Summary
There are multiple lines of evidence of a strong link between obstructive sleep apnoea and heart failure. First, obstructive sleep apnoea is associated with cardiovascular risk factors (hypertension and diabetes) and cardiovascular diseases (coronary artery disease, atrial fibrillation) which predispose to the development of heart failure. Second, obstructive sleep apnoea per se is associated with changes in cardiac structure and function. Third, the presence of obstructive sleep apnoea is related to an increased incidence of heart failure. Fourth, in patients with established heart failure with reduced ejection fraction (HFrEF), the presence of obstructive sleep apnoea is associated with worse outcomes. And fifth, mechanistic proof-of-concept studies have shown that treatment with continuous positive airway pressure (CPAP) not only successfully treats the obstructive sleep apnoea but also has the potential to reverse or at least to attenuate cardiac dysfunction in subjects with obstructive sleep apnoea without heart failure, as well as in patients with HFrEF and obstructive sleep apnoea. However, there are no randomised studies showing that treatment of obstructive sleep apnoea with CPAP improves clinical outcomes in patients with heart failure. In the present review, we summarise the current knowledge on the role of obstructive sleep apnoea as a risk factor predisposing to the development of heart failure, its role in patients with heart failure, the effects of CPAP on cardiac function in patients with and without heart failure, and the potential role of obstructive sleep apnoea as a disease modifier and therapeutic target in patients with heart failure.
Introduction
Obstructive sleep apnoea (OSA) is a sleep-related breathing disorder that, by recurrent collapse of the upper airway during sleep, causes intermittent airway obstruction with subsequent absence of airflow despite respiratory efforts of the diaphragm against the occluded pharynx. Thus, OSA is characterised by cycles of hypoxaemia followed by arousals, and thereby sleep fragmentation [1], which may result in poor sleep quality, increased daytime sleepiness and reduced quality of life [2]. Apart from the problem of daytime sleepiness and its potentially deleterious consequences, there is now a growing body of evidence suggesting an association between the presence and severity of OSA and cardiovascular diseases, in particular heart failure [1,3,4]. In this review, we provide a summary and critical discussion of the available evidence on the role of OSA as a risk factor predisposing to the development of cardiac dysfunction and heart failure, the importance of concomitant OSA in patients with heart failure, the effects of continuous positive airway pressure (CPAP) on cardiac function in patients with and without heart failure, and the potential role of OSA as a disease modifier and therapeutic target in patients with heart failure.
Definition and Prevalence of Obstructive Sleep Apnoea
The diagnosis of OSA is based on the apnoea-hypopnoea index (AHI)—the number of apnoeas and hypopnoeas per hour of sleep—which is assessed in a sleep study (polysomnography or polygraphy). An apnoea is defined as absence of airflow (reduction to less than 10% of baseline for ≥10 s), and a hypopnoea is defined as a reduction in airflow by ≥30% of baseline for ≥10 s and accompanied by a ≥3% decrease in oxygen saturation or an arousal [5]. The apnoea or hypopnoea is obstructive if any of the following criteria are fulfilled: (a) snoring during the event, (b) increased inspiratory flattening of the nasal pressure waveform, (c) associated paradoxical motion of the chest and abdominal respiratory inductance plethysmography excursions [5]. Otherwise the event is central. The differentiation between obstructive and central events is particularly important in heart failure patients as it has impact on therapeutic considerations, as discussed below. An AHI < 5 events per hour of sleep is normal. Mild, moderate and severe OSA are defined as AHI between 5 and 15 h−1, AHI between 15 and, which may result in poor sleep qualfined as AHI between 5 and 15 h−1, AHI between 15 and 30 h−1, and AHI >30 h−1. An OSA syndrome is present if OSA is associated with significant daytime sleepiness as assessed with the Epworth sleepiness scale [6,7]. Data from the Wisconsin Sleep Cohort Study published in 1993 suggested that at the age of 30 to 60 years, 9% of women and 24% of men had some form of sleep-disordered breathing (AHI ≥ 5 h−1), and 2% of women and 4% of men had OSA syndrome [8,9]. However, in a recent population-based study from Switzerland using contemporary recording technologies, a prevalence of 50% of moderate or severe OSA (AHI ≥ 15 h−1) in men and 23% in women was found, which suggests that (asymptomatic) OSA is very common [7]. Thus, the relationship of OSA with heart failure is of interest for the majority of sleep and heart failure specialists, and even from a public health point of view.
Pathophysiology of the Cardiovascular Effects of Obstructive Sleep Apnoea
The pathophysiology underlying the cardiovascular effects of OSA is complex and incompletely understood. However, it is very likely that intermittent hypoxia, hypercapnia, sleep fragmentation and intrathoracic pressure swings play an important role [3,4]. Three key mechanisms resulting from intermittent hypoxia/arousals are particularly relevant: sympathetic activation, oxidative stress and systemic inflammation [3,4,10]. Additional potentially important effects include massive intrathoracic pressure swings with generation of increased left ventricular afterload and increased right ventricular preload [11], and hypoxic pulmonary vasoconstriction with subsequently increased right ventricular afterload. Studies in healthy volunteers have demonstrated that simulation of obstructive apnoea by a Mueller manoeuvre (voluntary forced inspiration against a fixed resistance) leads to a reduction in left ventricular ejection fraction (LVEF) and strain [12], as well as a significant variation in left atrial volume [13] and, interestingly, to a reduction in right ventricular strain [12]. Notably, given that sleep restriction without OSA can induce adverse cardiovascular effects including left ventricular diastolic [14], left atrial and endothelial dysfunction [15] in healthy subjects, it is possible that sleep deprivation per se also contributes to the cardiovascular features associated with OSA.
Obstructive Sleep Apnoea, and Hypertension and Diabetes
The associations between OSA and the major cardiovascular risk factors hypertension and diabetes are now well established, although the causal relationships are less clear [1]. Several studies have clearly shown an association between the presence and severity of OSA and the prevalence of hypertension [1]. In the landmark study by Young et al. [16] in a cohort of 1060 subjects aged between 30 and 60 years, a linear rise in blood pressure with rising AHI was observed. Subjects with an AHI of 15 h−1 were 1.8 times more likely to have hypertension than those with an AHI of zero. Similarly, Heinzer et al. [7] found an independent association between high AHI, defined as the highest quartile (>20.6 h−1), and hypertension (odds ratio 1.6) in unselected subjects undergoing polysomnography (n = 2121; median AHI 6.9 h−1). These associations suggest, but do not prove, that OSA causes hypertension since there are many potentially confounding factors, in particular obesity. The findings on OSA and the incidence of hypertension are conflicting [17,18,19]. In an analysis of the Wisconsin Sleep Cohort Study among 709 participants without hypertension at baseline, those with AHI ≥15 h−1 were nearly three times more likely (odds ratio 2.89) to have hypertension (blood pressure >140/90 mm Hg or use of antihypertensive medication) at the 4-year follow-up than those with an AHI of zero [17]. Cano-Pumarega et al. [18] also found a higher risk of incident hypertension with increasing OSA severity in 1180 initially normotensive subjects after a follow-up of 7.5 years. However, after adjustment for confounders this relationship was no longer statistically significant. In contrast, Marin et al. [19] reported a higher incidence of hypertension compared with controls in subjects with OSA who were ineligible for continuous positive airway pressure (CPAP), declined CPAP, or were nonadherent to CPAP, and this was statistically significant after multivariate adjustment. It is generally accepted that, in patients with significant OSA, CPAP lowers blood pressure [20], but this effect is modest (weighted mean decrease in diurnal systolic and diastolic blood pressure of 2.6 and 2.0 mm Hg according to a meta-analysis [21]) and less than that of pharmacological antihypertensive therapy (valsartan) [22]. Importantly, the antihypertensive effect of CPAP depends on the hours of CPAP use and on several other patient characteristics. A trial has suggested that, in patients with resistant hypertension (3.8 antihypertensive drugs) and OSA (mean AHI 40 h−1), CPAP therapy may have an additional antihypertensive effect [23].
There is also a biologically plausible link between OSA and insulin resistance [24], and an association between the presence and severity of OSA and the prevalence [7,25] and incidence [25,26] of type 2 diabetes has been found in several studies. Data on the effect of CPAP on glycaemic control and other markers of the metabolic syndrome are very conflicting, however [1]. A trial evaluating the effect of CPAP (crossover CPAP vs. shamCPAP) in 86 patients with OSA (87% with the metabolic syndrome) revealed a reduction in glycosylated haemoglobin (by 0.2%) after 3 months of therapy [27]. However, in this study (which was later retracted [28]), the improvement in glycosylated haemoglobin was accompanied by a reduction in body mass index and visceral fat. In contrast, the majority of other trials found a weight gain in OSA patients after CPAP initiation [29]. Nonetheless, a recently published short-term (1 week) study in a small number (n = 19, 2:1 randomisation to CPAP or sham CPAP) of patients with type 2 diabetes and OSA (the majority with severe OSA) also revealed an improvement in glycaemic control after CPAP [30]. However, this is in contrast to a previous randomised sham CPAP-controlled study with a longer treatment duration (3 months) in patients with type 2 diabetes mellitus and newly diagnosed OSA, which showed no effect of CPAP on glycosylated haemoglobin and insulin resistance [31]. Thus, the role of CPAP for both the prevention and treatment of diabetes in patients with OSA has not yet been clarified.
Obstructive Sleep Apnoea and Global Cardiovascular Risk
Severe OSA per se is a risk marker for cardiovascular mortality and morbidity [32,33,34,35]. In their landmark long-term study (mean follow-up 10.1 years), Marin et al. [32] demonstrated that subjects with severe untreated OSA experienced significantly more fatal (adjusted hazard ratio 2.87) and nonfatal (hazard ratio 3.17) cardiovascular events than healthy controls, and that the event rate in these patients with untreated severe OSA was also higher than in simple snorers, patients with mild-to-moderate OSA, and CPAP-treated OSA patients [32]. In another study among nearly 1000 subjects aged ≥65 years, patients with severe OSA (AHI ≥ 30 h−1) had higher cardiovascular mortality (adjusted hazard ratio 2.25) than those with no OSA (AHI < 10 h−1), whereas outcomes in CPAP-treated OSA patients and controls did not differ [34]. Thus, the presence of severe OSA obviously indicates an increased cardiovascular risk, and good CPAP adherence (which may also be a surrogate for unmeasured confounding factors) seems to attenuate this risk. However, the mechanisms underlying the association between OSA and mortality (such as the occurrence of heart failure), on the one hand, and the beneficial impact of CPAP, on the other hand, remain to be defined.
Surrogate Markers of Autonomic Imbalance in Obstructive Sleep Apnoea Without Overt Cardiac Disease
The pathophysiological concept of sympathetic overactivity in OSA is supported by studies showing that surrogate markers of sympathetic tone, including muscle sympathetic nerve activity [36,37], heart rate variability [38] and heart rate recovery [39], are abnormal in OSA patients without overt cardiovascular disease compared with controls without OSA, and that CPAP improves these measures of autonomic dysfunction [40,41]. A detailed discussion of these findings is beyond the scope of this article, but can be found elsewhere [1]. These observations are particularly important with regards to the interaction between OSA and heart failure, since activation of the sympathetic nervous system is also a cardinal feature of the pathophysiology of heart failure.
The Cardiovascular System in OSA
Mechanistic studies in patients with OSA but without overt cardiovascular disease have revealed that OSA per se is associated with subtle abnormalities of cardiovascular structure and function including endothelial dysfunction [42], increased carotid intima-media thickness [43], coronary artery calcification [44,45], aortic dilation [46], left ventricular remodelling/hypertrophy [47,48], left ventricular systolic [49,50,51] and diastolic [49,52,53] dysfunction, left atrial dilation [49,53], right ventricular dilation and dysfunction [54,55] and pulmonary hypertension [56]. Key findings from these studies are summarised in Table 1. Figure 1 is a schematic representation of the cardiac features of OSA. Furthermore, OSA has been shown to attenuate or reverse several of these features (Table 1) [42,49,50,54,57,58]. In addition, some but not all studies have revealed an association between the presence and severity of OSA and both cardiac troponin [59,60] and natriuretic peptides [61], which are established markers of cardiovascular stress. A detailed summary of studies on biomarkers of cardiovascular stress in OSA is beyond the scope of this article, but has recently been published elsewhere [62]. Some of the abovementioned studies had limitations, in particular the difficulties in separating the effects of OSA, obesity and hypertension, and the lack of control groups in several studies assessing the effect of CPAP on cardiac function. Nonetheless, these studies led to the concept that OSA per se is associated with changes in cardiac structure and function that may predispose to the development of heart failure and of disease progression in patients with established heart failure.

Table 1.
Cardiovascular effects associated with obstructive sleep apnea without overt cardiac disease and effects of continuous positive airway pressure.
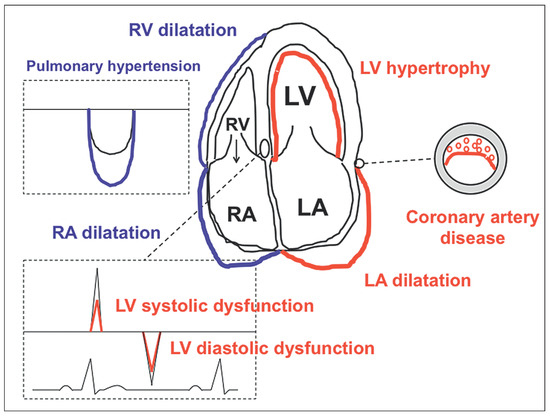
Figure 1.
Schematic representation of the OSA-related effects on cardiac structure and function. Red: effects on the left heart; blue: effects on the pulmonary circulation and right heart. For details please see text and Table 1. LA = left atrium/atrial; LV = left ventricle/ventricular; RA = right atrium/atrial; RV = right ventricle/ventricular Reproduced from Maeder MT et al. [1] with permission from Dove press.
Cardiac Diseases Predisposing to Heart Failure and Obstructive Sleep Apnoea
Epidemiological studies and cohort studies have also established a strong link between OSA and cardiac diseases that finally can lead to the syndrome of heart failure, in particular coronary artery disease (CAD) and atrial fibrillation.
Coronary Artery Disease
The prevalence of CAD is higher in patients with OSA than in those without OSA [63,64]. In the Sleep Heart Health Study including 1927 men (median AHI of 6.2 h−1) and 2495 women (median AHI of 2.7 h−1) aged ≥40 years and without known CAD, the incidence of CAD defined as a coronary event (myocardial infarction, revascularisation, or death due to CAD) was higher in men <70 years with OSA than in those without OSA (adjusted hazard ratio 1.68 for AHI ≥30 h−1 compared with AHI <5 h−1) [65]. However, such an association was not found in older men or women [65]. In patients with established CAD, OSA is common and seems to have an unfavourable prognostic impact [66,67,68], and CPAP seems to have the potential to attenuate these OSA-associated adverse effects [69]. In the recently published Sleep and Stent Study, the presence of OSA (AHI ≥15 h−1, present in 45% of all patients) was a predictor of the composite endpoint of cardiovascular mortality, nonfatal myocardial infarction, nonfatal stroke, and unplanned revascularisation, independent of age, sex, body mass index, diabetes mellitus, and hypertension (hazard ratio 1.57) after a median follow-up of 1.9 years [68]. An observational study revealed better outcomes (composite of cardiovascular death, acute coronary syndrome, coronary revascularisation, hospitalisation for heart failure) in a small number of patients with both CAD and OSA (AHI ≥15 h−1), if patients were treated for OSA (6/25 with endpoint; hazard ratio 0.24) compared with those not treated (17/29 with endpoint) after a median follow-up of 87 months [69]. However, in this study OSA treatment (CPAP or upper airway surgery) was not randomised but according to patient preference. Similarly, a lower risk of cardiac death (3% vs. 10%) after percutaneous coronary intervention was found in OSA patients (AHI ≥15 h−1) treated with CPAP (n = 175) compared with those without (n = 196) [67]. Very recently, the results of the Sleep Apnoea cardioVascular Endpoints (SAVE) study were reported. In this study, patients with moderate-to-severe OSA (definition based not on AHI but on oxygen desaturation index, ≥12 episodes per hour of desaturation by ≥4% was required) but without severe daytime sleepiness and with CAD or cerebrovascular disease (50% with CAD) were randomised to CPAP and usual care or usual care alone. after a mean follow-up of 3.7 years no effect was observed on the composite primary endpoint (death from cardiovascular causes, myocardial infarction, stroke, or hospitalisation for heart failure, unstable angina, and transient ischaemic attack). However, given the mean CPAP use of only 3.3 h per night (which is not considered to be sufficient), the results of this trial are hard to interpret. This is particularly relevant given that a recent, albeit significantly smaller trial, showed that patients with recently revascularised CAD and OSA (AHI ≥ 15 h−1) treated with CPAP for ≥4 h per night experienced fewer cardiovascular events that those who used CPAP <4 h per night or had no CPAP [70].
Atrial Fibrillation
Patients with OSA have both a substrate and triggers for atrial fibrillation. The substrate includes left ventricular hypertrophy, left ventricular diastolic dysfunction and left atrial dilatation (as discussed above), as well structural and electrical atrial remodelling [71]. Triggers include the sympathetic tone on the one hand, and exaggerated intrathoracic pressure changes with subsequent vagal activation on the other hand. Indeed, a higher prevalence [72] and incidence [73] of atrial fibrillation have been found in subjects with OSA compared with those without. However, given that there is also an association between obesity and incident atrial fibrillation, it is possible that the association between OSA and atrial fibrillation may be at least in part explained by the high burden of coexisting risk factors. The presence OSA is also linked with higher risk of recurrence of atrial fibrillation after electrical cardioversion [74] or catheter ablation [75,76], and epidemiological and cohort studies suggest that CPAP may reduce this risk of recurrent atrial fibrillation after cardioversion and pulmonary vein isolation [76,77]. Thus, OSA may be a factor initiating and maintaining atrial fibrillation in patients with OSA, and atrial fibrillation may be a mechanism by which OSA can lead to disease progression and development of heart failure in subjects with structural heart disease. The most recent guidelines on atrial fibrillation (AF) state that “obstructive sleep apnoea treatment should be optimized to reduce AF recurrence and AF treatment results” [78]. It should be noted, however, that there are no randomised trials showing the CPAP treatment in patients with OSA reduced the risk of atrial fibrillation, its recurrence or its complications.
Obstructive Sleep Apnoea and Heart Failure Epidemiology
OSA has been found to be associated with an increased prevalence and incidence of heart failure. In a crosssectional study (6424 subjects, median AHI 4.4 h−1), the prevalence of self-reported heart failure was more than doubled (adjusted odds ratio 2.38) in subjects in the highest AHI quartile compared with those in the lowest AHI quartile [63]. In the Sleep Heart Health Study, including 1927 men (median baseline AHI 6.2 h−1) and 2495 women (median baseline AHI 2.7 h−1) with age ≥40 years and no known heart failure at baseline, a higher risk of incident heart failure was found in men with severe OSA (AHI ≥30 h−1) compared with those without OSA (AHI <5 h−1; adjusted hazard ratio 1.58) [65]; this was not case in women [65]. In contrast, an analysis from the Atherosclerosis Risk In Communities and the Sleep Heart Health studies based on data from 752 men and 893 women without known cardiovascular disease and mean follow-up of 13.6 years revealed a significant association between OSA and incident heart failure or death only in women [79].
Sleep-disordered breathing is very common in patients with heart failure and reduced LVEF (HFrEF) as well as those with heart failure and preserved LVEF (HFpEF) [80]. Sleep-disordered breathing is also prevalent in patients with heart failure and mid-range LVEF, although detailed studies on patients with this novel entity are not yet available. In the entire LVEF spectrum, not only OSA but also central sleep apnoea are prevalent. As a general rule, central sleep apnoea is more common in those with more advanced heart failure [80]. Notably, obstructive and central sleep apnoea can coexist in the same patient.
Pathophysiological Concept of Sleep-Disordered Breathing in Heart Failure
When discussing the role of sleep-disordered breathing in heart failure patients, it is of utmost importance to differentiate between OSA and central sleep apnoea, although both forms of sleep-disordered breathing can occur in the same patient. It is though that OSA is either a coexisting entity, which may have contributed to the occurrence of heart failure as discussed above, and which may adversely influence the clinical course of heart failure as discussed later, or that OSA occurs or worsens due to a so-called nocturnal rostral fluid shift: swelling of the neck due to nocturnal fluid redistribution in patients with oedema [81,82]. In the following paragraphs, the role of OSA is discussed as a disease modifier and potential therapeutic target in heart failure. Central sleep apnoea is not the topic of this review, but is briefly discussed to put the data on OSA into context. Central sleep apnoea is most likely an epiphenomenon of advanced heart failure and a marker of the severity of heart failure. For example, the severity of central sleep apnoea expressed as AHI is correlated with higher B-type natriuretic peptide levels and higher pulmonary capillary wedge pressure in HFrEF. Although CPAP has been shown to improve AHI, minimum oxygen saturation during sleep and LVEF in patients with HFrEF and central sleep apnoea, CPAP did not improve clinical outcomes in a randomised trial [83]. Based on the post-hoc observation that patients with suppression of central sleep apnoea by CPAP had better outcomes than both those without this suppression and controls [84], further trials were designed. However, the SERVE-HF trial testing adaptive servoventilation versus no ventilation in HFrEF patients resulted in increased mortality in the device-treated patients [85], and therefore adaptive servoventilation is now contraindicated in HFrEF [86].
Heart Failure with Reduced Ejection Fraction and Obstructive Seep Apnoea
Chronic HFrEF is characterised by activation of the sympathetic nervous system, the severity of which is related to prognosis [87]. Antagonism of sympathetic overactivity by beta-blocker therapy is a mainstay of modern HFrEF therapy [86]. Patients with HFrEF and concomitant OSA have more pronounced sympathetic overactivity than HFrEF patients without OSA. Spaak et al. [88] have shown that in HFrEF patients (n = 60; mean LVEF 22%), those with sleep-disordered breathing (predominantly OSA, AHI ≥15 h−1, n = 43) had higher muscle sympathetic nerve activity than those without OSA (AHI <15 h−1, n = 17) although the difference was modest (58 vs. 50 bursts/min; p = 0.005). The same group of investigators has shown that CPAP has the potential to suppress this additional sympathetic overactivity. In a trial of 17 patients with HFrEF and OSA (AHI >20 h−1), patients treated with CPAP for 1 month (n = 9) experienced a significant reduction in muscle sympathetic nerve activity (from 58 to 48 bursts/min) compared with those not treated with CPAP (n = 8; 63 and 63 bursts/min) [89]. Other studies have shown an improvement in heart rate variability [90] and baroreflex sensitivity [91] with CPAP in HFrEF; that is, attenuation of sympathetic overactivity as assessed with measurements other than sympathetic muscle nerve activity. These findings led to the hypothesis that coexisting OSA may have an adverse prognostic impact in patients with HFrEF, and that CPAP may have the potential to influence the clinical course of HFrEF patients with OSA in a beneficial manner. Two small mechanistic studies evaluated the impact of CPAP therapy on LVEF in HFrEF patients with concomitant OSA [92,93]. Kaneko et al. [92] randomised 24 HFrEF patients (LVEF ≤45%) and OSA (AHI ≥20 h−1, at least 50% obstructive events) to CPAP in addition to optimal medical therapy or medical therapy alone for a study duration of 1 month. As expected, AHI was reduced in the CPAP group (average nightly use 6.2 h; AHI 37.1 at baseline and 8.3 h−1 at follow-up, p <0.001; no effect on central events) but not in the control group (AHI 45.2 vs. 44.7 h−1). Those undergoing CPAP therapy had significantly improved LVEF (from 25.0 to 30.8%; p <0.001), whereas no effect was observed in the control group (from 28.5 to 30%; p <0.001 for difference between changes) [92]. It should be noted that 20 of 22 (91%) patients were on an angiotensin converting enzyme inhibitor, 12 of 24 (50%) were on a beta-blocker, and that doses of these drugs and the use of mineralocorticoid receptor antagonists were not reported. Mansfield et al. [93] performed a similar study in a somewhat larger population (n = 55), but with a high drop-out rate: 27%; data from 40 patients available for final analysis. The study population had also better LVEF (LVEF ≤55% required for inclusion) and a longer treatment duration (3 months). Treatment with CPAP (average nightly use 5.6 h) resulted in a larger reduction in AHI in the CPAP group (n = 19; from 25.0 to 2.9 h−1) than in the control group (n = 21; from 26.6 to 18.2 h−1; p <0.001 for difference between changes between groups). As in the study by Kaneko et al. [92], a more favourable effect on LVEF was seen in the CPAP group (from 37.6 to 42.6%) compared with the control group (from 33.6 to 35.1%; p = 0.04 for difference between changes) [93]. In addition, a significant reduction in overnight urinary noradrenaline excretion and an improvement in quality of life were observed in the CPAP group compared with the control group [93]. There was no information on medical therapy in this study. These two studies have been criticised for not using a placebo-controlled design, with sham CPAP in the control group. In addition, from a 2016 perspective, medical therapy was not optimal, as in one study at least only 50% of patients were on a beta-blocker and the use of mineralocorticoid receptor antagonist was not established at the time.
Data on the impact of coexisting OSA on clinical outcomes in HFrEF are still relatively sparse. In an observational study in HFrEF patients (LVEF <45%, n = 164), a higher mortality was observed in patients with untreated moderate to severe OSA (mean AHI 33 h−1; n = 37) than in those with no/mild OSA (mean AHI 7 h−1, n = 113; 24 vs. 12%) [94]. Patients with moderate to severe OSA treated with CPAP (n = 14) tended to have lower mortality than those with untreated moderate to severe OSA (p = 0.07) [94]. However, numbers for this analysis were too small. A recent publication from a large cohort of patients with HFrEF (LVEF <45%) admitted with acute decompensation and undergoing polygraphy during the index hospitalisation revealed that not only central sleep apnea but also OSA was an independent predictor of mortality (hazard ratio 1.53) after a median follow-up of 3 years [95]. Notably, in contrast to central sleep apnoea there are no randomised trials of ventilation therapy for patients with HFrEF and OSA. Thus, although the role of OSA as a mediator of adverse effects and a therapeutic target in HFrEF is very plausible from several lines of evidence, there is a paucity of clinical data proving this concept, and there is currently no convincing evidence for the application of CPAP to improve prognosis in patients with HFrEF. It should be emphasised that optimal control of fluid balance by diuretics and fluid restriction is highly important, not only for heart failure management in general as it may also have an effect on OSA by the prevention of significant nocturnal rostral fluid shifts and therefore worsening of OSA. In addition, exercise training has been shown to have beneficial effects in patients with HFrEF [96] (and also HFpEF [97]) on the one hand, and in patients with OSA on the other hand [98]. Thus, exercise training is likely to be particularly beneficial in patients with both HFrEF and OSA. Treatment of OSA in patients with HFrEF seems to be safe, which is important for HFrEF patients who suffer from symptoms of OSA (i.e., daytime sleepiness) and thereby have an indication for CPAP. Treatment for patients with HFrEF is currently well defined and includes several drugs and devices that improve their mortality and morbidity [86]. Only a well-designed and adequately powered trial evaluating CPAP versus no CPAP on top of optimal medical and device therapy will have the potential to convince doctors and patients of the usefulness of CPAP for HFrEF with OSA without symptoms of daytime sleepiness.
Heart failure with preserved ejection fraction and obstructive sleep apnoea
In contrast to HFrEF, there is still no established therapy for patients with HFpEF [86,99]. In particular, studies evaluating the usefulness of drugs with proven benefit in HFrEF patients, including angiotensin converting enzyme inhibitors, angiotensin receptor blockers and mineralocorticoid receptor antagonists, have been neutral with respect to mortality [86]. Research over the last decade has revealed that, apart from diastolic dysfunction, the pathophysiology of HFpEF is characterised by a number of additional mechanisms including systolic left ventricular dysfunction, left atrial dysfunction, pulmonary hypertension and abnormal right ventricular/pulmonary artery coupling; the importance of these factors may vary between patients [100]. In addition, comorbidities also play an important role, and it has been proposed that there are different HFpEF phenotypes that require different treatment approaches [100,101]. Current guidelines therefore emphasise the importance of identifying the predominant cardiovascular features, as well as noncardiovascular comorbidities in HFpEF patients [86]. A subset of HFpEF patients is characterised by the combination of hypertension, obesity, diabetes and atrial fibrillation. Sleep-disordered breathing, predominantly OSA, is also common in HFpEF [102] and may play a role in this subset of patients. A recent study among patients with atrial fibrillation and mainly preserved LVEF undergoing cardiac magnetic resonance imaging prior to catheter ablation revealed that sleep apnoea (probably OSA in most cases) was an independent predictor of death and hospitalisation for heart failure [103]. However, although a role for OSA in the pathophysiology of HFpEF and as therapeutic target in these patients is plausible, prospective studies evaluating the impact of OSA and OSA severity on cardiac function and outcomes HFpEF, and role of CPAP on surrogate markers and the clinical endpoints in patients with HFpEF and concomitant OSA are lacking.
Summary and Conclusions
There is good evidence for a causal link between OSA and cardiovascular risk factors, cardiac dysfunction, and cardiac diseases predisposing to heart failure including CAD and atrial fibrillation. OSA is associated with the major risk factors hypertension and diabetes, but may also directly contribute to adverse changes in cardiac structure and function, the occurrence and/or progression of CAD, and the incidence of atrial fibrillation. By these mechanisms, OSA may contribute to HFrEF or HFpEF, probably depending on the predominant mechanism, i.e., HFrEF in a CAD-dominant phenotype, and HFpEF in a phenotype with predominant hypertensive heart disease/atrial fibrillation. This hypothesis is schematically depicted in Figure 2. In small prospective studies with surrogate endpoints, promising data on the effect of CPAP in HFrEF have been obtained. However, randomised studies with clearly defined study populations and clinical endpoints will be required to define to role of OSA, not only as a risk marker but also as a therapeutic target in heart failure patients. In the meantime, an indication for CPAP in heart failure and OSA exists only for patients with OSA-related symptoms of daytime sleepiness, where improvement of symptoms can be expected.
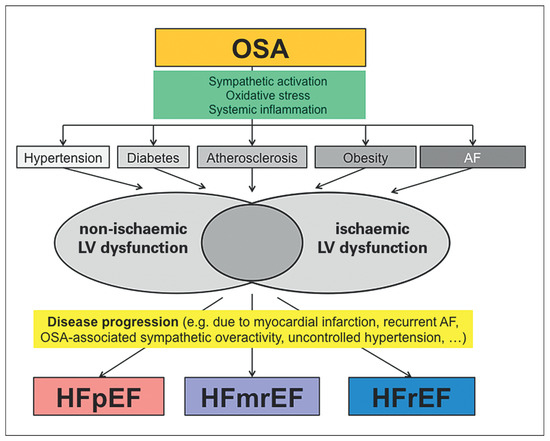
Figure 2.
Illustration of the possible contribution of obstructive sleep apnoea (OSA) in the pathogenesis of heart failure (HF). The figure emphasises that OSA may contribute to the occurrence of and/or may coexist with several risk factors for the development of left ventricular dysfunction through non-ischaemic and ischaemic mechanisms. In addition, OSA may among other factors contribute to the transition from asymptomatic structural heart disease with left ventricular dysfunction to the clinical syndrome of HF. OSA may be important for all HF phenotypes. For details please see text. AF = atrial fibrillation; HFpEF = heart failure with preserved ejection fraction; HFmrEF = heart failure with mid-range ejection fraction; HFrEF = heart failure with reduced ejection fraction.
Disclosure statement
No financial support and no other potential conflict of interest relevant to this article was reported.
References
The full list of references is included in the online article at www.cardiovascmed.ch

© 2017 by the author. Attribution - Non-Commercial - NoDerivatives 4.0.