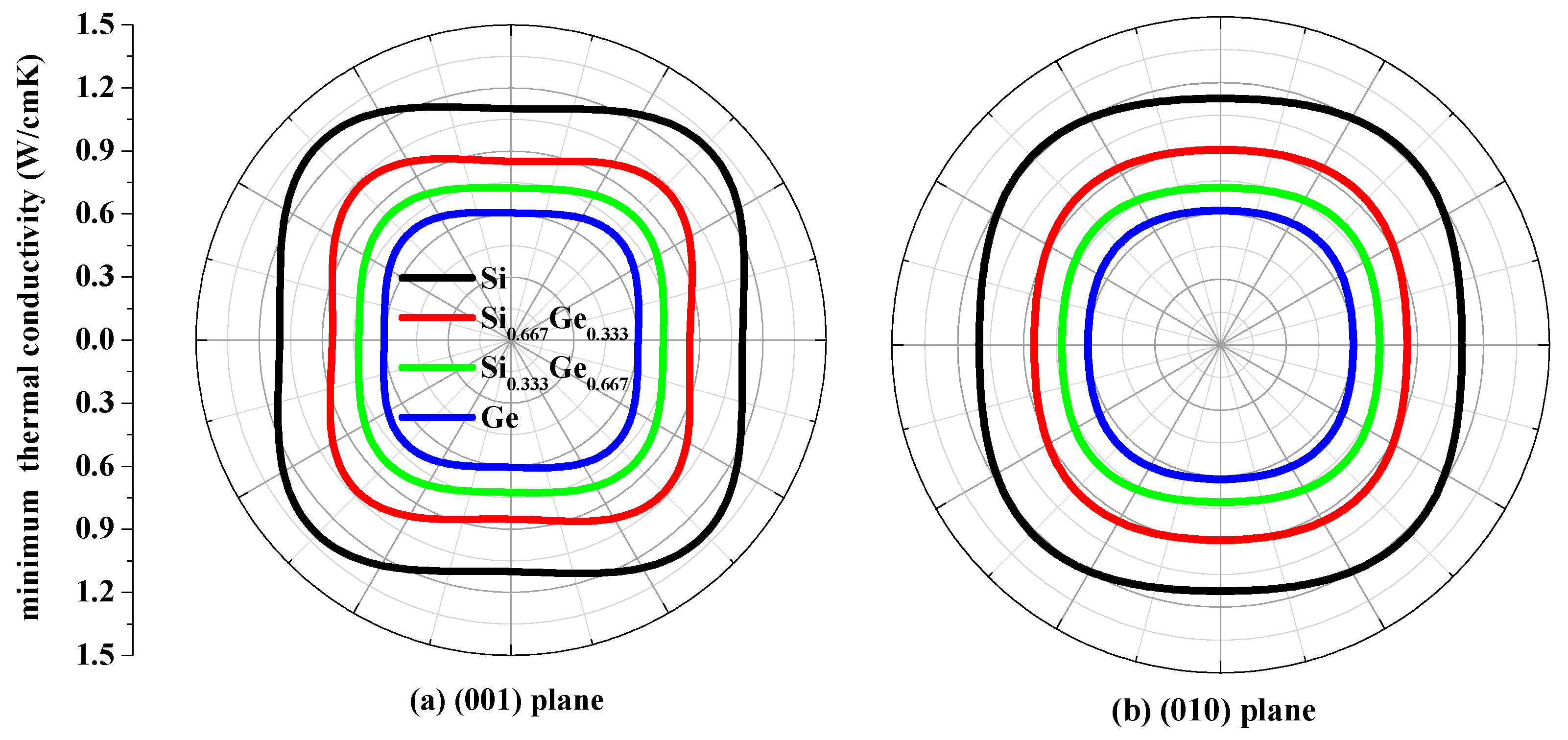
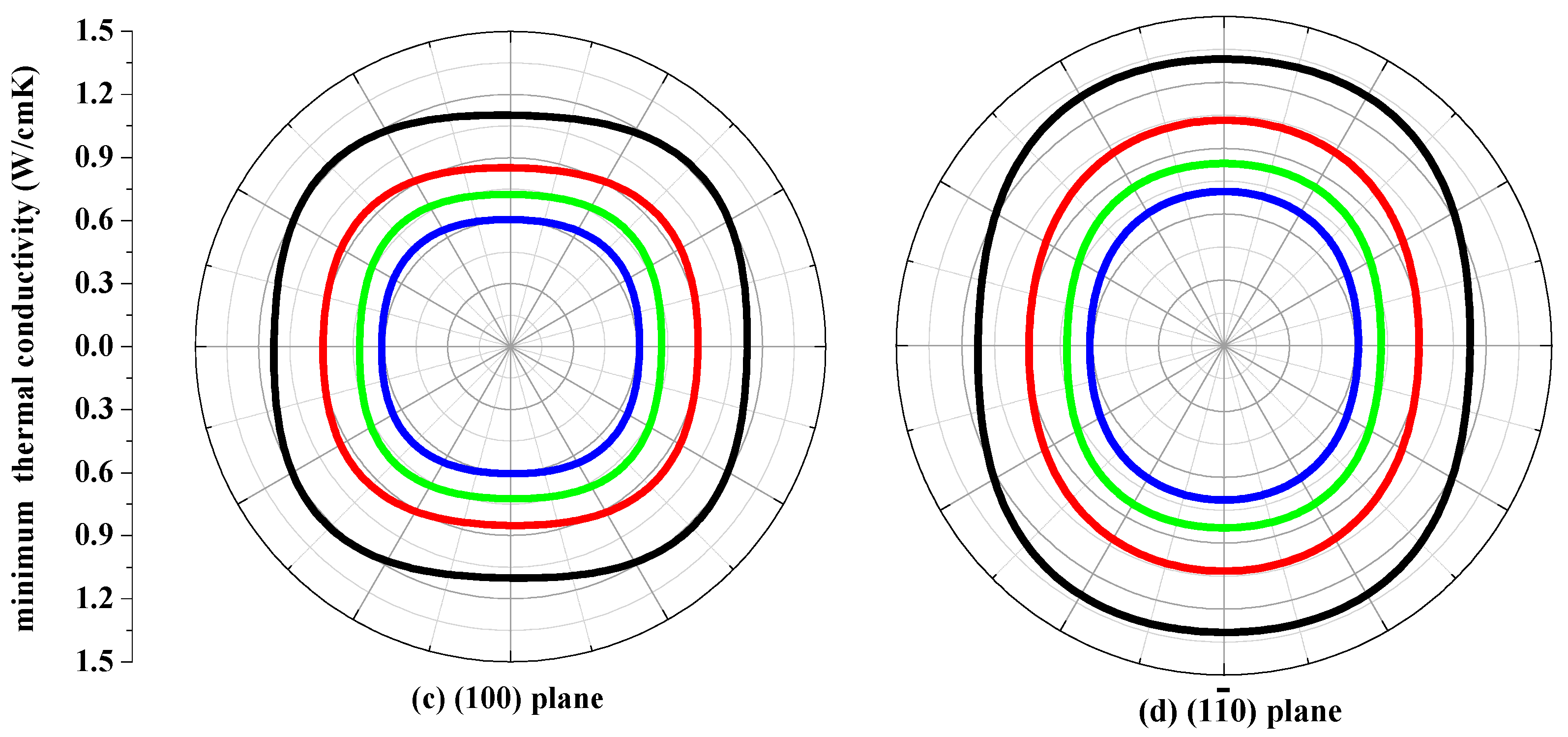
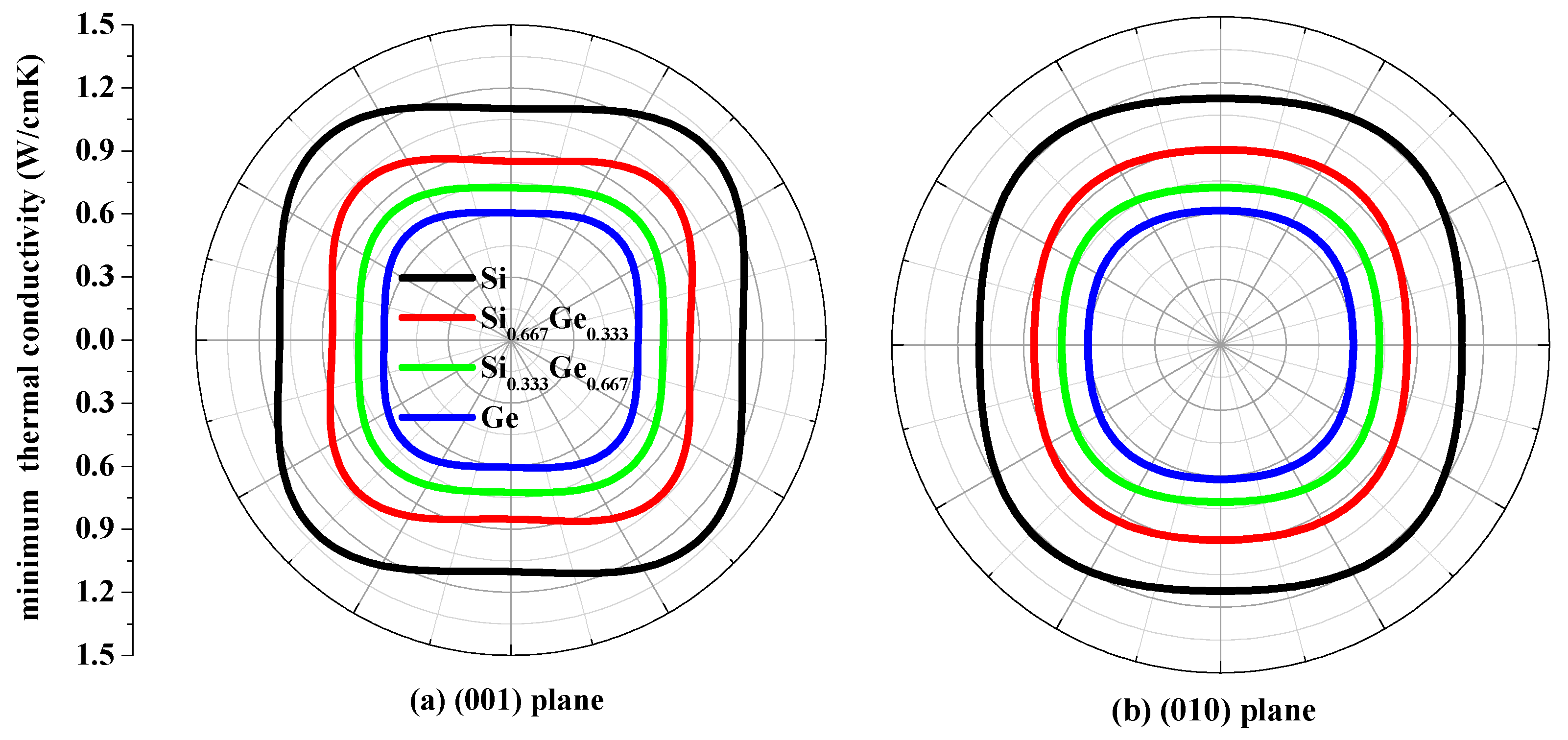
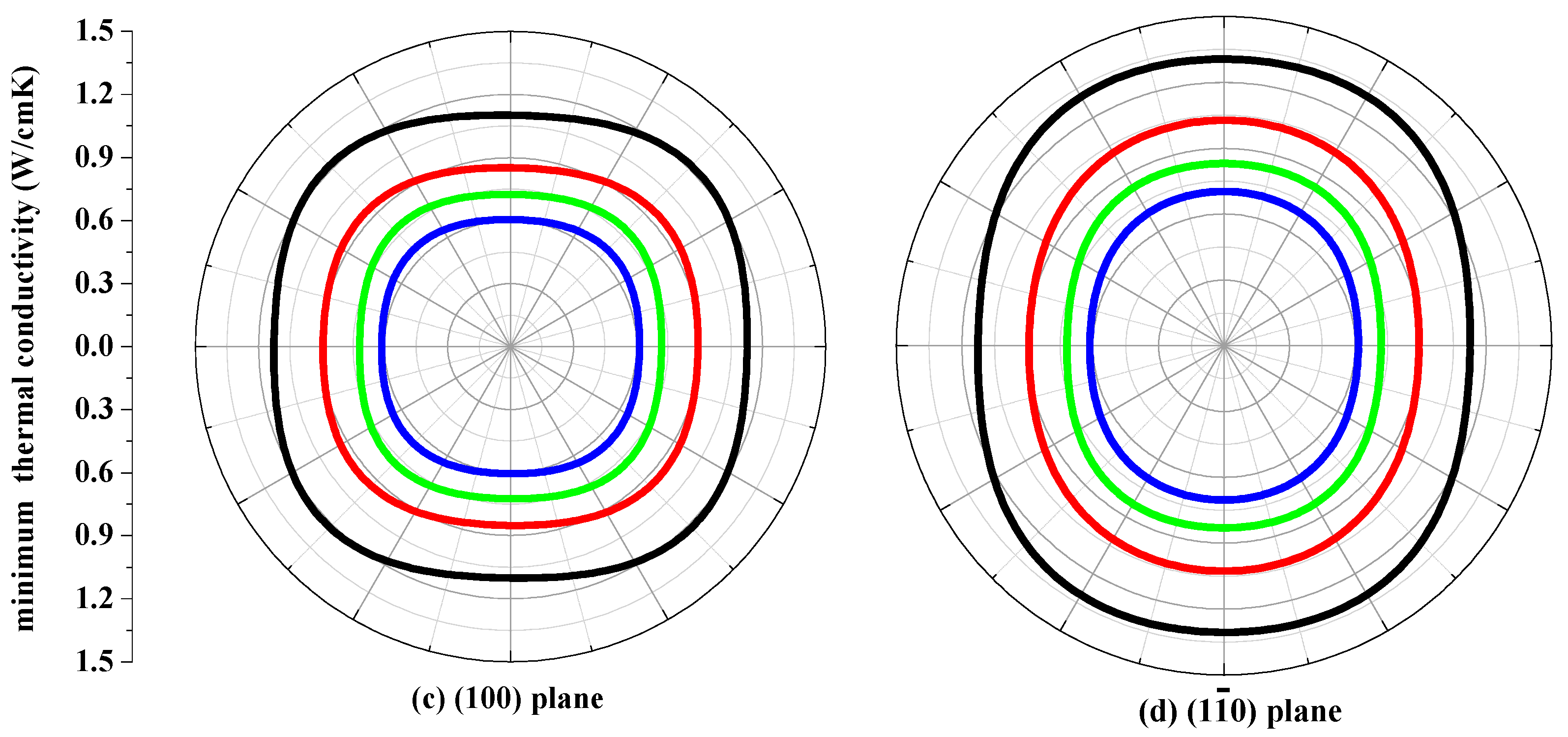
3.4. The Minimum Thermal Conductivity κmin
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wu, F.; Jun, D.; Kan, E.J.; Li, Z.Y. Density functional predictions of new silicon allotropes: Electronic properties and potential applications to Li-battery anode materials. Solid State Commun. 2011, 151, 1228–1230. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Koretsune, T.; Saito, S.; Miyake, T.; Oshiyama, A. A new crystalline phase of four-fold coordinated silicon and germanium. New J. Phys. 2008, 10, 083001. [Google Scholar] [CrossRef]
- De, A.; Pryor, C.E. Electronic structure and optical properties of Si, Ge and diamond in the lonsdaleite phase. J. Phys. Condens. Matter 2014, 26, 045801. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.D.; Sau, J.D.; Cohen, M.L. Ab initio survey of the electronic structure of tetrahedrally bonded phases of silicon. Phys. Rev. B 2008, 78, 035210. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yan, H.Y.; Zhao, Y.B.; Yang, Y.T.; Yu, X.H.; Liu, Y.; Xing, M.J.; Zhang, J.Q.; et al. Novel silicon allotropes: Stability, mechanical, and electronic properties. J. Appl. Phys. 2015, 118, 185704. [Google Scholar] [CrossRef]
- Xiang, H.J.; Huang, B.; Kan, E.J.; Wei, S.H.; Gong, X.G. Towards direct-gap silicon phases by the inverse band structure design approach. Phys. Rev. Lett. 2013, 110, 118702. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.Q.; Xu, B.; Sun, J.; Liu, H.Y.; Zhao, Z.S.; Yu, D.L.; Fan, C.Z.; He, J.L. Direct band gap silicon allotropes. J. Am. Chem. Soc. 2014, 136, 9826–9829. [Google Scholar] [CrossRef] [PubMed]
- He, C.Y.; Zhang, C.X.; Li, J.; Peng, X.Y.; Meng, L.J.; Tang, C.; Zhong, J.X. Direct and quasi-direct band gap silicon allotropes with remarkable stability. Phys. Chem. Chem. Phys. 2016, 18, 9682–9686. [Google Scholar] [CrossRef] [PubMed]
- Amsler, M.; Flores-Livas, J.A.; Lehtovaara, L.; Balima, F.; Ghasemi, S.A.; Machon, D.; Pailhes, S.; Willand, A.; Caliste, D.; Botti, S.; et al. Crystal Structure of Cold Compressed Graphite. Phys. Rev. Lett. 2012, 108, 065501. [Google Scholar] [CrossRef] [PubMed]
- Pfrommer, B.G.; Cote, M.; Louie, S.G.; Cohen, M.L. Ab initio study of silicon in the R8 phase. Phys. Rev. B 1997, 56, 662–668. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T.; Yang, Q.; Chen, P.Y.; Xing, M.J.; Zhang, J.Q.; Yao, R.H. Prediction of novel phase of silicon and Si–Ge alloys. J. Solid State Chem. 2016, 233, 471–483. [Google Scholar] [CrossRef]
- Zhang, X.D.; Ying, C.H.; Quan, S.Y.; Shi, G.M.; Li, Z.J. A first principles investigation on the structural, phonon, elastic and thermodynamic properties of the Si0.5Sn0.5 cubic alloy. Solid State Commun. 2012, 152, 955–959. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Xiang, G.; Gu, G.X.; Li, R.; He, D.W.; Zhang, X. Nonlinear concentration-dependent electronic and optical properties of Si1–xGex alloy nanowires. J. Phys. Chem. C 2012, 116, 17934–17938. [Google Scholar] [CrossRef]
- Zhang, X.D.; Ying, C.H.; Li, Z.J.; Shi, G.M. First-principles calculations of structural stability, elastic, dynamical and thermodynamic properties of SiGe, SiSn, GeSn. Superlattices Microstruct. 2012, 52, 459–469. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, X.Y.; Zhang, S.H.; Sun, X.W.; Wang, L.M.; Ma, M.Z.; Liu, R.P. First-principles investigations on thermodynamic properties of the ordered and disordered Si0.5Ge0.5 alloys. Appl. Phys. A 2014, 115, 667. [Google Scholar] [CrossRef]
- Bautista-Hernandez, A.; Rangel, T.; Romero, A.H.; Rignanese, G.M.; Salazar-Villanueva, M.; Chigo-Anota, E. Structural and vibrational stability of M and Z phases of silicon and germanium from first principles. J. Appl. Phys. 2013, 113, 193504. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Chai, C.C.; Fan, Q.Y.; Yang, Y.T.; Xing, M.J. Mechanical and electronic properties of Si-Ge alloy in Cmmm structure. Chin. J. Phys. 2016, 54, 298–307. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Q.; Zhou, P.K.; Xing, M.J.; Yang, Y.T. Mechanical and electronic properties of Si, Ge and their alloys in P42/mnm structure. Mater. Sci. Semicond. Process. 2016, 43, 187–195. [Google Scholar] [CrossRef]
- Zhao, Z.S.; Tian, F.; Dong, X.; Li, Q.; Wang, Q.Q.; Wang, H.; Zhong, X.; Xu, B.; Yu, D.; He, J.L.; et al. Tetragonal allotrope of group 14 Elements. J. Am. Chem. Soc. 2012, 134, 12362–12365. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892R. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–568. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef]
- Gueorguiev, G.K.; Pacheco, J.M. Shapes of cagelike metal carbide clusters: First-principles calculations. Phys. Rev. B 2003, 68, 241401R. [Google Scholar] [CrossRef]
- Gueorguiev, G.K.; Broitman, E.; Furlan, A.; Stafström, S.; Hultman, L. Dangling bond energetics in carbon nitride and phosphorus carbide thin films with fullerene-like and amorphous structure. Chem. Phys. Lett. 2009, 482, 110–113. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T. Two novel silicon phases with direct band gaps. Phys. Chem. Chem. Phys. 2016, 18, 12905–12913. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 73rd ed.; Chemical Rubber: Boca Raton, FL, USA, 1992. [Google Scholar]
- Cohen, E.R.; Taylor, B.N. The 1986 adjustment of the fundamental physical constants. Rev. Mod. Phys. 1987, 59, 1121–1148. [Google Scholar] [CrossRef]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T. Two novel C3N4 phases: Structural, mechanical and electronic properties. Materials 2016, 9, 427. [Google Scholar] [CrossRef]
- Gomez-Abal, R.; Li, X.Z.; Scheffler, M.; Ambrosch-Draxl, C. Influence of the core-valence interaction and of the pseudopotential approximation on the electron self-energy in semiconductors. Phys. Rev. Lett. 2008, 101, 106404. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.Y.; Han, Z.; Liu, X.H.; Yu, X.H.; Wang, D.Y.; Tian, Y. Pnma-BN: Another Boron Nitride polymorph with interesting physical properties. Nanomaterials 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.C.; Liu, Y.; Li, D.J.; Zeng, X.Q.; Xu, C.S. First-principles study of structural and electronic properties of C14-type Laves phase Al2Zr and Al2Hf. Comput. Mater. Sci. 2014, 83, 27–34. [Google Scholar] [CrossRef]
- Marmier, A.; Lethbridge, Z.A.D.; Walton, R.I.; Smith, C.W.; Parker, S.C.; Evans, K.E. ElAM: A computer program for the analysis and representation of anisotropic elastic properties. Comput. Phys. Commun. 2010, 181, 2102–2115. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef]
- Long, J.P.; Shu, C.Z.; Yang, L.J.; Yang, M. Predicting crystal structures and physical properties of novel superhard p-BN under pressure via first-principles investigation. J. Alloys Compd. 2015, 644, 638–644. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | PBE | CA-PZ | Experimental | ||
|---|---|---|---|---|---|
| a | c | a | c | a | |
| Si | 5.237 | 9.274 | 5.132 | 9.125 | - |
| - | - | 5.135 1 | 9.167 | - | |
| Si0.667Ge0.333 | 5.306 | 9.397 | 5.185 | 9.163 | - |
| Si333Ge0.667 | 5.380 | 9.635 | 5.222 | 9.361 | - |
| Ge | 5.462 | 9.779 | 5.281 | 9.396 | - |
| - | - | 5.292 1 | 9.346 | - | |
| Diamond-Si | 5.441 | - | 5.419 | - | 5.431 2 |
| Diamond-Ge | 5.699 | - | 5.580 | - | 5.660 2 |
| Materials | C11 | C12 | C13 | C33 | C44 | C66 | B | G | E | v | AU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P42/ncm-Si | 141 | 61 | 49 | 138 | 61 | 73 | 82 | 55 | 135 | 0.226 | 0.237 |
| Si0.667Ge0.333 | 130 | 56 | 43 | 130 | 56 | 68 | 75 | 51 | 125 | 0.220 | 0.225 |
| Si0.333Ge0.667 | 122 | 45 | 38 | 115 | 53 | 61 | 66 | 48 | 117 | 0.207 | 0.156 |
| P42/ncm-Ge | 108 | 41 | 33 | 106 | 47 | 56 | 59 | 44 | 105 | 0.205 | 0.158 |
| P42/mnm-Si 1 | 123 | 47 | 48 | 146 | 49 | 61 | 75 | 48 | 119 | 0.236 | 0.134 |
| Si0.667Ge0.333 1 | 111 | 36 | 42 | 130 | 43 | 38 | 66 | 40 | 100 | 0.252 | 0.035 |
| Si0.333Ge0.667 1 | 100 | 32 | 37 | 110 | 39 | 44 | 58 | 38 | 94 | 0.231 | 0.056 |
| P42/mnm-Ge 1 | 88 | 26 | 32 | 100 | 35 | 40 | 50 | 34 | 83 | 0.223 | 0.054 |
| Diamond-Si | 160 | 62 | - | - | 79 | - | 95 | 65 | 159 | 0.221 | 0.279 |
| 166 2 | 64 | - | - | 80 | - | 102 | - | - | - | - | |
| Diamond-Ge | 123 | 47 | - | - | 62 | - | 72 | 51 | 124 | 0.213 | 0.293 |
| 129 2 | 48 | - | - | 67 | - | 77 | - | - | - | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Z.; Liu, X.; Yu, X.; Shi, C.; Yan, F. Theoretical Investigations of Si-Ge Alloys in P42/ncm Phase: First-Principles Calculations. Materials 2017, 10, 599. https://doi.org/10.3390/ma10060599
Ma Z, Liu X, Yu X, Shi C, Yan F. Theoretical Investigations of Si-Ge Alloys in P42/ncm Phase: First-Principles Calculations. Materials. 2017; 10(6):599. https://doi.org/10.3390/ma10060599
Chicago/Turabian StyleMa, Zhenyang, Xuhong Liu, Xinhai Yu, Chunlei Shi, and Fang Yan. 2017. "Theoretical Investigations of Si-Ge Alloys in P42/ncm Phase: First-Principles Calculations" Materials 10, no. 6: 599. https://doi.org/10.3390/ma10060599
APA StyleMa, Z., Liu, X., Yu, X., Shi, C., & Yan, F. (2017). Theoretical Investigations of Si-Ge Alloys in P42/ncm Phase: First-Principles Calculations. Materials, 10(6), 599. https://doi.org/10.3390/ma10060599