Unraveling Racial Disparities in Papillary Thyroid Cancer: A Comparative Bulk RNA-Sequencing Gene Expression Analysis



Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Sample Selection
2.2. Total RNA Isolation from FFPE Samples
2.3. RNA Sequencing
2.4. Data Analysis
2.4.1. Data Acquisition and Quality Control
2.4.2. Alignment and Feature Counting
2.4.3. Differential Expression Analysis
2.4.4. DESeq2 Analysis
2.4.5. Visualization and Functional Analysis
2.4.6. Gene Set Enrichment Analysis
2.4.7. TCGA Data Validation
3. Results
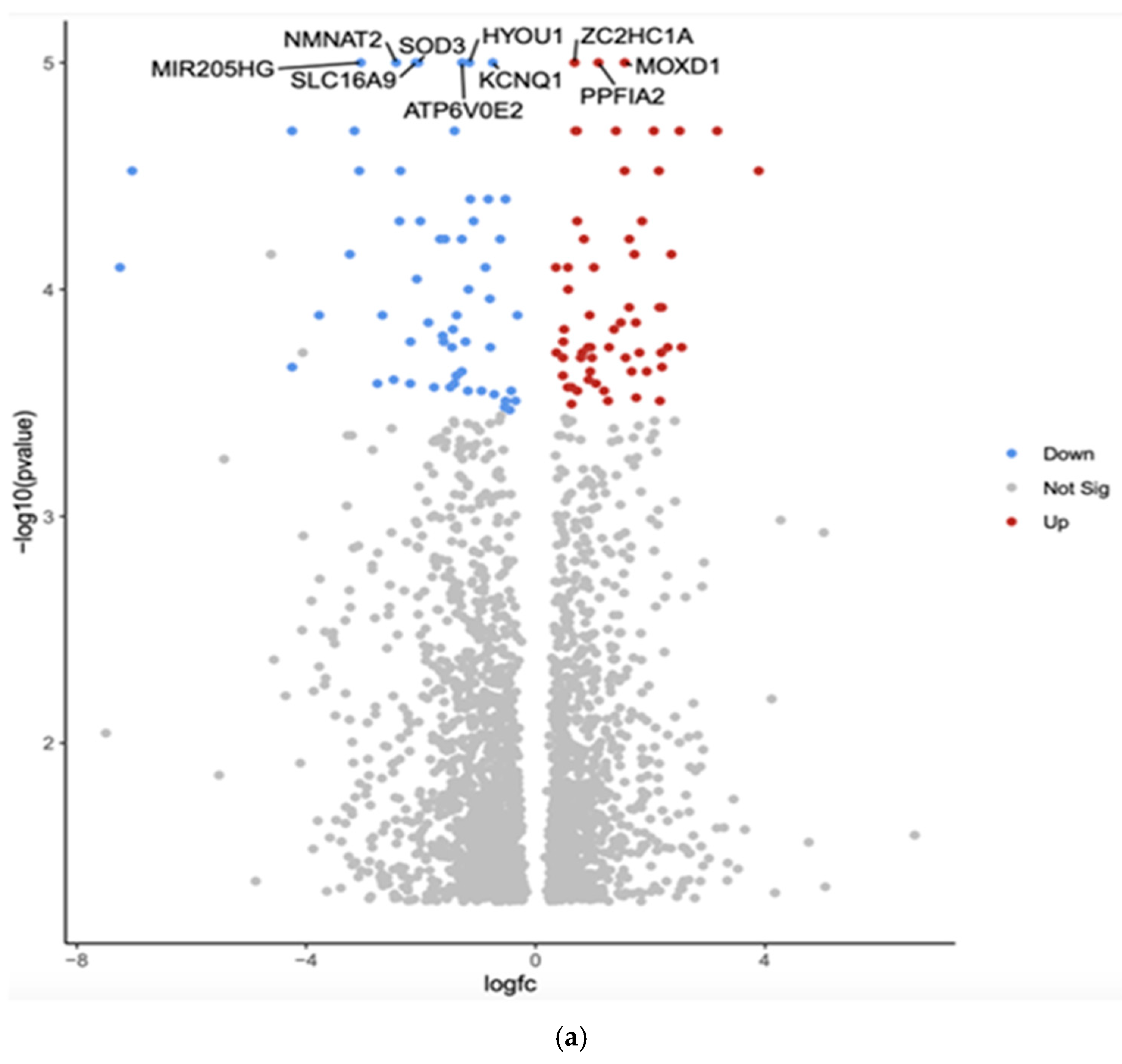
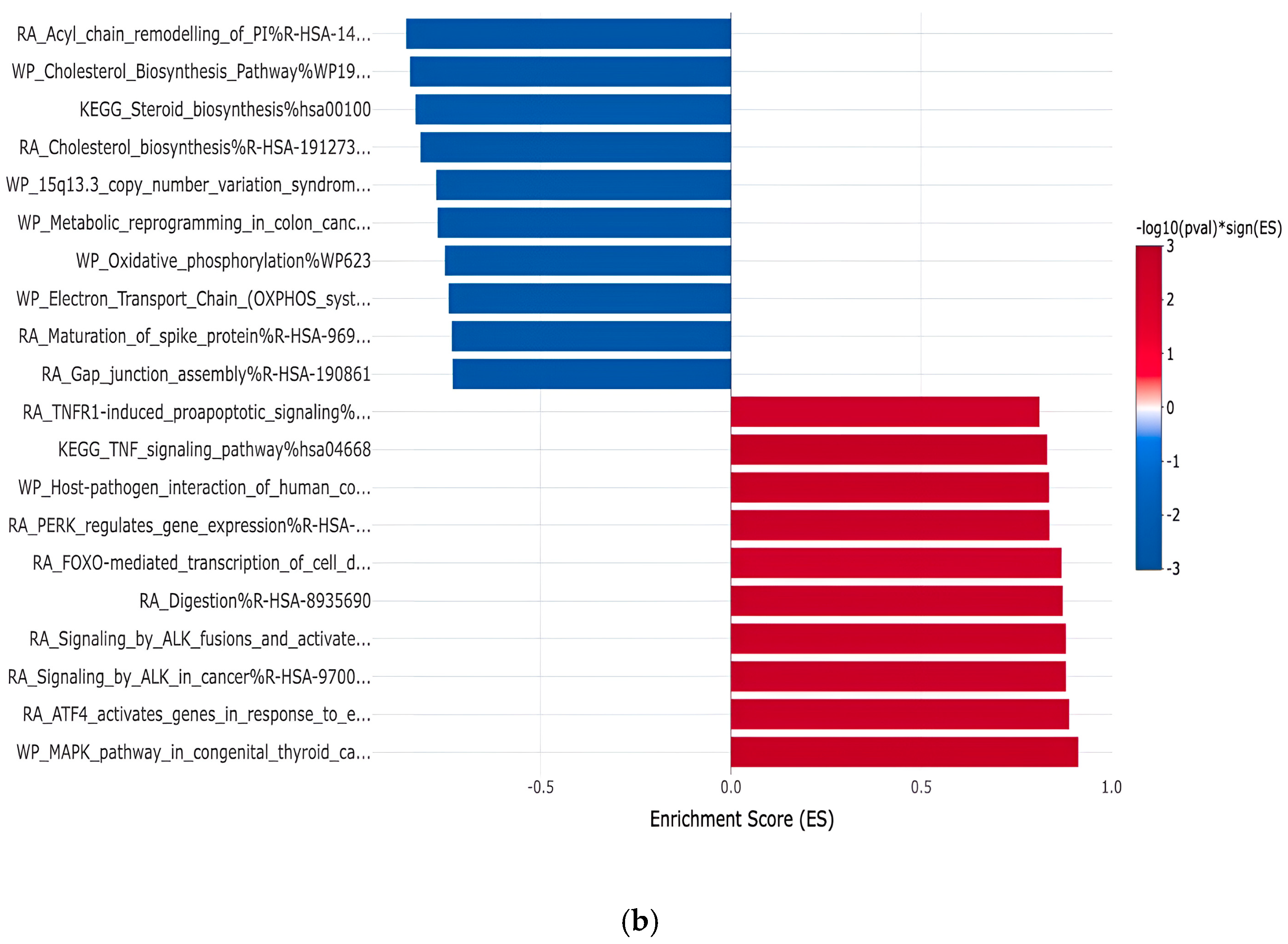
3.1. Gene Expression Profiles: Comparison Between White and Asian Papillary Thyroid Cancer Tissue Samples
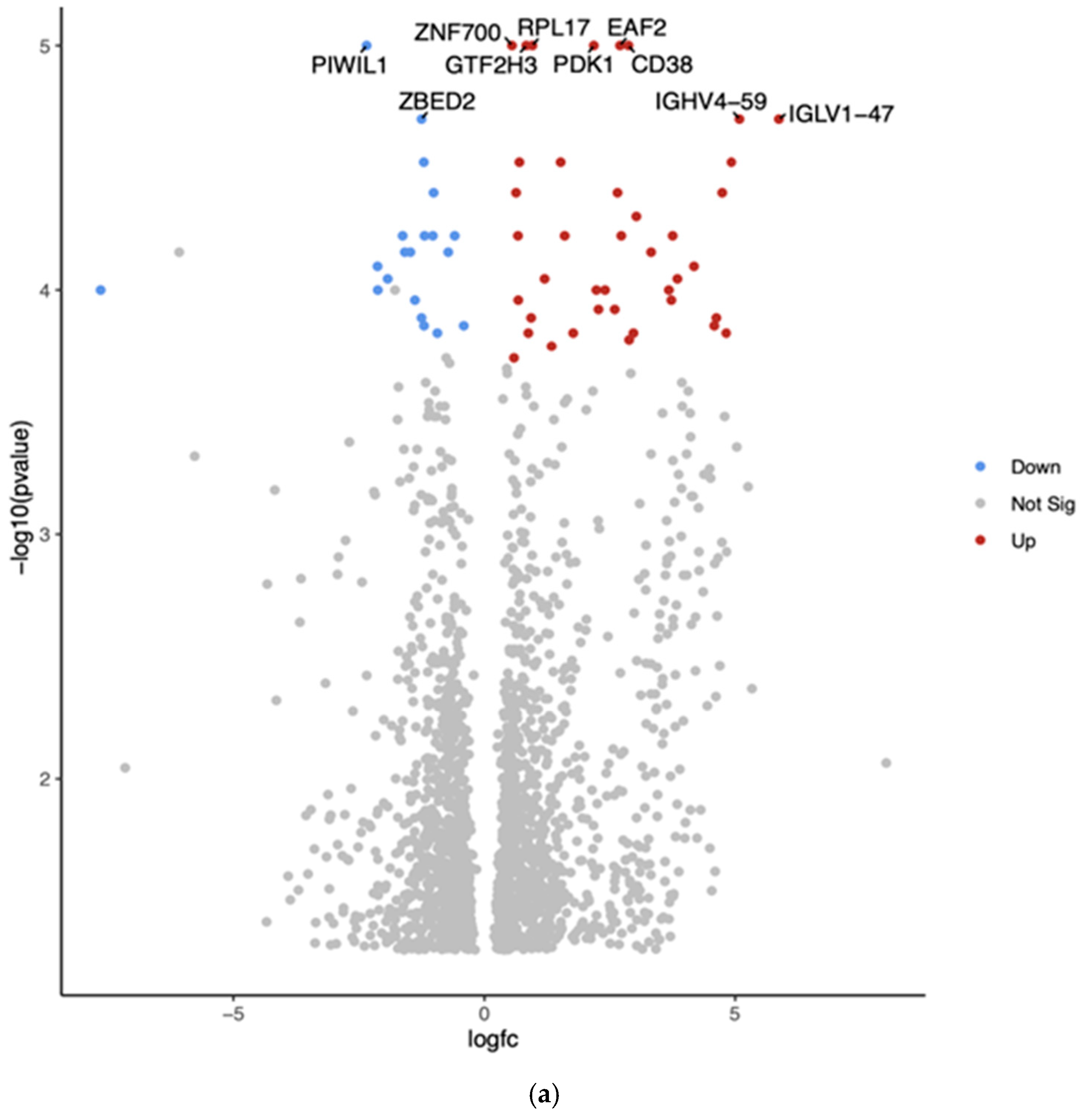
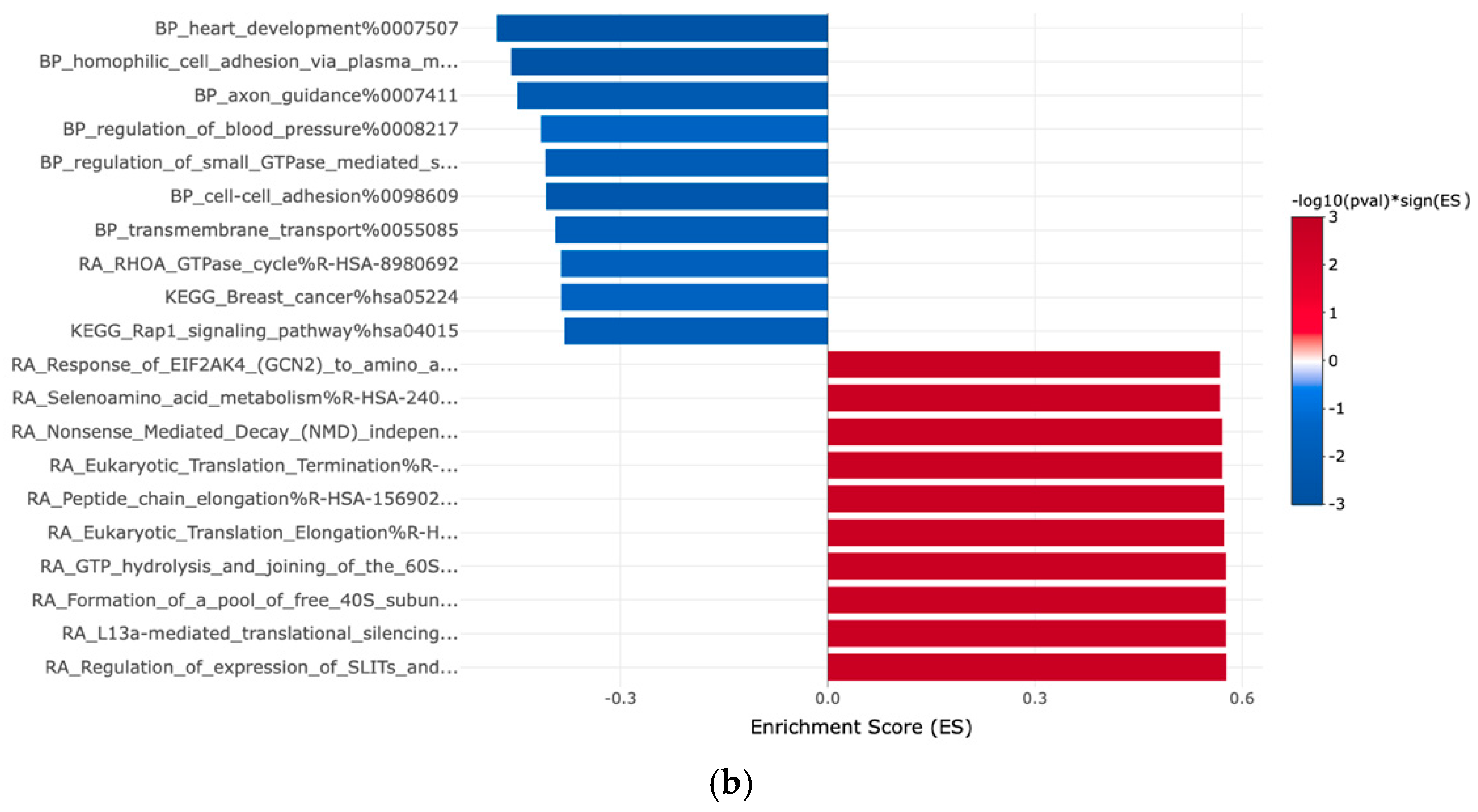
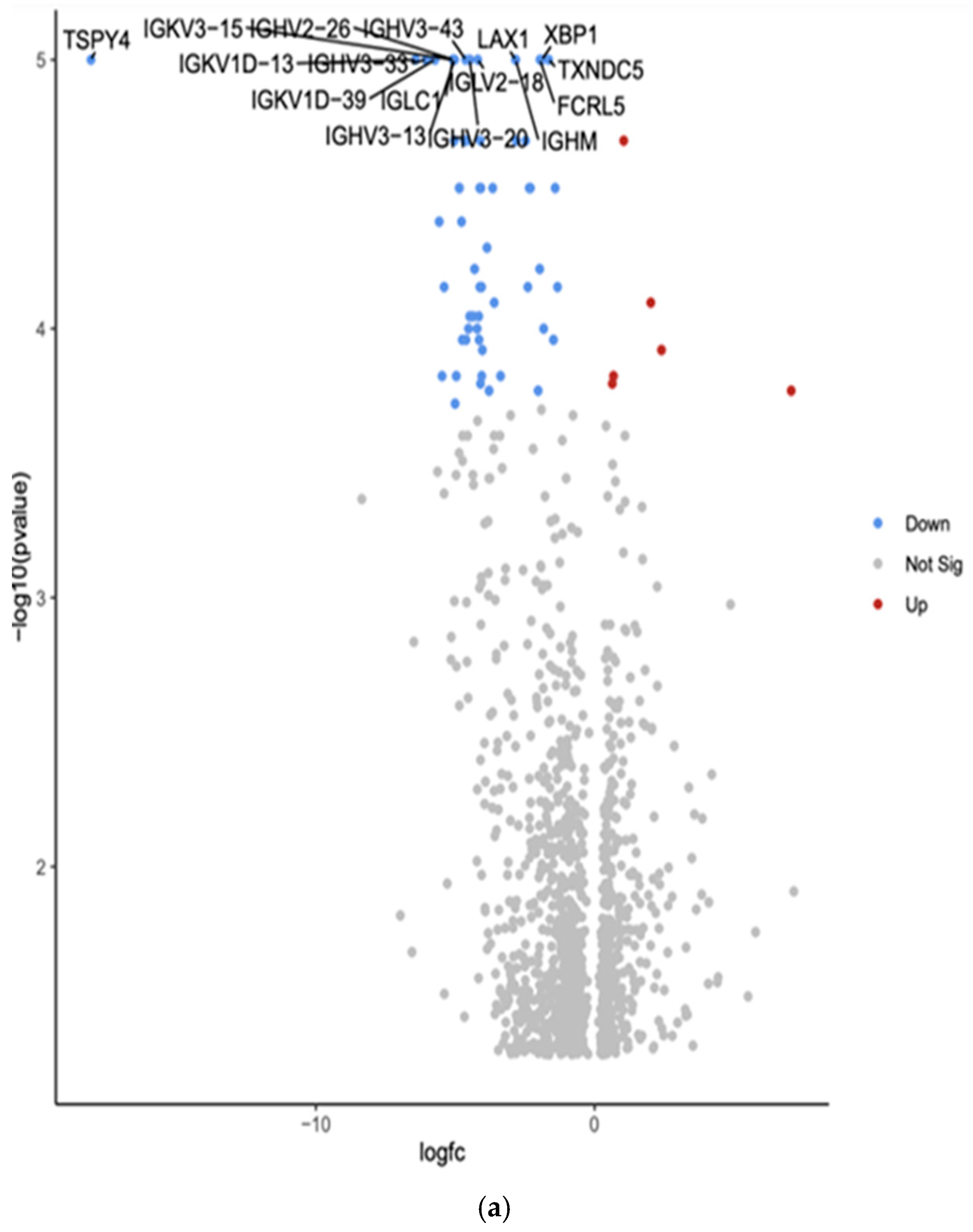
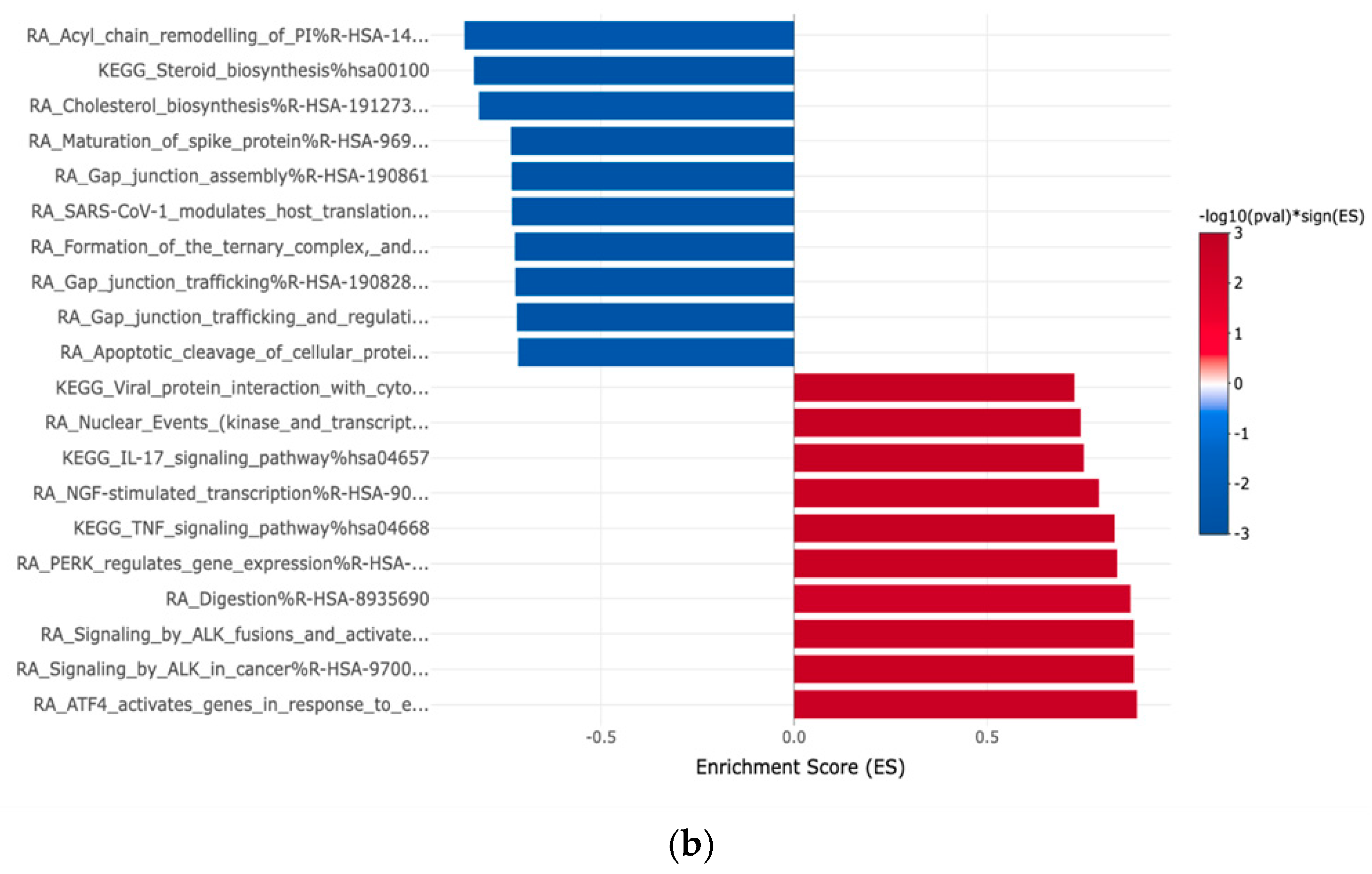
3.2. Gene Expression Profiles: Comparison Between Hispanic and African American Papillary Thyroid Cancer Tissue Samples
3.3. Gene Expression Profiles: Comparison Between White and African American Papillary Thyroid Cancer Tissue Samples
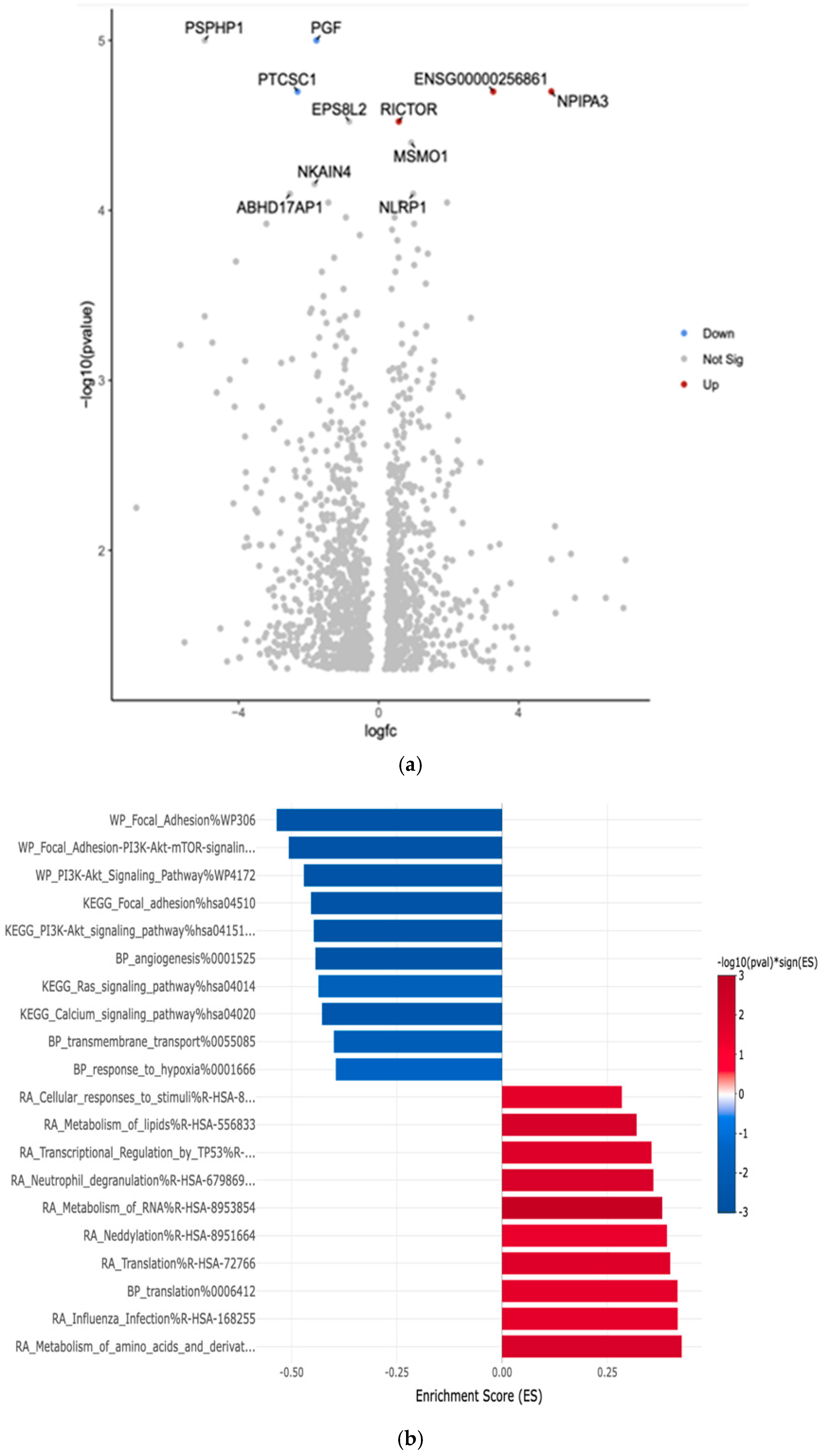
3.4. Gene Expression Profiles: Comparison Between White and Hispanic Papillary Thyroid Cancer Tissue Samples
3.5. Correlation of Differentially Expressed Genes from the LLU Cohort to the TCGA Cohort in African Americans, Whites, and Asians
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP6V0E2 | Component of vacuolar ATPase |
| BAM | Binary Alignment/Map |
| BRAF | B-Rapidly Accelerated Fibrosarcoma |
| DESeq2 | Differential Expression analysis for Sequence count data version 2 |
| EAF2 | ELL-associated factor 2 |
| EMT | Epithelial-to-mesenchymal transition |
| eVITTA | electronic Visualization of Text-based Tumor Annotations |
| FastQC | Fast Quality Control |
| FCRL5 | Fc receptor-like 5 |
| FDR | False Discovery Rate |
| FFPE | Formalin-fixed paraffin-embedded |
| GHV4-59 | Immunoglobulin heavy variable 4-59 |
| GSEA | Gene Set Enrichment Analysis |
| GTF2H3 | General transcription factor IIH subunit 3 |
| HYOU1 | Hypoxia upregulated protein 1 |
| IGHM | Immunoglobulin heavy constant mu |
| IGHV2-26 | Immunoglobulin heavy variable 2-26 |
| IGHV3-13 | Immunoglobulin heavy variable 3-13 |
| IGHV3-20 | Immunoglobulin heavy variable 3-20 |
| IGHV3-33 | Immunoglobulin heavy variable 3-33 |
| IGHV3-43 | Immunoglobulin heavy variable 3-43 |
| IGKV1D-13 | Immunoglobulin kappa variable 1D-13 |
| IGKV1D-39 | Immunoglobulin kappa variable 1D-39 |
| IGKV3-15 | Immunoglobulin kappa variable 3-15 |
| IGLC1 | Immunoglobulin lambda constant 1 |
| IGLV1-47 | Immunoglobulin lambda variable 1-47 |
| IGLV2-18 | Immunoglobulin lambda variable 2-18 |
| JAK-STAT | Janus Kinase-Signal Transducer and Activator of Transcription |
| KCNQ1 | Potassium voltage-gated channel |
| LAX1 | Lymphocyte transmembrane adapter 1 |
| MAPK | Mitogen-Activated Protein Kinase |
| MIR205HG | MicroRNA 205 Host Gene |
| MOXD1 | Monooxygenase DBH-like 1 |
| NGS | Next Generation Sequencing |
| NMNAT2 | Nicotinamide Mononucleotide Adenylyl Transferase 2 |
| NPIPA3 | Nuclear Pore Complex Interacting Protein Family Member A3 |
| PDK1 | Pyruvate dehydrogenase kinase 1 |
| PGF | Placental Growth Factor |
| PIWIL1 | P-element Induced Wimpy Testis Protein 1 |
| PPFIA2 | Protein tyrosine phosphatase, receptor type, f polypeptide, interacting protein alpha 2 |
| PTC | Papillary thyroid cancer |
| PTCSC1 | Papillary Thyroid Carcinoma Susceptibility Candidate 1 |
| RAS | Rat Sarcoma Virus |
| RET | Rearranged during Transfection |
| RICTOR | Rapamycin-insensitive companion of mTOR |
| RPL17 | Ribosomal protein L17 |
| SLC16A9 | Solute carrier family 16 member 9 |
| SOC3 | Suppressor of Cytokine Signaling |
| TPM | Transcript per million |
| TSPY4 | Testis-specific protein Y-encoded 4 |
| TXNDC5 | Thioredoxin domain containing 5 |
| XBP1 | X-box binding protein 1 |
| ZBED2 | Zinc finger BED-type containing 2 |
| ZC2HC1A | Zinc finger C2HC-type |
| ZNF700 | Zinc finger protein |
References
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Davies, L.; Welch, H.G. Current thyroid cancer trends in the United States. JAMA Otolaryngol. Head. Neck Surg. 2014, 140, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Devesa, S.S.; Sosa, J.A.; Check, D.; Kitahara, C.M. Trends in Thyroid Cancer Incidence and Mortality in the United States, 1974–2013. JAMA 2017, 317, 1338–1348. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yip, L.; Stang, M.T.; Carty, S.E. Thyroid carcinoma: The surgeon’s perspective. Radiol. Clin. N. Am. 2011, 49, 463–471, vi. [Google Scholar] [CrossRef]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Xing, M.; Alzahrani, A.S.; Carson, K.A.; Viola, D.; Elisei, R.; Bendlova, B.; Yip, L.; Mian, C.; Vianello, F.; Tuttle, R.M.; et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA 2013, 309, 1493–1501. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nikiforov, Y.E. Role of molecular markers in thyroid nodule management: Then and now. Endocr. Pract. 2017, 23, 979–988, Erratum in: Endocr. Pract. 2017, 23, 1362. https://doi.org/10.4158/1934-2403-23.11.1362. [Google Scholar] [CrossRef] [PubMed]
- American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer; Cooper, D.S.; Doherty, G.M.; Haugen, B.R.; Kloos, R.T.; Lee, S.L.; Mandel, S.J.; Mazzaferri, E.L.; McIver, B.; Pacini, F.; et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009, 19, 1167–1214, Erratum in: Thyroid 2010, 20, 942; Erratum in Thyroid 2010, 20, 674–675. [Google Scholar] [CrossRef] [PubMed]
- Rood, K.; Begum, K.; Wang, H.; Wangworawat, Y.C.; Davis, R.; Yamauchi, C.R.; Perez, M.C.; Simental, A.A.; Laxa, R.T.; Wang, C.; et al. Differential Expression of Non-Coding RNA Signatures in Thyroid Cancer between Two Ethnic Groups. Curr. Oncol. 2021, 28, 3610–3628. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rood, K.; Yamauchi, C.R.; Sharma, U.; Laxa, R.T.; Robins, C.; Lanza, G.; Sánchez-Ruiz, K.; Khan, A.; Kim, H.S.; Shields, A.; et al. Regulatory and Interacting Partners of PDLIM7 in Thyroid Cancer. Curr. Oncol. 2023, 30, 10450–10462. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [PubMed]
- Mull, B.; Davis, R.; Munir, I.; Perez, M.C.; Simental, A.A.; Khan, S. Differential expression of Vitamin D binding protein in thyroid cancer health disparities. Oncotarget 2021, 12, 596–607. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, J.H.; Sun, S.S.; Li, N.; Lv, P.; Xie, S.Y.; Wang, P.Y. MiR-205 as a promising biomarker in the diagnosis and prognosis of lung cancer. Oncotarget 2017, 8, 91938–91949. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Griess, B.; Tom, E.; Domann, F.; Teoh-Fitzgerald, M. Extracellular superoxide dismutase and its role in cancer. Free Radic. Biol. Med. 2017, 112, 464–479. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gallegos-Arreola, M.P.; Garibaldi-Ríos, A.F.; Magaña-Torres, M.T.; Figuera, L.E.; Gómez-Meda, B.C.; Zúñiga-González, G.M.; Puebla-Pérez, A.M.; Carrillo-Dávila, I.A.; Rosales-Reynoso, M.A.; Dávalos-Rodríguez, I.P.; et al. Association Between the rs13306703 and rs8192288 Variants of the SOD3 Gene and Breast Cancer and an In Silico Analysis of the Variants’ Impact. Diseases 2024, 12, 276. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeng, M.-S. Noncoding RNAs in Cancer Diagnosis. In The Long and Short Non-coding RNAs in Cancer Biology; Song, E., Ed.; Springer Singapore: Singapore, 2016; pp. 391–427. [Google Scholar]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Javed, Z.; Ahmed Shah, F.; Rajabi, S.; Raza, Q.; Iqbal, Z.; Ullah, M.; Ahmad, T.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; et al. LncRNAs as Potential Therapeutic Targets in Thyroid Cancer. Asian Pac. J. Cancer Prev. 2020, 21, 281–287. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, N.; Ko, Y.; Shin, Y.; Park, J.; Lee, A.J.; Kim, K.W.; Pyo, J. Comprehensive Analysis for Anti-Cancer Target-Indication Prioritization of Placental Growth Factor Inhibitor (PGF) by Use of Omics and Patient Survival Data. Biology 2023, 12, 970. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tabatabaeian, H.; Peiling Yang, S.; Tay, Y. Non-Coding RNAs: Uncharted Mediators of Thyroid Cancer Pathogenesis. Cancers 2020, 12, 3264. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Fold Change | Function | Relation to Cancer |
|---|---|---|---|
| MIR205HG | Downregulated −3.05 (***, p = 1 × 10−5) | Long non-coding RNA that hosts microRNA-205; involved in epithelial-to-mesenchymal transition (EMT) | Associated with various cancers, including breast and prostate cancer; can act as both a tumor suppressor and an oncogene |
| SOD3 | Downregulated −2.05 (*** p = 1 × 10−5) | SOD3 has anti-oxidative, anti-inflammatory, anti-apoptotic, and growth-promoting characteristics in cardiovascular and cancer models | Tumor suppression, increased expression in a benign thyroid tumor goiter model and gradually downregulated in cell lines that model advanced papillary and anaplastic thyroid cancers |
| HYOU1 | Downregulated −1.16 (***, p = 1 × 10−5) | Hypoxia upregulated protein 1; involved in protein folding and secretion under stress conditions | Overexpression is associated with poor prognosis in some cancers; promotes tumor growth and metastasis |
| SLC16A9 | Downregulated −2.09 (***, p = 1 × 10−5) | Solute carrier family 16 member 9; monocarboxylate transporter | Involved in metabolic reprogramming in cancer cells; may contribute to tumor growth |
| KCNQ1 | Downregulated −0.75 (***, p = 1 × 10−5) | Potassium voltage-gated channel; involved in cardiac action potential | Acts as a tumor suppressor in some cancers; its downregulation may promote cancer progression |
| ATP6V0E2 | Downregulated −1.28 (***, p = 1 × 10−5) | Component of vacuolar ATPase; involved in cellular pH regulation | May contribute to cancer cell survival and metastasis by regulating pH |
| ZC2HC1A | Upregulated 0.68 (***, p = 1 × 10−5) | Zinc finger C2HC-type containing 1A; function not well characterized | Limited information on its role in cancer |
| MOXD1 | Upregulated 1.55 (***, p = 1 × 10−5) | Monooxygenase DBH-like 1; involved in catecholamine metabolism | Limited information on its role in cancer |
| PPFIA2 | Upregulated 1.09 (***, p = 1 × 10−5) | Protein tyrosine phosphatase, receptor type, f polypeptide, interacting protein alpha 2; involved in cell adhesion | May play a role in cancer cell invasion and metastasis |
| NMNAT2 | Downregulated −2.43 (*** p = 1 × 10−5) | Nicotinamide Mononucleotide Adenylyltransferase 2 (NMNAT2) is an enzyme that helps maintain axons and protects neurons; it is encoded by the NMNAT2 gene in humans | NMNAT2 exerts a cancer-promoting role in solid tumors, including colorectal cancer, lung cancer, ovarian cancer, and glioma, and is closely related to tumor occurrence and development |
| Gene | Fold Change | Function | Relation to Cancer |
|---|---|---|---|
| PIWIL1 | Downregulated −2.35 (***, p = 1.00 × 10−5) | PIWI-like RNA-mediated gene-silencing protein | Potential tumor suppressor |
| ZNF700 | Upregulated 0.55 (***, p = 1.00 × 10−5) | Zinc finger protein | Limited information on its role in cancer |
| GTF2H3 | Upregulated 0.83 (***, p = 1.00 × 10−5) | General transcription factor IIH subunit 3 | May play a role in DNA repair and genomic stability |
| ZBED2 | Downregulated −1.25 (***, p = 2.00 × 10−5) | Zinc finger BED-type containing 2 | Limited information on its role in cancer |
| RPL17 | Upregulated 0.95 (***, p = 1.00 × 10−5) | Ribosomal protein L17 | Often dysregulated in cancer |
| EAF2 | Upregulated 2.70 (***, p = 1.00 × 10−5) | ELL-associated factor 2 | Potential tumor suppressor in some cancers |
| PDK1 | Upregulated 2.18 (***, p = 1.00 × 10−5) | Pyruvate dehydrogenase kinase 1 | Often upregulated in cancer; contributes to metabolic reprogramming |
| CD38 | Upregulated 2.87 (***, p = 1.00 × 10−5) | Cyclic ADP-ribose hydrolase | Plays various roles in different cancers |
| IGHV4-59 | Upregulated 5.09 (***, p = 2.00 × 10−5) | Immunoglobulin heavy variable 4-59 | May be involved in immune responses to cancer |
| IGLV1-47 | Upregulated 5.87 (***, p = 2.00 × 10−5) | Immunoglobulin lambda variable 1-47 | May be involved in immune responses to cancer |
| Gene | Fold Change | Function | Relation to Cancer |
|---|---|---|---|
| PGF | Downregulated −1.78 (***, p = 1.00 × 10−5) | Placental Growth Factor | Often upregulated in various cancers; promotes tumor angiogenesis and metastasis |
| PTCSC1 | Downregulated −2.32 (***, p = 2.00 × 10−5) | Papillary Thyroid Carcinoma Susceptibility Candidate 1 | Associated with increased risk of papillary thyroid cancer; potential tumor suppressor |
| RICTOR | Upregulated 0.58 (***, p = 3.00 × 10−5) | Rapamycin-insensitive companion of mTOR | Often dysregulated in cancer; involved in cell growth, survival, and metabolism |
| NPIPA3 | Upregulated 4.93 (***, p = 2.00 × 10−5) | Nuclear Pore Complex Interacting Protein Family Member A3 | Limited information on its direct role in cancer |
| Gene | Fold Change | Function | Relation to Cancer |
|---|---|---|---|
| TSPY4 | Downregulated −18.07 (***, p = 1.00 × 10−5) | Testis-specific protein Y-encoded 4; involved in cell proliferation and differentiation | May be involved in testicular cancer |
| IGKV3-15 | Downregulated −5.09 (***, p = 1.00 × 10−5) | Immunoglobulin kappa variable 3-15; part of antibody light chains | May be involved in immune responses to cancer |
| IGHV3-43 | Downregulated −4.63 (***, p = 1.00 × 10−5) | Immunoglobulin heavy variable 3-43; part of antibody heavy chains | May be involved in immune responses to cancer |
| LAX1 | Downregulated −2.84 (***, p = 1.00 × 10−5) | Lymphocyte transmembrane adaptor 1; involved in T cell signaling | May play a role in immune responses to cancer; potential tumor suppressor in some cancers |
| XBP1 | Downregulated −1.89 (***, p = 1.00 × 10−5) | X-box binding protein 1; transcription factor involved in unfolded protein response | Often dysregulated in cancer; plays a role in tumor cell survival and drug resistance |
| IGKV1D-13 | Downregulated −4.54 (***, p = 1.00 × 10−5) | Immunoglobulin kappa variable 1D-13; part of antibody light chains | May be involved in immune responses to cancer |
| IGHV3-33 | Downregulated −6.04 (***, p = 1.00 × 10−5) | Immunoglobulin heavy variable 3-33; part of antibody heavy chains | May be involved in immune responses to cancer |
| IGLV2-18 | Downregulated −4.19 (***, p = 1.00 × 10−5) | Immunoglobulin lambda variable 2-18; part of antibody light chains | May be involved in immune responses to cancer |
| TXNDC5 | Downregulated −1.65 (***, p = 1.00 × 10−5) | Thioredoxin domain containing 5; involved in protein folding and redox regulation | Often upregulated in various cancers, may promote tumor growth and metastasis |
| IGKV1D-39 | Downregulated −5.73 (***, p = 1.00 × 10−5) | Immunoglobulin kappa variable 1D-39; part of antibody light chains | May be involved in immune responses to cancer |
| IGLC1 | Downregulated −5.09 (***, p = 1.00 × 10−5) | Immunoglobulin lambda constant 1; part of antibody light chains | May be involved in immune responses to cancer |
| FCRL5 | Downregulated −1.96 (***, p = 1.00 × 10−5) | Fc receptor-like 5; involved in B cell activation and differentiation | May play a role in B cell malignancies; potential therapeutic target in some cancers |
| IGHV3-13 | Downregulated −5.02 (***, p = 1.00 × 10−5) | Immunoglobulin heavy variable 3-13; part of antibody heavy chains | May be involved in immune responses to cancer |
| IGHV3-20 | Downregulated −4.48 (***, p = 1.00 × 10−5) | Immunoglobulin heavy variable 3-20; part of antibody heavy chains | May be involved in immune responses to cancer |
| IGHM | Downregulated −2.83 (***, p = 1.00 × 10−5) | Immunoglobulin heavy constant mu; part of IgM antibodies | May be involved in immune responses to cancer; altered expression in some B cell malignancies |
| RNA-Seq | African American (n = 27) | White (n = 334) | Asian (n = 51) | Survival |
|---|---|---|---|---|
| MIR205HG | - | ⬆ *** p = 0.000006 | ⬆ ** p = 0.004 | No Significance |
| SOD3 | ⬇ *** p = 0.000098 | ⬇ *** p = 0.000000000001 | ⬇ *** p = 0.000000000001 | No Significance |
| HYOU1 | ⬇ *** p = 0.00002 | ⬇ *** p = 0.000000000001 | ⬇ *** p = 0.000000000001 | ** p = 0.0039 |
| SLC16A9 | ⬆ *** p = 0.0001 | ⬆ * p = 0.03 | ⬆ * p = 0.02 | No Significance |
| KCNQ1 | ⬇ *** p = 0.000000000009 | ⬇ *** p = 0.000000000001 | ⬇ *** p = 0.000000000001 | No Significance |
| ATP6V0E2 | ⬇ * p = 0.01 | ⬇ *** p = 0.000003 | ⬇ *** p = 0.0000002 | No Significance |
| PPFIA2 | ⬇ ** p = 0.006 | ⬇ *** p = 0.000001 | ⬇ *** p = 0.00002 | * p = 0.036 |
| PIWIL1 | ⬆ * p = 0.02 | ⬆ *** p = 0.000000000001 | ⬆ *** p = 0.0009 | No Significance |
| ZNF700 | ⬇ ** p = 0.009 | ⬇ *** p = 0.000001 | ⬇ * p = 0.03 | No Significance |
| GTF2H3 | ⬇ * p = 0.04 | ⬇ *** p = 0.000002 | ⬇ *** p = 0.00003 | No Significance |
| ZBED2 | ⬇ ** p = 0.003 | ⬇ *** p = 0.0003 | ⬇ * p = 0.01 | No Significance |
| RPL17 | ⬇ ** p = 0.001 | ⬇ *** p = 0.00000001 | ⬇ *** p = 0.00003 | No Significance |
| EAF2 | ⬇ ** p = 0.0009 | ⬇ ** p = 0.004 | ⬇ * p = 0.01 | No Significance |
| PDK1 | ⬇ * p = 0.03 | - | - | * p = 0.026 |
| CD38 | ⬇ * p = 0.01 | ⬇ * p = 0.01 | - | No Significance |
| PGF | ⬇ ** p = 0.003 | ⬇ *** p = 0.0001 | ⬇ *** p = 0.0000008 | No Significance |
| PTCSC1 | ⬇ * p = 0.01 | ⬇ *** p = 0.000001 | ⬇ ** p = 0.009 | No Significance |
| RICTOR | ⬇ *** p = 0.000003 | ⬇ *** p = 0.000000000001 | ⬇ *** p = 0.000000001 | No Significance |
| LAX1 | ⬇ * p = 0.01 | ⬇ * p = 0.04 | - | No Significance |
| XBP1 | ⬇ *** p = 0.000000000009 | ⬇ *** p = 0.000000000001 | ⬇ *** p = 0.000000000005 | No Significance |
| TXNDC5 | ⬇ ** p = 0.005 | ⬇ * p = 0.02 | - | No Significance |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barseghyan, L.; Chan, S.; Yamauchi, C.R.; Shields, A.; Perez, M.C.; Simental, A.A.; Khan, S. Unraveling Racial Disparities in Papillary Thyroid Cancer: A Comparative Bulk RNA-Sequencing Gene Expression Analysis. Curr. Oncol. 2025, 32, 315. https://doi.org/10.3390/curroncol32060315
Barseghyan L, Chan S, Yamauchi CR, Shields A, Perez MC, Simental AA, Khan S. Unraveling Racial Disparities in Papillary Thyroid Cancer: A Comparative Bulk RNA-Sequencing Gene Expression Analysis. Current Oncology. 2025; 32(6):315. https://doi.org/10.3390/curroncol32060315
Chicago/Turabian StyleBarseghyan, Luiza, Samuel Chan, Celina R. Yamauchi, Andrea Shields, Mia C. Perez, Alfred A. Simental, and Salma Khan. 2025. "Unraveling Racial Disparities in Papillary Thyroid Cancer: A Comparative Bulk RNA-Sequencing Gene Expression Analysis" Current Oncology 32, no. 6: 315. https://doi.org/10.3390/curroncol32060315
APA StyleBarseghyan, L., Chan, S., Yamauchi, C. R., Shields, A., Perez, M. C., Simental, A. A., & Khan, S. (2025). Unraveling Racial Disparities in Papillary Thyroid Cancer: A Comparative Bulk RNA-Sequencing Gene Expression Analysis. Current Oncology, 32(6), 315. https://doi.org/10.3390/curroncol32060315